
Abstract
Adolescent idiopathic scoliosis (AIS) is the most common type of spinal deformity and has a significant genetic background. Genome-wide association studies (GWASs) identified several susceptibility loci associated with AIS. Among them is a locus on chromosome 6q24.1 that we identified by a GWAS in a Japanese cohort. The locus is represented by rs6570507 located within GPR126. To ensure the association of rs6570507 with AIS, we conducted a meta-analysis using eight cohorts from East Asia, Northern Europe and USA. The analysis included a total of 6,873 cases and 38,916 controls and yielded significant association (combined P = 2.95 × 10−20; odds ratio = 1.22), providing convincing evidence of the worldwide association between rs6570507 and AIS susceptibility. In silico analyses strongly suggested that GPR126 is a susceptibility gene at this locus.
Similar content being viewed by others

Introduction
Adolescent idiopathic scoliosis (AIS) is defined as a lateral spinal curvature that occurs without obvious cause between age 10 and skeletal maturity. The prevalence of AIS in the adolescent population is approximately 2–3%1 and AIS occurs predominantly in females2,3. AIS has been regarded as a multifactorial disease and a number of population, family and twin studies strongly suggest the importance of genetic factors in its etiology and pathogenesis4,5,6,7.
Genome-wide association study (GWAS) is a powerful tool to detect genetic variants associated with common diseases. Recently, several GWASs have identified several loci associated with AIS such as chromosome 10q24.31, 6q24.1, 20p11.22, and 1p36.32 loci8,9,10,11. Our initial GWAS identified rs11190870, a common variant on chromosome 10q24.31 that showed significant association with AIS in a Japanese cohort8 and subsequent multi-ethnic meta-analysis provided convincing evidence of the association12. In addition, recent validation studies in different populations have also reported its significant associations13,14,15. Thus, the association of the 10q24.31 locus with AIS has been confirmed in multiple studies; however, other AIS loci have not been fully investigated.
In our previous GWAS, we identified an additional genetic variant, rs6570507 on chromosome 6q24.1, associated with AIS in a Japanese cohort16. The association was replicated in independent cohorts of Chinese and Caucasian in the USA16; however, additional studies would be necessary to confirm the association and to identify the susceptibility gene at the locus.
In this study, we performed a meta-analysis using multi-ethnic cohorts of ~46,000 subjects. The result provided convincing evidence of the association of rs6570507 with AIS susceptibility, suggesting that the chromosome 6q24.1 locus is related to the global risk of AIS. In silico analyses strongly suggested that GPR126 is a susceptibility gene at this locus.
Results
Association of rs6570507 and AIS susceptibility
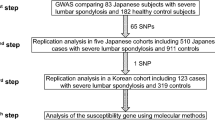
We conducted the meta-analysis of rs6570507 using eight cohorts (Table 1, Fig. 1). Three cohorts were previously reported16, and the other five were recruited for this study that included cohorts from Guangzhou (case 647 and control 1,048), Hong Kong (case 300 and control 788), Beijing (case 482 and control 861), USA (case 1,360 and control 7,267), and Scandinavia (case 1,522 and control 1,804). Finally, 6,873 cases and 38,916 controls were included in the meta-analysis. The analysis showed a convincing association between rs6570507 and AIS: combined P = 2.95 × 10−20; odds ratio (OR) = 1.22; 95% confidence interval (CI) = 1.17–1.27 (Table 1, Supplementary Fig. S1). ORs were >1 in all eight cohorts, with little difference between ethnic groups. The analysis did not show any significant heterogeneity, suggesting no statistical difference between studies. Because AIS has clinical evidence of sexual dimorphism17, we performed gender-stratified analyses to determine whether a genetic difference existed between males and females (Table 2, Supplementary Fig. S1). However, no gender difference was observed in the association.

The flow diagram of the meta-analysis using eight cohorts. Three cohorts (Japanese GWAS, replication study and Nanjing study) were previously reported16, and the other five were recruited for this analysis.
