
Abstract
Small-conductance Ca2+-activated K+ (SK) channels mediate medium afterhyperpolarization in the neurons and play a key role in the regulation of neuronal excitability. SK channels are potential drug targets for ataxia and Amyotrophic Lateral Sclerosis (ALS). SK channels are activated exclusively by the Ca2+-bound calmodulin. Previously, we identified an intrinsically disordered fragment that is essential for the mechanical coupling between Ca2+/calmodulin binding and channel opening. Here, we report that substitution of a valine to phenylalanine (V407F) in the intrinsically disordered fragment caused a ~6 fold increase in the Ca2+ sensitivity of SK2-a channels. This substitution resulted in a novel interaction between the ectopic phenylalanine and M411, which stabilized PIP2-interacting residue K405, and subsequently enhanced Ca2+ sensitivity. Also, equivalent valine to phenylalanine substitutions in SK1 or SK3 channels conferred Ca2+ hypersensitivity. An equivalent phenylalanine substitution in the Caenorhabditis elegans (C. elegans) SK2 ortholog kcnl-2 partially rescued locomotion defects in an existing C. elegans ALS model, in which human SOD1G85R is expressed at high levels in neurons, confirming that this phenylalanine substitution impacts channel function in vivo. This work for the first time provides a critical reagent for future studies: an SK channel that is hypersensitive to Ca2+ with increased activity in vivo.
Similar content being viewed by others


Introduction
Calcium (Ca2+) mediates a variety of cellular signaling processes, including regulation of enzymatic activities, gene expression, synaptic transmission and ion channel activities1,2. Small conductance Ca2+-activated K+ (SK) channels are a unique group of ion channels that are activated exclusively by intracellular Ca2+ levels. The Ca2+-binding protein calmodulin (CaM) is constitutively associated with SK channels and Ca2+-binding by CaM activates these channels3. SK channels play a key role in the regulation of membrane excitability of neurons by Ca2+. In the central nervous system, activation of SK channels mediates the medium afterhyperpolarization (mAHP) and reduces the firing frequency of action potentials, thus contributing to regulation of neuronal excitability4,5.
In neurons, the Ca2+ sensitivity of SK channels is subject to negative modulation by neurotransmitters such as acetylcholine6 and norepinephrine7. Increased signaling by these neurotransmitters results in increased CaM phosphorylation at threonine 79 (T79). This phosphorylation can decrease the Ca2+ sensitivity of SK channels and, thus, increase neuronal excitability3,8. On the other hand, small molecules can increase the Ca2+ sensitivity of SK channels. For example, 1-ethyl-2-benzimidazolinone (1-EBIO)9 increases Ca2+ sensitivity of SK channels and reduces neuronal excitability10. And, NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) increases Ca2+ sensitivity of SK channels with a higher potency11.
ALS has commonalities with Spinal Muscular Atrophy (SMA), as both diseases result in spinal cord motor neuron degeneration and these diseases share other phenotypic, genetic, and molecular similarities. A missense mutation in the vesicle-associated membrane protein/synaptobrevin-associated membrane protein B (VAPB) gene causes both late-onset SMA and ALS12. These disorders may share a common neurodegenerative pathway and respond to similar treatments (e.g. riluzole). Previously, we reported that SK channels are genetic modifiers in vertebrate and invertebrate models of SMA13 and that SK channels are likely a critical target for the neuroprotective effects of riluzole in these models14.
Because of their critical roles in neuronal excitability, SK channels have been proposed as a drug target for motor neuron diseases and movement disorders13,14,15,16,17,18. Both SK2 and SK3 channel subtypes are expressed in the mammalian spinal motor neurons19. They play a critical role in the mAHP and the excitability of motor neurons20,21,22. Positive modulators of SK channels can also be used to regulate firing rates of cerebellar Purkinje cells. SK channel positive modulators Chlorzoxazone (CHZ), 1-EBIO and CyPPA normalize Purkinje cell firing and exert beneficial effects in mouse models of ataxia16,17,23,24,25. Riluzole showed promising results in recent phase II studies in a mixed population of ataxia patients26 and in inherited ataxia patients27; it was suggested that the ability of riluzole to facilitate SK channel activity was responsible for the beneficial impacts observed28.
