
Abstract
With the extension of the egg-laying cycle, the rapid decline in egg quality at late laying period has aroused great concern in the poultry industry. Herein, we performed a genome-wide association study (GWAS) to identify genomic variations associated with egg quality, employing chicken 600 K high-density SNP arrays in a population of 1078 hens at 72 and 80 weeks of age. The results indicated that a genomic region spanning from 8.95 to 9.31 Mb (~0.36 Mb) on GGA13 was significantly associated with the albumen height (AH) and the haugh unit (HU), and the two most significant SNPs accounted for 3.12 ~ 5.75% of the phenotypic variance. Two promising genes, MSX2 and DRD1, were mapped to the narrow significant region, which was involved in embryonic and ovary development and found to be related to egg production, respectively. Moreover, three interesting genes, RHOA, SDF4 and TNFRSF4, identified from three significant loci, were considered to be candidate genes for egg shell colour. Findings in our study could provide worthy theoretical basis and technological support to improve late-stage egg quality for breeders.
Similar content being viewed by others

Introduction
Eggs are rich in protein, fatty acids, vitamins and minerals, etc. and are considered as an excellent source of animal protein. With the improvement of laying hens’ production performance, the laying age of commercial laying hens has been extended from the original 72 weeks to 80 weeks, and some breeding companies have even extended the laying cycle to 100 weeks, proposing the breeding programme “Breeding for 500 eggs in 100 weeks”1,2. However, the rapid decline in egg quality at the end of the laying cycle (such as the low commercialization rate along with enlarged egg size, declined egg shell quality, increased egg broken rate (%cracks), decreased albumen height, and shortened egg storage time) has badly hindered the achievement of this goal. Therefore, improving egg quality plays an important role in realizing this programme and extending the laying cycle.
Egg quality is a comprehensive concept that encompasses both internal and external quality and includes many aspects. External quality is composed of eggshell colour, egg shape index, eggshell thickness and eggshell strength, while internal quality refers to albumen height, egg yolk colour and haugh unit. All of these are quantitative traits. With the development of molecular genetics, many studies have been carried out to reveal the genetic determination for egg quality. Microsatellite markers were first employed, and many QTLs were reported through linkage analysis3,4,5,6, mostly based on F2 crossed populations. Currently, there are 430 QTLs reported to be associated with egg quality in the Animal QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/GG/index)7. Although many studies identified QTLs, they had wide confidence intervals for position and were rarely replicated8,9. A new era began with the subsequent advance in SNP chip and sequencing technology, and the genome wide association study (GWAS) has become one of the most effective methods to detect genetic variations in livestock. Liu et al. carried out the first GWAS with the Illumina 60 K SNP array10 to uncover the genetic associations with egg quality traits in chickens11. Wolc et al. and Sun et al. subsequently reported many genes related to egg quality that were identified by genome-wide association studies12,13. Despite all this, the genetic improvement of egg quality is slow because of relatively low heritabilities and intensively artificial selection on egg production.
To our knowledge, little research reported the genetic analysis for egg quality in the late laying period of chickens. In the present study, we employed the commercial chicken 600 K SNP chip to detect the genetic variations associated with egg quality in a population of 1078 hens at 72 and 80 weeks of age by genome-wide association analysis (GWAS) to provide a theoretical basis and technological support for improving late-stage egg quality.
Results
Phenotypic statistics and estimation of genetic parameter
The descriptive statistics for eggshell colour (ESC), egg shape index (ESI), eggshell thickness (EST), eggshell strength (ESS), albumen height (AH), yolk colour (YC) and haugh unit (HU) at 2 age points were presented in Table 1. With the extension of the laying period, the phenotypic values of ESS, AH and HU at 80 weeks of age decreased compared to the values at 72 weeks of age. Both eggshell strength and albumen height at the two points had higher phenotypic variation (19~30%) than the other traits. The pedigree-based hereditability were high for ESC, ESI, AH and HU (0.32~0.46) and moderate for EST, ESS and YC (0.14~0.28).
The estimates of SNP-based heritability and genetic or phenotypic correlations among egg quality traits at the two points are shown in Table 2 and Supplementary Table S1 (at 80 weeks of age). The estimates of SNP-based heritability were lower than those for pedigree-based heritability for all traits except EST (0.20 vs 0.14). Regardless of genetic relation or phenotypic correlation, HU has a strong correlation with AH (0.97 ± 0.02, 0.94), while ESC has very weak correlation with the other traits.