Fine mapping
The sentinel SNP rs6570507 is present in an intron of GPR126 (encoding G protein-coupled receptor 126), and GPR126 is the only gene contained within the linkage disequilibrium (LD) block (r2 > 0.8) represented by rs6570507 (ref.16). To identify the candidate susceptibility gene in the locus, we evaluated the topologically associated domains (TADs) around the associated SNPs. Hi-C data18 (http://promoter.bx.psu.edu/hi-c/view.php) revealed that GPR126 and VTA1 (encoding vesicle trafficking 1) were included in the TAD that contained the LD block (Fig. 2). Using expression quantitative trait loci (eQTL) data from the Genotype-Tissue Expression (GTEx) project19, we investigated the target genes regulated by rs6570507. We found that the expression level of GPR126 in subcutaneous adipose tissue and sun-exposed skin was significantly associated with rs6570507 and its risk allele rs6570507-A decreased the expression (Supplementary Fig. S2). These data strongly suggested that GPR126 is the most plausible AIS susceptibility gene at this locus.

The Hi-C interaction surrounding the AIS associated region on chromosome 6q24.1. Hi-C interaction in H1-mesenchymal stem cell was generated by using the Interactive Hi-C Data Browser. GPR126 and VTA1 lie within the topologically associated domain (black triangle) that contains the linkage disequilibrium (LD) block (bold line) of AIS associated SNPs. An arrow indicates the location of rs6570507.
Target sequencing
Recently, numerous sequencing studies of candidate genes in GWAS loci identified rare large-effect variants20,21,22. To assess the potential contribution of rare variants at the chromosome 6q24.1 locus to AIS, we carried out targeted sequencing. We screened the coding regions and exon-intron boundaries of VTA1 (chr.6: 142468421–142539785) and GPR126 (chr.6: 142623463–142764657) across 10,121 individuals (2,721 cases and 7,400 controls). The coverage rate of the target region, presented as an average in all individuals covered with ≥20 reads at each position, was 99.96%. We identified 183 single nucleotide variant (SNVs), 2 insertions and 9 deletions. The single-variant association analysis for each nonsynonymous variant (including missense, splice site, nonsense, and frameshift variants) with MAF <0.05 demonstrated no rare variant with significant associations (Supplementary Table S1). Subsequently, we performed the gene-based association analyses by the cohort allelic sums test (CAST)23 and the sequence kernel association test (SKAT)24, using the nonsynonymous variants. Only GPR126 showed a nominal significant association (P = 4.65 × 10−2) with AIS by CAST; however, it did not reach the threshold for significance after Bonferroni correction (Supplementary Table S2).
Discussion
In our previous study, we identified the significant association between rs6570507 and AIS in Japanese and the association is replicated in the independent cohorts of Chinese and Caucasian in the USA. However, as the numbers of Chinese and Caucasian subjects were both limited, the association between rs6570507 and AIS in these populations does not provide convincing evidence. In the current study, we have conducted a meta-analysis using eight independent multi-ethnic-cohorts and showed significant and consistent association of rs6570507 with AIS across the different ethnic groups. Recently, evidence for association of rs6570507 with AIS was reported in a Chinese cohort of Zhejiang25; however, we did not use the data for our meta-analysis because the allele frequency of rs6570507 in the control group of the study was quite different from the frequency for the Chinese in the public database (for example, the 1000 Genomes Project) and the minor allele was opposite. Nevertheless, our meta-analysis demonstrates convincing association between rs6570507 and AIS susceptibility.
For some common complex diseases, rare large-effect variants have recently been identified by targeted exon sequencing of genes within GWAS associated loci26,27. This approach is helpful to identify causal genes and to explain a further portion of disease variance. In the present study, we assessed the contribution of rare variants in the candidate genes at the locus; however, we could not find any statistically significant association in individual variants. GPR126 showed a nominally significant association by a gene-based study. The frequency of rare and low-frequency nonsynonymous variants was increased in cases, suggesting that AIS is caused by functional impairment of the GPR126 gene. However, in the current study, the evidence for association did not surpass Bonferroni correction. This negative result may be due to the insufficient sample size in the present study and further larger studies are required to evaluate the effect of rare and low-frequency variants in GPR126.