Yet, how SK channel Ca2+ sensitivity is modulated remains largely unclear, despite previous work29,30,31,32. Functional SK channels are tetrameric and composed of 4 channel subunits, like other voltage-dependent potassium channels. Each channel subunit contains six transmembrane α-helical domains that are denoted S1–S6. The Ca2+-sensor CaM associates with a channel CaM-binding domain (CaMBD), which is located within the channel C-terminus. Ca2+ binding to CaM induces conformational changes in both CaM and the channel CaMBD; these subsequently trigger opening of the channel pore33,34,35. A simplified gating scheme for Ca2+-dependent SK channel activation includes two steps: (1) binding of Ca2+ to CaM associated with the SK channel and (2) mechanical coupling between Ca2+ binding to CaM, CaMBD conformation change, and consequent channel opening (Fig. 1a). Thus, the Ca2+ sensitivity of SK channels could theoretically be modulated at either one of these two steps. Neither SK channel positive modulators (e.g. NS309) nor phosphorylation of CaM T79 influences the first step of the simplified gating scheme; i.e., Ca2+ binding to CaM-channel complex30. Instead, both SK channel positive modulators and phosphorylation of CaM T79 exert their modulation through the second step; i.e., mechanical coupling to channel opening. Previous mutagenesis and MD simulations work also established phosphatidylinositol 4,5-biphosphate (i.e. PIP2) as a critical factor in SK channel opening, modulation of SK channels by SK positive modulators32 and phosphorylation of CaM Thr7931.

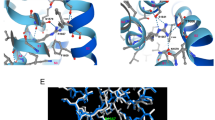
The SK channels IDF connects the CaMBD and transmembrane domain S6. (a) A simplified gating scheme for Ca2+-dependent SK channel activation may include two steps: (1) binding of Ca2+ to CaM associated with the SK channel and (2) mechanical coupling between Ca2+ binding to CaM and subsequent channel opening. (b) Amino acid sequence alignment of mammalian SK channels at the region connecting the CaMBD and the S6 transmembrane domain. Seven amino acid residues from the IDF shown in magenta are the focus of this study. Human and rat sequences are identical at this region. (c) Seven residues from the IDF (in magenta) are located between the CaMBD and the transmembrane S6 domain.
We previously identified a flexible region (E404-M412) in the SK2-a channel that is essential for the second step of the hypothesized gating scheme and, thus, Ca2+ sensitivity of the SK2-a channel30,31,32. Flexible or intrinsically disordered fragments (IDFs) in proteins can be important for modulation of protein function36,37,38. We considered the possibility that the SK2-a IDF might play a critical role in regulation of SK channel Ca2+ sensitivity. Here, we report a valine to phenylalanine mutation in the IDF results in ~6 fold increase in SK2-a channel Ca2+ sensitivity. A combination of electrophysiological, X-ray crystallographic and computational approaches was utilized to determine the structural basis of SK2-a channel Ca2+ hypersensitivity. We found that the ectopic phenylalanine residue forms a novel interaction with methionine 411 in the IDF, stabilizes a putative PIP2-interacting residue lysine 405, and consequently increases channel Ca2+ sensitivity. We found that the equivalent mutation in the orthologous Caenorhabditis elegans (C. elegans) KCNL-2 protein partially rescues locomotion defects in a previously described model of superoxide dismutase (SOD1)-associated ALS. This result suggests that the valine to phenylalanine substitution increases Ca2+ sensitivity of SK channels in vivo. Combined, the studies reported here use biophysical, structural and genetic techniques, to develop a mutant SK channel that is hypersensitive to Ca2+.