Genome-wide association study (GWAS)
The analysis of the population structure was shown in Fig. 1. As shown in the 3D plot, the samples had a slight population stratification. Therefore, we treated the first five principal components as covariates and included them in the linear mixed model of GWAS as fixed effects to adjust for the population structure effects.

Principal component plot (3D) of chickens using SNP markers. Purple points represent different individuals.
The Manhattan and Quantile-Quantile (QQ) plots of seventeen separate GWASs using the univariate model or the bivariate model for egg quality traits are shown in Fig. 2 and Supplementary Fig. S1. The univariate analyses revealed 99 and 86 significant SNPs (Supplementary Table S2) associated with AH and HU (Fig. 2), respectively. Combined with the bivariate model analyses, all these significant SNPs were identified as being located in an ~0.36 Mb region spanning from 8.95 to 9.31 Mb on GGA13 because of the strong genetic correlations. We calculated the genomic control inflation factors (λ) of AH and HU, which ideally equal 1. In fact, they were slightly greater than 1 (1.01, 1.03), which reflected the slight population stratification. These results were also consistent with the previous principal component analyses (PCA). Linkage disequilibrium (LD) analysis uncovered SNPs in GGA13 from 8.95 to 9.31 Mb that showed strong LD (Fig. 3B, Supplementary Fig. S2). To find the independent SNPs, stepwise conditional analyses were carried out. The locus rs315953420 significantly associated with AH and HU at the two periods was subsequently added to the model to examine the independent associations (Fig. 3Aa). The level of all significant or suggestive loci around the rs315953420 SNP decreased below the genome-wide suggestive threshold after treating the genotype of the locus as a covariate in the conditional GWAS (Fig. 3Ab)). Another SNP in the significant region, rs15695238, was identified as being associated with AH and HU.

Manhattan plots and Q-Q plots of genome-wide association studies for AH and HU. Each dot represents an SNP in the dataset. The horizontal red and black lines indicate the genome-wide significant threshold (1.58e–6) and genome-wide suggestive significant threshold (3.17e–5), respectively. The Manhattan plots indicate −log10 (observed P-values) for genome-wide SNPs (y-axis) against their corresponding position on each chromosome (x-axis), while the Q-Q plots show the expected -log10-transformed P-values vs. the observed -log10-transformed P-values. AH and HU denote albumen height and haugh unit, respectively. GIF represents genomic inflation factor.

Conditional GWAS of AH (A) and LD analysis of loci in the significant region (B). Conditional association analyses were carried out by fitting the most significant SNP rs315953420 as a covariate. The strong LD block is defined as D’ ≥ 0.8.
As shown in Fig. 4, there were 24 and 37 hits significantly associated with ESC on GGA12 and GGA21, respectively (Supplementary Table S2). LD analysis showed that 24 SNPs on GGA12 were clustered into two neighbour blocks, Block 1 (42 kb) and Block 2 (191 kb) (Supplementary Fig. S2). All the significant SNPs on GGA21 were in a strong LD block (Supplementary Fig. S2). Then, we separately added the two most significant SNPs (rs315306430 and rs315046589) into the model of conditional analysis. The significant difference in levels is displayed in Fig. 5 (upper panel was before the addition of the SNPs, while the lower panel was after). In addition, four SNPs (rs14283106, rs317521562, rs13603074 and rs16179081) were identified by the bivariate analysis as affecting phenotypes at the two time points. Unfortunately, no genome-wide significant locus was screened for ESI, EST, ESS or YC.

Manhattan plots and Q-Q plots of genome-wide association study for ESC at 72 weeks of age. The horizontal red and black lines represent the genome-wide significant threshold (1.58e–6) and genome-wide suggestive significant threshold (3.17e–5), respectively. ESC denotes eggshell colour. GIF indicates genomic inflation factor.

Regional association plots of two loci associated with ESC72. Each plot shows the −log10 (observed P-values) against their chromosomal positions. The horizontal red line represents the genome-wide significance level (1.58e–6). (A) Depicts the results for ESC72 before (upper) and after (lower) the inclusion of rs315306430. (B) Depicts the results for ESC72 before (upper) and after (lower) condition on rs315046589.