To identify the susceptibility gene at the chromosome 6q24.1 locus, we evaluated the TADs around the associated SNPs using the Hi-C database. The data showed that GPR126 and VTA1 were involved in the TAD that contained the LD block of the AIS associated SNPs. All SNPs strongly correlated with rs6570507 (r2 > 0.8) were present in the GPR126 gene region. The eQTL data indicated that these SNPs were significantly associated with the expression level of GPR126 in some tissues, such as subcutaneous adipose tissue and sun-exposed skin. It is well known that AIS patients are associated with low body weight and low body mass index (BMI)28,29, and are also reported to be significantly associated with lower body fat30. These observations suggest that GPR126 has a potential to affect AIS susceptibility through the adipose tissues. However, tissues used in GTEx project are limited, and the sample size is different by tissue. Therefore, it may fail to identify tissues contributing to the disease pathology. In order to more optimally determine the effect of the associated SNPs on scoliosis, eQTL analysis in AIS-related tissues such as intervertebral disc, cartilage and bone should be performed. Moreover, our previous experiments indicated that GPR126 is highly expressed in the cartilage of human and proliferating chondrocytes of the vertebral body in mouse embryo16. Recently, Karner et al. reported that the mice with a conditional deletion of Gpr126 in the cartilage displayed the clinical features of AIS including postembryonic onset of spine curvatures with rotation and absence of vertebral malformations at birth31. These lines of evidence strongly suggested that the GPR126 is a susceptibility gene for AIS and its loss of function is implicated in the pathogenesis of AIS. Further studies are necessary to identify the causal variant at the locus and clarify its functional impact on the function of GPR126.
Materials and Methods
Subjects and genotyping
Informed consent was obtained from all subjects participating in this study. This study was approved by the ethics committee of the RIKEN center for Integrative Medical Sciences and all experiments were performed in accordance with relevant guidelines and regulations. All AIS subjects met clinical criteria including scoliosis with Cobb angle of 10° or more on standing spinal postero-anterior (P-A) radiographs and excluding other non-idiopathic forms of scoliosis. The subjects in the Japanese and Nanjing-Chinese cohorts were recruited and genotyped as previously described16. The details of additional studies: i.e., Guangzhou, Hong Kong, Beijing, USA, and Scandinavia studies were described as below.
Guangzhou study
AIS subjects were recruited from the First Affiliated Hospital and Sun Yat-sen Memorial Hospital of Sun Yat-sen University. They provided detailed histories, accepted physical examinations, underwent standard up-standing P-A radiography of the whole spine, and other testing such as magnetic resonance imaging (MRI), computed tomography (CT) and nuclear scintigraphy, when necessary. All AIS subjects were diagnosed at ages 10–16 years. Control subjects were recruited from individuals who received scoliosis screening at middle and primary schools in Guangzhou and fracture patients selected from the participating hospitals. All subjects were confirmed for not having AIS by x-ray scans of the spine. Routine history and physical examinations were also conducted to exclude other relevant diseases. Genomic DNA was extracted from blood using DNA Blood Mini-kit (Tiangen Biotech, Beijing, China). Primer extension sequencing (SNaPshot) assay (Applied Biosystems, CA, USA) was used for genotyping and the results were analyzed by GeneMarker software (SoftGenetics LLC, PA, USA) at Beijing Genomics Institute (Shenzhen, China) and checked by visual inspection of I.K. and H.D.
Hong Kong study
AIS subjects were recruited from the Duchess of Kent Children’s Hospital in Hong Kong. The inclusion criteria were as previously described32. Control subjects were randomly selected from the subjects recruited for the Genetic Study of Degenerative Disc Disease project33. All were confirmed not to have AIS by MRI examination of the spine. Genomic DNA was extracted from peripheral blood lymphocytes using standard procedures. The PCR-based invader assay (Third Wave Technologies, WI, USA) was used for genotyping.
Beijing Study
AIS subjects were recruited from Peking Union Medical College Hospital. All subjects underwent clinical and radiologic examination such as whole spine X-ray, CT and MRI by expert spinal surgeons. Control subjects were recruited from in-house control bank. Genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The MassARRAY system (Sequenom; BioMiao Biological Technology, Beijing, China) was used for genotyping.
USA Study
AIS subjects were recruited at Texas Scottish Rite Hospital for Children as previously described10 and genotyped using the Illumina HumanCoreExome Beadchip array. For controls, we utilized a single dataset of individuals downloaded from the dbGaP web site (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap) from Geisinger Health System-MyCode, eMERGE III Exome Chip Study under phs000957.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000957.v1.p1). The dbGaP controls were previously genotyped on the same microarray platform used for cases. Only subjects of self-reported Non-Hispanic White were included in the present study. Phenotypes of all controls were reviewed to exclude any with musculoskeletal or neurological disorders. We applied initial per sample quality control measures and excluded sex inconsistencies and any with missing genotype rate per person more than 0.03. Remaining samples were merged using the default mode in PLINK.1.9 (ref.34). Duplicated or related individuals were removed as previously described35. We used principal component analysis (PCA)36 on the merged data projected onto HapMap3 samples to correct possible stratification37. After quality controls, 8,647 subjects (1,360 AIS subjects and 7,287 controls) were included for the current study. We applied initial per-SNPs quality control measures using PLINK including genotyping call-rate per marker (>95%), minor allele frequency (>0.01) and deviation from Hardy-Weinberg equilibrium (cutoff p-value = 10−4). We noted that the SNP success rate was 99.99% for the rs6570507 and there was no significant difference in the missingness rate between cases and controls after quality controls.