Results
Mutagenesis of the residues in the IDF of SK2-a channel
In our previous study, we identified an IDF region (Fig. 1b, E404-M412) in the SK2-a channel that is essential for modulation of channel Ca2+ sensitivity30,31,32. IDF conformation is flexible and, thus, invisible in the apo-crystal structure, whereas the drug NS309 stabilizes IDF, resulting in a well-defined conformation in the crystal structure (Fig. 1c)30. One of the nine residues of the IDF, the negatively charged E404, forms a salt bridge with K75 of CaM. Mutation of E404 to other amino acids results in loss of SK2-a channel function30. K405 may be involved in the channel-PIP2 interactions and consequent modulation of SK2-a channel Ca2+ sensitivity31,32. Here, we investigate the remaining seven residues (Fig. 1b,c, H406-M412, highlighted in magenta) of the IDF, using site-directed mutagenesis, electrophysiological recordings, crystallography and molecular dynamic (MD) simulations.
We first determined if the side chains of these residues are important for the Ca2+ sensitivity of SK2-a channels, using an alanine scanning strategy (Fig. 2a). The effect of substituting alanine at each of the IDF seven residues on the Ca2+-dependent channel activation was examined using inside-out patch clamp recordings in into TsA201 cells. Compared with wild type (WT) SK2-a channel (EC50 = 0.30 ± 0.016 μM, n = 8), the alanine substitution at H406, V407, H408, N409, M411 or M412 did not change Ca2+-dependent activation of SK2-a currents. EC50 values for Ca2+ were 0.36 ± 0.058 μM (n = 7, P = 0.67), 0.34 ± 0.037 μM (n = 5, P = 0.95), 0.38 ± 0.045 μM (n = 4, P = 0.65), 0.36 ± 0.025 μM (n = 8, P = 0.73), 0.32 ± 0.021 μM (n = 7, P = 0.99) and 0.31 ± 0.020 μM (n = 7, P = 0.99), respectively. However, replacing aromatic amino acid F410 with an alanine (F410A) effectively reduced the SK2-a channel Ca2+ sensitivity (Fig. 2b). The F410A mutant channel was significantly less responsive to Ca2+, with an EC50 of 0.52 ± 0.036 μM (n = 5, P < 0.0001).

Mutations in the IDF change SK2-a channel Ca2+ sensitivity. (a) Dose-dependent activation by Ca2+ of the WT and alanine mutant SK2-a channels. (b) EC50 values for activation by Ca2+ of the WT and alanine mutant channels. (c) Dose-dependent activation by Ca2+ of the WT and phenylalanine mutant SK2-a channels. (d) EC50 values for activation by Ca2+ of the WT and phenylalanine mutant channels. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc tests. All data are presented as mean ± s.e.m.
The identification of a mutant SK2-a channel hypersensitive to Ca2+
As mutation of the only phenylalanine residue (F410) in the IDF resulted in reduced Ca2+ sensitivity, we hypothesized that introduction of an additional aromatic residue into the IDF might also change SK2-a channel Ca2+ sensitivity. We undertook phenylalanine scanning of the six IDF residues excluding F410 (Fig. 2c) and tested the effect of the phenylalanine substitutions on Ca2+-dependent channel activation using inside-out patches. Phenylalanine substitution at H406, H408, N409 and M412 did not change the activation of the SK2-a channel by Ca2+, compared to the WT channel, with EC50 values of 0.27 ± 0.036 μM (n = 6, P = 0.95), 0.25 ± 0.023 μM (n = 7, P = 0.55), 0.27 ± 0.037 μM (n = 7, P = 0.92) and 0.32 ± 0.013 μM (n = 7, P = 0.95), respectively. Changing M411 to phenylalanine modestly enhanced SK2–a channel Ca2+ sensitivity from 0.30 ± 0.016 μM (n = 8) to 0.15 ± 0.011 μM (n = 8, P < 0.0001). But, substituting a phenylalanine for hydrophobic V407 dramatically enhanced SK2-a channel Ca2+ sensitivity by almost six-fold, from 0.30 ± 0.016 μM (n = 8) to 0.051 ± 0.0024 μM (n = 8, P < 0.0001) (Fig. 2d).