SNP annotation and promising genes associated with egg quality
The annotation of significant SNPs using the Variant Effect Predictor (VEP) tool supplied by Ensembl could help us identify promising genes associated with egg quality traits. The detailed information about the genes was summarized in Table 3. For AH and HU, two significant loci in an ~0.36 Mb (8.95 to 9.31 Mb) significant region on GGA13 were detected to have substitutions of C to A and T to C, respectively. The SNP rs315953420 was located 60 Kb upstream of msh homeobox 2 (MSX2), while the SNP rs15695238 was located 170 Kb downstream of dopamine receptor D1 (DRD1). Egg shell colour has high heritability and is affected by polygenes. Five candidate genes, namely, ras homologue family member A (ROHA), stromal cell derived factor 4 (SDF4), TNF receptor superfamily member 4 (TNFRSF4), tubulin tyrosine ligase-like 10 (TTLL10), and LOC419425, and one microRNA (MIR429) were found close to the significant SNPs associated with ESC (Table 3).
SNP contribution to phenotypic variation
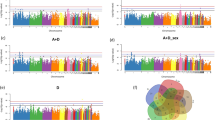
As shown in Table 4, we extracted six meaningful loci which reached genome-wide significance in the univariate and bivariate GWAS for further analysis: rs315953420, rs15695238, rs14283106, rs317521562, rs13603074 and rs16179081 in MSX2, DRD1, SDF4, TNFRSF4, TTLL10 and LOC419425, respectively. The effect allele frequency (EAF) ranged from 0.356 to 0.465, which meant that these SNPs were separate. For AH and HU, the phenotypic variance explained by two significant SNPs ranged from 3.12 to 5.75%. The substitution of one copy of EA at the rs31594320 site caused the highest decrease (0.339 SD/allele) in haugh unit at 72 weeks of age. The remaining four SNPs accounted for 3.28~4.15% of the variance in ESC. In addition, we compared actual phenotypic differences among the three genotypes of the above SNPs (Fig. 6). The results revealed that the phenotypes corresponding to the different genotypes displayed significant segregation. The homozygotes of the effect allele or the alternative allele resulted in the lowest and highest individual phenotypes, respectively, while the heterozygotes were intermediate.

Boxplots of SNP effects on AH, HU and ESC at 72 and 80 weeks of age. The figure shows the genotypes of six representative SNPs (x-axis) versus the phenotypic values of the corresponding traits (y-axis). AH, HU and ESC represent albumen height, haugh unit and eggshell colour, respectively.
Discussion
Egg quality is an important economic trait. With the extension of the laying cycle, the decline in egg quality during the late laying period, such as the increased %cracks and the large variation in eggshell colour, has aroused great concern2. The present research was designed to uncover the key genes that affect egg quality using egg quality data from chickens with laying ages of 72 and 80 weeks. This study is the first to conduct GWASs of egg quality at the late laying period, employing the chicken 600 K high-density SNP chip.
The population employed in this study was a purebred brown egg-type chicken line, rather than a cross between two or more distant populations. This decreased the power to detect QTLs for egg quality traits that differ between the crossed populations14, but this was compensated for by the numerous genotypes and phenotypes of the individuals and the effective methods used15. The phenotypic values of AH and HU at 80 weeks of age decreased compared to those at 72 weeks of age. Previous research has indicated that the rapid reduction in egg quality during the late laying period could lead to a decrease in hatchability2. The pedigree-based heritability estimates of ESC, AH and YC were slightly lower than those in the Hy-line resource population, which were evaluated at 42 and 46 weeks of age12. The estimates of ESS and EST were similar to those in the F2 population at 72 weeks of age13. The genetic and phenotypic correlation of all egg quality traits was significantly reduced compared with the correlations found in previous studies16 because of the larger coefficient of variance at late laying period. Moreover, the pedigree-based heritability estimates were larger than the SNP-based estimates. This may be caused by “missing heritability”17 which refers to the fact that the eligible SNPs in the Affymetrix 600 K SNP chip do not represent the complete genomic variation in chickens.