Scandinavia study
AIS subjects were recruited from six hospitals in Sweden and one in Denmark as described previously to the Scoliosis and Genetics in Scandinavia (ScoliGeneS) study15,38,39,40. Individuals with a history or clinical sign of a non-idiopathic scoliosis and with neural abnormalities in a MRI of the spine were excluded. All control subjects were females and recruited from the Osteoporosis Prospective Risk Assessment cohort and PEAK-25 cohort41,42. Dual-energy X-ray absorptiometry (DXA) scan was performed in both cohorts and subjects showing any sign of a curved spine on DXA were excluded. Genomic DNA was extracted from blood or saliva using the QIAamp 96 DNA Blood Kit and the Autopure LS system (Qiagen). iPLEX Gold chemistry and the MassARRAY system (Agena Bioscience, CA, USA) were used for genotyping. Genotype calls were checked by two persons individually using the MassARRAY Typer v4.0 Software (Agena Bioscience).
Statistical analysis
The association between rs6570507 and AIS in each study was evaluated by the Cochrane-Armitage trend test. Data from the eight studies were combined using the inverse-variance method assuming a fixed-effects model in the METAL software package (http://csg.sph.umich.edu//abecasis/Metal/)43. The heterogeneity among studies was tested using Cochran’s Q test based upon inverse variance weights using METAL.
Target sequencing
Cases were recruited from collaborating hospitals of Japan Scoliosis Clinical Research Group (JSCRG)8,9, and controls were recruited from the BioBank Japan project, the Osaka-Midosuji Rotary Club and the PharmaSNP Consortium44,45. Multiplex PCR-based target sequencing was carried out as previously described45. Primers were designed using the Primer 3 software (ver. 2.3.4) to obtain 180–300 bp PCR products. We amplified 9,847 bp consisting of 47 genomic fractions using three multiplex-PCR products with dual barcodes for each individual. After purifying of each library using Agencourt AMPure XP (Beckman Coulter, CA, USA), the library was applied to the bioanalyzer (Agilent Technologies, CA, USA) to check the size distribution and quantified using the KAPA library quantification kit (Kapa Biosystems, MA, USA) on an ABI Prism 7700 sequence detection system (Life Technologies, CA, USA). Sequencing was carried out using the HiSeq. 2500 instrument (Illumina) with 2 × 150-bp paired-end reads and dual 8-bp barcode sequences. Sequence reads were aligned to the human reference sequence (hg19) by Burrows-Wheeler Aligner (ver. 0.7.9a) and then applied to the RealignerTargetCreator and IndelRealigner tools using GATK (ver. 3.2.2) for each bam file. The quality control was performed as previously described45. We selected variants that clearly showed three peaks corresponding to three genotypes and two peaks if they were considered rare variants by visual inspection.
References
Weinstein, S. L. Natural history. Spine (Phila Pa 1976). 24, 2592–2600 (1999).
Ueno, M. et al. A 5-year epidemiological study on the prevalence rate of idiopathic scoliosis in Tokyo: school screening of more than 250,000 children. J. Orthop. Sci. 16, 1–6 (2011).
Konieczny, M. R., Senyurt, H. & Krauspe, R. Epidemiology of adolescent idiopathic scoliosis. J. Child. Orthop. 7, 3–9 (2013).
Kesling, K. L. & Reinker, K. A. Scoliosis in twins. A meta-analysis of the literature and report of six cases. Spine (Phila Pa 1976). 22, 2009–2014 (1997).
Ward, K. et al. Polygenic inheritance of adolescent idiopathic scoliosis: a study of extended families in Utah. Am. J. Med. Genet. A. 152A, 1178–1188 (2010).
Tang, N. L. et al. Genetic epidemiology and heritability of AIS: A study of 415 Chinese female patients. J. Orthop. Res. 30, 1464–1469 (2012).
Grauers, A., Rahman, I. & Gerdhem, P. Heritability of scoliosis. Eur. Spine J. 21, 1069–1074 (2012).
Takahashi, Y. et al. A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 43, 1237–1240 (2011).
Ogura, Y. et al. A Functional SNP in BNC2 Is Associated with Adolescent Idiopathic Scoliosis. Am. J. Hum. Genet. 97, 337–342 (2015).
Sharma, S. et al. A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun. 6, 6452, https://doi.org/10.1038/ncomms7452 (2015).
Zhu, Z. et al. Genome-wide association study identifies new susceptibility loci for adolescent idiopathic scoliosis in Chinese girls. Nat. Commun. 6, 8355, https://doi.org/10.1038/ncomms9355 (2015).
Londono, D. et al. A meta-analysis identifies adolescent idiopathic scoliosis association with LBX1 locus in multiple ethnic groups. J. Med. Genet. 51, 401–406 (2014).
Cao, Y., Min, J., Zhang, Q., Li, H. & Li, H. Associations of LBX1 gene and adolescent idiopathic scoliosis susceptibility: a meta-analysis based on 34,626 subjects. BMC Musculoskelet. Disord. 17, 309 (2016).
Chettier, R., Nelson, L., Ogilvie, J. W., Albertsen, H. M. & Ward, K. Haplotypes at LBX1 have distinct inheritance patterns with opposite effects in adolescent idiopathic scoliosis. PLoS One. 10, e0117708 (2015).
Grauers, A. et al. Candidate gene analysis and exome sequencing confirm LBX1 as a susceptibility gene for idiopathic scoliosis. Spine J. 15, 2239–2246 (2015).
Kou, I. et al. Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 45, 676–679 (2013).
Raggio, C. L. Sexual dimorphism in adolescent idiopathic scoliosis. Orthop. Clin. North Am. 37, 555–558 (2006).
Dixon, J. R. et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 518, 331–336 (2015).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 348, 648–660 (2015).
Hsu, Y. H. et al. Targeted sequencing of genome wide significant loci associated with bone mineral density (BMD) reveals significant novel and rare variants: the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) targeted sequencing study. Hum. Mol. Genet. 25, 5234–5243 (2016).
Cuyvers, E. et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol. 14, 814–822 (2015).
Raychaudhuri, S. et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 43, 1232–1236 (2011).
Morgenthaler, S. & Thilly, W. G. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutat. Res. 615, 28–56 (2007).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Xu, J. F. et al. Association of GPR126 gene polymorphism with adolescent idiopathic scoliosis in Chinese populations. Genomics. 105, 101–107 (2015).
Bonnefond, A. et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 44, 297–301 (2012).
Rivas, M. A. et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073 (2011).
Hershkovich, O. et al. Association between body mass index, body height, and the prevalence of spinal deformities. Spine J. 14, 1581–1587 (2014).
Qiu, Y. et al. Decreased circulating leptin level and its association with body and bone mass in girls with adolescent idiopathic scoliosis. Spine (Phila Pa 1976). 32, 2703–2710 (2007).
Tam, E. M. et al. Lower Muscle Mass and Body Fat in Adolescent Idiopathic Scoliosis Are Associated With Abnormal Leptin Bioavailability. Spine (Phila Pa 1976). 41, 940–946 (2016).
Karner, C. M., Long, F., Solnica-Krezel, L., Monk, K. R. & Gray, R. S. Gpr126/Adgrg6 deletion in cartilage models idiopathic scoliosis and pectus excavatum in mice. Hum. Mol. Genet. 24, 4365–4373 (2015).
Fan, Y. H. et al. SNP rs11190870 near LBX1 is associated with adolescent idiopathic scoliosis in southern Chinese. J. Hum. Genet. 57, 244–246 (2012).
Song, Y. Q. et al. Lumbar disc degeneration is linked to a carbohydrate sulfotransferase 3 variant. J. Clin. Invest. 123, 4909–4917 (2013).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4, 7 (2015).
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564–1573 (2010).
Price, A. L., Zaitlen, N. A., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–463 (2010).
Mitchell, B. D. et al. Using previously genotyped controls in genome-wide association studies (GWAS): application to the Stroke Genetics Network (SiGN). Front Genet. 5, 95 (2014).
Grauers, A., Danielsson, A., Karlsson, M., Ohlin, A. & Gerdhem, P. Family history and its association to curve size and treatment in 1,463 patients with idiopathic scoliosis. Eur. Spine J. 22, 2421–2426 (2013).