Genome-wide association analysis of egg quality traits were carried out separately. A significant genomic region of 0.36 Mb on chromosome 13 (GGA13) that harboured 85 unique SNPs (Supplementary Table S2) was identified as being associated with albumen height (AH) and haugh unit (HU). The results of the conditional GWAS and the linkage disequilibrium (LD) analysis revealed that the SNPs were closely linked together in this region. As listed in the chicken QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/GG/index), a total of 67 QTL_IDs were reported for AH and HU. For example, four putative QTL regions were detected on GGA 1, 9, 13 and 23 that were associated with albumen height11, one was detected in the centre of GGA 7, identified by its proximity to a candidate gene18, and 5 regions were reported on GGA2, 3, 4, 9 and Z12. The published QTL regions on GGA13 were close to the genomic region in our study. By annotating the significant SNPs, two promising genes (MSX2 and DRD1) near this region were found to be associated with AH and HU. The MSX2 gene is a member of the msh homeobox family and is expressed in many embryonic tissues. In the developing chick, MSX2 is expressed in the apical ectodermal ridge and the ectoderm of the genital tubercle, and it plays a crucial role in the growth and patterning of the limb mesoderm19. In addition, the expression of MSX2 could influence the function of TNF-α (tumour necrosis factor-α), which is known to suppress adipocyte differentiation and to activate the Wnt/β-catenin pathway20,21. The Wnt signalling pathway regulates crucial aspects of cell fate determination, cell migration, and cell polarity during embryonic and ovary development22. In addition, a previous proteomic study reported that, in eggs, a portion of the functions of the proteins were associated with embryonic development13. We know that the egg proteins are secreted by the magnum during the egg-laying process. Therefore, we believe that MSX2 gene has an indirect effect on AH and HU during the process of protein formation. Another gene, DRD1, belongs to the D1-like type of dopamine receptor. In birds, dopamine has been revealed to be involved in both stimulating and inhibiting prolactin (PRL) secretion, which has been demonstrated to play a crucial role in the onset and maintenance of incubation behaviour23,24,25. Dopamine stimulates PRL secretion by activating DRD1 at the hypothalamus level and inhibits PRL secretion through DRD2 at the pituitary level26,27,28,29. All these studies have indicated that the dopamine receptor participates in the system of regulating avian reproductive behaviour. Like other D1-like members, the chicken DRD1 is an intronless gene and belongs to the rhodopsin family. Recently, Schnell et al. and Chaiseha et al. demonstrated that the DRD1 gene is widely expressed in the hypothalamus and pituitary and that the expression is associated with the functioning of the reproductive system in turkeys30,31. Moreover, previous findings have revealed that the DRD1 gene and its haplotypes are associated with some egg production traits in chickens32. We suggest that DRD1 could be treated as a candidate gene related to egg quality in further analyses.
There is extensive scientific literature regarding eggshell colour, and some studies have concluded that the green colour of the eggshell is controlled by a single dominant gene (An EAV-HP Insertion in 5′ Flanking Region of SLCO1B3 Causes Blue Eggshell in the Chicken)33,34, but there still exists much uncertainty about the molecular mechanism underlying brown eggshell colour, even though it has been reported to be controlled by multiple minor genes5,12,35,36. Li et al. used quantitative PCR to determine the expression levels of 8 genes encoding enzymes in the liver and shell gland in a Rhode Island Red pure line. The interesting gene ABCG2 might facilitate the accumulation of protoporphyrin IX, which is the main pigment resulting in the brown coloration of eggshell36. In addition, Wolc et al. found two genomic regions located on GGA4 and GGA12 that affect the early and late eggshell colour, respectively, in a pureline Hy-line population12. In the present study, one gene (RHOA) on GGA12 was related to ESC at 72 weeks of age, and four genes, namely, SDF4, TNFRSF4, TTLL10 and LOC419425, were identified by GWAS as being associated with egg shell colour (ESC) at the two periods. The SNP rs315306430 located 4.73 Kb upstream of RHOA overlapped with a previously identified QTL12. RHOA is a small GTPase and belongs to the ras homologue (Rho) family. The Rho family of small GTPases are molecular switches that control a wide variety of cell functions, including cytoskeletal reorganization, cell motility, and gene expression37. In addition, it has been revealed that the RHOA signalling system plays a role in the modulation of actin stress fibres and chondrogenesis38. SDF4 is the stromal cell derived factor 4, and its human orthologue is known as Cab45. A 45-kDa Ca2+-binding protein, Cab45 is important because it can regulate cell migration through various molecular mechanisms39. Another gene, TNFRSF4, encodes proteins and can be used to specifically modulate the expression of other genes that directly stimulate effector T-cell activity40. All the above three genes are associated with body growth and phylogenesis. Recently, a very interesting study reported that eggshell pigment deposition and eggshell coloration were strongly and positively correlated with phylogenesis41. Therefore, we conclude that RHOA, SDF4, and TNFRSF4 affect eggshell colour through the process of phylogenesis. Unfortunately, little information about TTLL10 and LOC419425 could be found in the NCBI GenBank.