Andersen, M. O., Christensen, S. B. & Thomsen, K. Outcome at 10 years after treatment for adolescent idiopathic scoliosis. Spine (Phila Pa 1976). 31, 350–354 (2006).
Grauers, A. et al. Prevalence of Back Problems in 1069 Adults With Idiopathic Scoliosis and 158 Adults Without Scoliosis. Spine (Phila Pa 1976). 39, 886–892 (2014).
Gerdhem, P. & Akesson, K. Rates of fracture in participants and non-participants in the Osteoporosis Prospective Risk Assessment study. J. Bone Joint Surg. Br. 89, 1627–1631 (2007).
McGuigan, F. E. et al. Variation in the BMP2 gene: bone mineral density and ultrasound in young adult and elderly women. Calcif. Tissue Int. 81, 254–262 (2007).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 26, 2190–2191 (2010).
Nagai, A. et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Momozawa, Y. et al. Low-frequency coding variants in CETP and CFB are associated with susceptibility of exudative age-related macular degeneration in the Japanese population. Hum. Mol. Genet. 25, 5027–5034 (2016).
Acknowledgements
We thank all participating subjects and clinical staff at collaborating institutes. We specially thank Nobumasa Suzuki, Masashi Saito, Michihiro Kamata and Hitoshi Hase for patient recruitment, and Yoshie Takahashi, Sayaka Tominaga, Tomomi Oguma and the members of Laboratory for Genotyping Development for technical assistance. We also thank Dr. Jianguo Zhang, Jianxiong Shen, Shugang Li, Yipeng Wang, Hong Zhao and Yu Zhao from Peking Union Medical College Hospital for patient enrollment and clinical evaluation. This work was conducted as part of the BioBank Japan Project supported by the Japan Agency for Medical Research and Development and by the Ministry of Education, Culture, Sports, Sciences and Technology of the Japanese government and was supported by JSPS KAKENHI Grant Number JP16H05453 (to MM), Hong Kong Health and Medical Research Fund (HMRF No. 04152256) (to YQS), the Swedish Research Council (number K-2013-52X-22198-01-3) (to AG and PG), National Natural Science Foundati on of China (81501852), Beijing Natural Science Foundation (7172175), Beijing nova program (Z161100004916123), Beijing nova program interdisciplinary collaborative project (xxjc201717) and the 2016 Milstein Medical Asian American Partnership Foundation Fellowship Award in Translational Medicine (to NW) and the Scoliosis Research Society, the NIH (P01 HD084387) and the Texas Scottish Rite Hospital Research Fund (to CAW).
Author information
Authors and Affiliations
Consortia
Contributions
S.I. designed the project and provided overall project management. I.K. performed the multi-ethnic meta-analysis and I.K. and S.I. prepared the manuscript. I.K., K.W., Y.T., K.T., Y.O., J.S.C.R.G. and M.M. collected and managed Japanese samples; Y.Q. collected and managed Nanjing samples; H.Z., T.Z., D.H. and P.S. collected and managed Guangzhou samples and I.K. and H.Z. analyzed the Guangzhou data; Y.T., Y.H.F. and Y.Q.S. collected and managed Hong Kong samples; G.L., Z.W., G.Q. and N.W. collected and managed Beijing samples; A.K., T.S.R.H.C.C.G. and C.A.W. collected and managed USA samples; A.G., E.E., J.K. and P.G. collected and managed Scandinavia samples; I.K., Y.M., Y.I. and M.K. designed and performed target sequencing and analyzed the data. All authors read and approved the final manuscript before submission.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A comprehensive list of consortium members appears at the end of the paper
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kou, I., Watanabe, K., Takahashi, Y. et al. A multi-ethnic meta-analysis confirms the association of rs6570507 with adolescent idiopathic scoliosis. Sci Rep 8, 11575 (2018). https://doi.org/10.1038/s41598-018-29011-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29011-7
This article is cited by
-
Genetic variants associated with the occurrence and progression of adolescent idiopathic scoliosis: a systematic review protocol
Systematic Reviews (2022)
-
Genetic animal modeling for idiopathic scoliosis research: history and considerations
Spine Deformity (2022)
-
Adhesion G protein-coupled receptor gluing action guides tissue development and disease
Journal of Molecular Medicine (2022)
-
Genome-wide association study identifies 14 previously unreported susceptibility loci for adolescent idiopathic scoliosis in Japanese
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