In summary, we carried out univariate, bivariate and conditional GWASs for egg quality at late laying period, employing the chicken 600 K high density SNP array. A genomic region spanning from 8.95 to 9.31 Mb (~0.36 Mb) in GGA13 was detected to be significantly associated with albumen height and haugh unit. Two promising genes, MSX2 and DRD1, were mapped to that narrow region. Furthermore, three interesting genes, RHOA, SDF4 and TNFRSF4, identified from three significant loci, were considered candidate genes related to egg shell colour. However, further functional validation needs to be performed in chickens. Findings in our study could provide worthy theoretical basis and technological support to improve late-stage egg quality for breeders.
Materials and Methods
Ethics statements
All the blood sample collections were performed in accordance with the Guidelines for Experimental Animals established by the Ministry of Agriculture of China (Beijing, China). The whole study was approved by the Animal Welfare Committee of China Agricultural University (Permit Number: SYXK 2007-0023).
Resource Population
An 11th generation population of Rhode Island Red chickens from the Beijing Huadu Yukou Poultry Breeding Co., Ltd. were the experimental animals used in this study. This pure line has been selected for egg production over many generations. Thus, a total of 1,078 hens with accurate pedigrees were chosen for SNP genotyping. The hens were housed in individual cages in the same area with free access to feed and water.
Phenotypic measurements and evaluation of heritability
Egg quality traits including eggshell colour (ESC), egg shape index (ESI), eggshell thickness (EST), eggshell strength (ESS), albumen height (AH), yolk colour (YC) and haugh unit (HU) were measured at 72 and 80 weeks of age. Due to the late laying period, we collected fresh eggs for three successive days to ensure one egg per hen. ESC was measured with a CM-2600D reflectometer (Konica Minolta, Tokyo, Japan) using the three following parameters: L* represents lightness, a* measures the balance of red and green, and b* describes hue of blue-yellow scale. Here, we only used the L* as the eggshell colour phenotypic value. The long and short diameters of each egg were measured with a Vernier calliper, and the egg shape index was calculated as the ratio of the long and short diameters. The eggshell strength (pole to pole) of each egg was measured vertically using an EFG-0502 gauge (Robotmation, Tokyo, Japan). Then, we broke the eggs to collect the internal contents, and the AH, YC and HU were measured with the EMT-5200 multi-functional egg analyser (Robotmation, Tokyo, Japan). And the HU is also a calculation from AH with egg weight. Finally, we cleaned the eggshell membrane and measured the EST of each egg with an eggshell thickness gauge (FHK, Tokyo, Japan). Descriptive statistics of all phenotypic records were handled with R version 3.3.1 software (https://www.r-project.org/).
Pedigree-based hereditability for ESC, ESI, EST, ESS, AH, YC and HU in the two periods was calculated with the average information restricted maximum likelihood (AI-REML) method supplied by the DMU v6.0 software42. The multi-traits general animal model was adopted in our analysis as follows:
where y is the phenotypic value, 1 is an n x 1 vector of all 1’s, µ is the population mean (fixed effect), Z is an incidence matrix for random additive effects, a and e are the additive effect and random residual, respectively.
Genotyping, quality control and imputation
Genomic DNA was extracted from whole blood samples using standard phenol/chloroform methods and the 1078 qualified hens were genotyped with the Affymetrix 600 K chicken SNP chip (Affymetrix, Inc. Santa Clara, CA, USA). From a preliminary set of 580,961 SNPs43, 6,550 SNPs with unknown physical position and 43 markers with repeated genomic coordinates were excluded. The genotype calling and quality control were carried out by the Affymetrix Power Tools v1.19.0 (APT) software, following the pipeline of the Axiom Genotyping Solution. Only individuals with dish quality control (DQC) >0.82 and call rate >97% were included in the downstream analyses. A package of ps-metrics supplied by the APT software was run to calculate the SNP quality, and the lower quality SNPs were filtered out using an R script. After the above QC steps, 1063 individuals and 517,856 SNPs remained. We also discarded SNPs on the sex chromosomes because of the low detection power for associations between phenotypes and sex chromosome genotypes. Moreover, PLINK v1.90 software was used for further quality control (minor allele frequencies (MAF) >0.01, Hardy Weinberg equilibrium (HWE) <1e–6)44. The remaining SNPs were used to impute some missing genotypes with the Beagle v4.0 procedure45. Finally, 1063 individuals and 294,705 SNPs distributed among 28 autosomes and two linkage groups (Table 5) were eligible for the subsequent genome-wide analyses.
Population structure and association analysis
Prior to the genome-wide association study (GWAS), a principal component analysis (PCA) supplied by the PLINK package was conducted to evaluate the population stratification. We pruned all SNPs to obtain independent SNPs via the option of -indep-pairwise 25 5 0.2. In addition, the kinship matrix was built through the independent SNP markers. The principal components were calculated from the linear combination of markers by the eigenvectors of the kinship matrix, which were treated as covariates and included in model of GWAS as fixed effects to control the population structure effects. We adjusted the P-value threshold of genome-wide significance using the simpleM package46, considering the over-conservation of the Bonferroni method. After the simpleM test, a total of 31,589 effective independent tests were obtained. Then, the genome-wide significance and suggestive significance were calculated as 1.58e–6 (0.05/31,589) and 3.17e–5 (1.00/31,589), respectively.
The univariate GWAS was first implemented using a linear mixed model to account for the associations between each trait and the effective SNPs, which was supplied by GEMMA software47. The statistical model was as follows:
where y is the phenotypic values of n individuals in the population of interest; W is a matrix of covariates (fixed effects: top five principal components and a column of 1 s) controlling for population structure; α is a vector of corresponding effects that compose the intercept; x is the marker genotypes; β is the corresponding marker’s effect; u is a vector of random polygenic effects with a covariance structure as u~N (0, KVg), where K represents a known n x n genetic relatedness matrix derived from SNP markers and Vg is the polygenic additive variance; and ε is vector of random residuals. The Wald statistical test was applied to test the alternative hypothesis H1:β ≠ 0 against the null hypothesis H0: β = 0 for each SNP, as Fwald = \({\hat{\beta }}^{2}/Var(\hat{\beta })\).
The Manhattan and quantile-quantile (Q-Q) plots were generated for each trait using the “gap” (https://cran.r-project.org/web/packages/gap/) and “qqman” (https://cran.r-project.org/web/packages/qqman/) packages in R software, which described the transformed −log10 of the observed P-values against the marker locations on the genome or the expected −log10 (P-values). Moreover, the genomic inflation factor (λ) was calculated to judge the extent of false positive signals by the “GenABEL” package in R48.
Furthermore, we carried out a bivariate association analysis to directly account for the effects of the genetic variants on the dynamic egg quality traits along with the two late laying periods. The mixed model was also supplied by the GEMMA software47.
Linkage disequilibrium (LD) analysis and gene identification
We performed linkage disequilibrium (LD) analysis in order to characterize causal SNPs in a strong LD region where many significant SNPs were identified by the solid spine algorithm in Haploview version 4.2 as being clustered49. In addition, we added the genotypes of the most significant SNPs (coded as 0, 1 or 2) as covariates into the univariate and multivariate models to elucidate independent signals in the step-wise conditional analysis. The information of significant SNPs were obtained with the annotation of Gallus-gallus 4.0, and candidate genes within 500 kb regions flanking the associated SNPs were identified using Variant Effect Predictor (VEP)50 supplied by Ensembl (http://www.ensembl.org).
Estimation of genetic parameters and contribution to phenotypic variance (CPV)
All SNP-based heritability (h2snp)51 and pair-wise genetic correlations of egg quality traits were calculated using the restricted maximum likelihood (REML) method with GCTA v1.24 software52. A matrix of genetic relationships was constructed from all eligible SNPs on autosomes and two linkage groups. We then calculated the phenotypic variance contribution of those genome-wide significant SNPs based on the genetic matrix.
References
Schulte-Drüggelte, R. & Thiele, H.-H. Determining the optimum replacement schedule for commercial layers:does molting pay off? Lohmann Information 48(1), 47 (2013).
Bain, M. M., Nys, Y. & Dunn, I. C. Increasing persistency in lay and stabilising egg quality in longer laying cycles. What are the challenges? British poultry science 57, 330–338 (2016).
WARDÊCKA, B. et al. Relationship between microsatellite marker alleles on chromosomes 1–5 originating from the Rhode Island Red and Green-legged Partrigenous breeds and egg production and quality traits in F2 mapping population. J. Appl. Genet. 43(3), 319–329 (2002).
Tuiskula-Haavisto, M. et al. Mapping of Quantitative Trait Loci Affecting Quality and Production Traits in Egg Layers. Poultry science 81, 919–927 (2002).
Schreiweis, M. A., Hester, P. Y., Settar, P. & Moody, D. E. Identification of quantitative trait loci associated with egg quality, egg production, and body weight in an F2 resource population of chickens. Animal genetics 37, 106–112 (2006).
Tuiskula-Haavisto, M. et al. Quantitative trait loci affecting eggshell traits in an F(2) population. Animal genetics 42, 293–299 (2011).
Hu, Z. L., Park, C. A. & Reecy, J. M. Developmental progress and current status of the Animal QTLdb. Nucleic Acids Res 44, D827–833 (2016).
Ball, A. D. et al. A comparison of SNPs and microsatellites as linkage mapping markers: lessons from the zebra finch (Taeniopygia guttata). BMC genomics 11, 218 (2010).
Fernández, M. E. et al. Comparison of the effectiveness of microsatellites and SNP panels for genetic identification, traceability and assessment of parentage in an inbred Angus herd. Genetics and Molecular Biology 36(2), 185–191 (2013).
Groenen, M. A. et al. The development and characterization of a 60K SNP chip for chicken. BMC genomics 12, 274 (2011).
Liu, W. et al. A genome-wide SNP scan reveals novel loci for egg production and quality traits in white leghorn and brown-egg dwarf layers. PloS one 6, e28600 (2011).
Wolc, A. et al. Genome-wide association study for egg production and quality in layer chickens. Journal of animal breeding and genetics = Zeitschrift fur Tierzuchtung und Zuchtungsbiologie 131, 173–182 (2014).
Sun, C. et al. Genome-wide association study revealed a promising region and candidate genes for eggshell quality in an F2 resource population. BMC genomics 16, 565 (2015).
Cavero, D., Schmutz, M. & Preisinger, R. Genetic evaluation of pure-line and cross-line performance in layers. Lohmann Information 45(2), 18 (2010).
Zhang, S., Meng, L., Wang, J. & Zhang, L. Background controlled QTL mapping in pure-line genetic populations derived from four-way crosses. Heredity 119, 256–264 (2017).
Alipanah, M., Deljo, J., Rokouie, M. & Mohammadnia, R. Heritabilities and genetic and phenotypic correaltions of egg quality traits in KHAZAK layers. Trakia Journal of Sciences, 175–180 (2013).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Zhang, Y. et al. Low-density lipoprotein receptor-related protein 2 gene is associated with egg-quality traits in dwarf layers. Poultry science 90, 2718–2722 (2011).
Sumoy, L. et al. Identification of a spatially specific enhancer element in the chicken Msx-2 gene that regulates its expression in the apical ectodermal ridge of the developing limb buds of transgenic mice. Development Biology 170, 230–42 (1995).
Lee, H. L. et al. Msx2 mediates the inhibitory action of TNF-alpha on osteoblast differentiation. Experimental & molecular medicine 42, 437–445 (2010).
Qadir, A. S. et al. Msx2 is required for TNF-alpha-induced canonical Wnt signaling in 3T3-L1 preadipocytes. Biochemical and biophysical research communications 408, 399–404 (2011).
Komiya, Y. & Habas, R. Wnt signal transduction pathways. Organogenesis 4(2), 68–75 (2008).
Sharp, P., Macnamee, M., Sterling, R., Lea, R. & Pedersen, H. Relationships between prolactin, LH and broody behaviour in bantam hens. J Endocrinol 118(2), 279–86 (1988).
March, J., Sharp, P., Wilson, P. & Sang, H. Effect of active immunization against recombinant-derived chicken prolactin fusion protein on the onset of broodiness and photoinduced egg laying in bantam hens. J Reprod Fertil 101(1), 227–33 (1994).
Youngren, O. M., Pitts, G. R., Phillips, R. E. & Halawani, M. E. E. The Stimulatory and Inhibitor Effects of Dopamine on Prolactin Secretion in the Turkey. General And Comparative Endocrinology 98, 111–117 (1995).
Porter, T. E. et al. Evidence that stimulatory dopamine receptors may be involved in the regulation of prolactin secretion. Endocrinology 134(3), 1263–68 (1994).
Youngren, O. et al. Regulation of Prolactin Secretion by Dopamine at the Level of the Hypothalamus in the Turkey. Neuroendocrinology 75(3), 185–192 (2002).
Youngren, O., Chaiseha, Y. & Halawani, M. E. Regulation of prolactin secretion by dopamine and vasoactive intestinal peptide at the level of the pituitary in the turkey. Neuroendocrinology 68(5), 319–325 (1998).
Kahtane, A. A., Chaiseha, Y. & Halawani, M. E. Dopaminergic regulation of avian prolactin gene transcription. J Mol Endocrinol 31(1), 185–196 (2003).
Schnell, S., You, S. & Halawani, M. E. D1 and D2 dopamine receptor messenger ribonucleic acid in brain and pituitary during the reproductive cycle of the turkey hen. Biol Reprod 60(6), 1378–1383 (1999).
Chaiseha, Y., Youngren, O., Al-Zailaie, K. & Halawani, M. E. Expression of D1 and D2 dopamine receptors in the hypothalamus and pituitary during the turkey reproductive cycle: colocalization with vasoactive intestinal peptide. Neuroendocrinology 77(2), 105–118 (2003).
Xu, H. et al. The genetic effects of the dopamine D1 receptor gene on chicken egg production and broodiness traits. BMC genetics 11, 17 (2010).
Punnett, R. C. Genetic studies in poultry IX The blue egg. Genetic Studies in Poultry 59–62 (1933).
Wang, Z. et al. An EAV-HP insertion in 5′ Flanking region of SLCO1B3 causes blue eggshell in the chicken. PLoS genetics 9, e1003183 (2013).
Rome, H. et al. GWAS analyses reveal QTL in egg layers that differ in response to diet differences. Genetics, selection, evolution: GSE 47, 83 (2015).
Li, G. et al. Comparison of protoporphyrin IX content and related gene expression in the tissues of chickens laying brown-shelled eggs. Poultry science 92, 3120–3124 (2013).
Nobes, C. & Hall, A. Regulation and function of the Rho subfamily of small GTPases. Curr Opin Genet Dev 4(1), 77–81 (1994).
Kim, M. J., Kim, S., Kim, Y., Jin, E. J. & Sonn, J. K. Inhibition of RhoA but not ROCK induces chondrogenesis of chick limb mesenchymal cells. Biochemical and biophysical research communications 418, 500–505 (2012).
Luo, J. et al. A Novel Role of Cab45-G in Mediating Cell Migration in Cancer Cells. International journal of biological sciences 12, 677–687 (2016).
Kumari, A. & Garnett-Benson, C. Effector function of CTLs is increased by irradiated colorectal tumor cells that modulate OX-40L and 4-1BBL and is reversed following dual blockade. BMC research notes 9, 92 (2016).
Brulez, K. et al. Eggshell pigment composition covaries with phylogeny but not with life history or with nesting ecology traits of British passerines. Ecol Evol 6, 1637–1645 (2016).
Madsen, P. & Jensen, J. A User’s Guide to DMU:A Package for Analysing Multivariate Mixed Models (2013).
Andreas Kranis et al. Development of a high density 600K SNP genotyping array for chicken. BMC genomics 14, 59 (2013).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575 (2007).
Browning, B. L. & Browning, S. R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 84, 210–223 (2009).
Gao, X., Becker, L. C., Becker, D. M., Starmer, J. D. & Province, M. A. Avoiding the high Bonferroni penalty in genome-wide association studies. Genetic epidemiology 34, 100–105 (2010).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nature genetics 44, 821–824 (2012).
Devlin, B. Genomic control for association studies. Biometrics 55, 997–1004 (1999).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010).
Lee, S. H., Yang, J., Goddard, M. E., Visscher, P. M. & Wray, N. R. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28, 2540–2542 (2012).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011).
Acknowledgements
This work was funded in part by grants of National High Technology Development Plan of China (2013AA102501), the Programs for Changjiang Scholars and Innovative Research in University (IRT_15R62), and China Agriculture Research Systems (CARS-40).
Author information
Authors and Affiliations
Contributions
N.Y. conceived the study and designed the project. Z.L. performed genetic and bioinformatics analyses. Z.L., Y.Y., G.L., F.S., G.W. and A.L. contributed to collecting samples and measuring the phenotypic data. Z.L. wrote the manuscript, N.Y. and C.S. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Z., Sun, C., Yan, Y. et al. Genetic variations for egg quality of chickens at late laying period revealed by genome-wide association study. Sci Rep 8, 10832 (2018). https://doi.org/10.1038/s41598-018-29162-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29162-7
This article is cited by
-
Insights into genetic diversity and phenotypic variations in domestic geese through comprehensive population and pan-genome analysis
Journal of Animal Science and Biotechnology (2023)
-
Identification of candidate genomic regions for egg yolk moisture content based on a genome-wide association study
BMC Genomics (2023)
-
Genetic parameters, reciprocal cross differences, and age-related heterosis of egg-laying performance in chickens
Genetics Selection Evolution (2023)
-
Association analysis of production traits of Japanese quail (Coturnix japonica) using restriction-site associated DNA sequencing
Scientific Reports (2023)
-
Species-wide genomics of kākāpō provides tools to accelerate recovery
Nature Ecology & Evolution (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