We next examined the relationship between the size of the side chain at the residue 407 and SK2-a channel Ca2+ sensitivity. Amino acids of different sizes were introduced at position 407 by site-directed mutagenesis and their impact on SK2-a channel Ca2+ sensitivity was tested with inside-out patch recordings (Supplementary Fig. S1A). Neither alanine nor leucine substitution changed SK2-a channel Ca2+ sensitivity, with EC50 values of 0.34 ± 0.037 μM (n = 5, P = 0.49) and 0.30 ± 0.023 μM (n = 6, P = 0.97), respectively (Supplementary Fig. S1B). On the other hand, phenylalanine V407F or tryptophan V407W substitution strongly increased SK2-a channel Ca2+ sensitivity, with EC50 values of 0.051 ± 0.0024 μM (n = 8, P < 0.0001) and 0.059 ± 0.0062 μM (n = 6, P < 0.0001), respectively (Supplementary Fig. S1B). The impact of V407F or V407W was indistinguishable (P value of 0.99), suggesting that these substitutions at residue V407 were equally effective in enhancing SK2-a channel Ca2+ sensitivity.
The structural insight into the Ca2+ hypersensitivity of the V407F mutant SK2-a channel
Next, we addressed how mutation of V407F might increase SK2-a channel Ca2+ sensitivity. First, we determined if the V407F substitution altered global conformation of the CaM–SK2 fragment complex using X-ray crystallography. CaM was co-crystallized with the mutant V407F SK2-a channel fragment, in the presence of Ca2+. The crystallographic data collection and refinement statistics are summarized in Supplementary Table S1.
In the previously determined WT structure, the electron density for the IDF region was very poor (Supplementary Fig. S2A). The IDF was missing from the WT structure (PDB Code: 4J9Y). In the V407F mutant structure, the electron density for the IDF region was remarkably improved as a result of the stabilization (Supplementary Fig. S2B). Previously, we found that the IDF conformation in the WT structure can be stabilized by a small molecule called NS309 and determined through crystallography (PDB Code: 4J9Z). Similarly, in the V407F mutant structure, the electron density of the IDF was further improved by NS309 (Supplementary Fig. S2C). This V407F mutant structure with NS309 (PDB Code: 6ALE) was then compared with the WT structure with NS309 (PDB Code: 4J9Z). The V407F substitution did not dramatically shift global structure of complex, when compared the WT complex (Fig. 3a), with a root mean square deviation (RMSD) of 0.246 Å and 0.262 Å for CaM and the SK2 fragment, respectively. However, local differences exist between the two structures in the conformation of the IDF, with an RMSD of 0.798 Å.

V407F stabilizes K405. (a) Overlaid crystal structures of WT (grey) and V407F (salmon) protein complexes show similar global conformation. (b) In MD simulations, the RMSF plot shows the difference in the structural flexibility of the residues between the WT and V407F mutant structures. (c) The difference RMSF plot (ΔRMSF = RMSF(V407F) − RMSF(WT)) shows the reduced structural flexibility of K405 in the IDF of the V407F mutant structure. (d) Neutralizing mutation K405N right-shifts the dose-dependent activation of the V407F mutant SK2-a channel by Ca2+. Statistical analysis was performed using two-tailed t-test. All data are presented in mean ± s.e.m.
We undertook MD simulations to compare the conformational dynamics of these two structures. The structural flexibility of the WT and V407F structures is shown as a plot of root mean square fluctuation (RMSF) results in Fig. 3b. Similar structural flexibility was observed in the rigid part (e.g. α-helices) of the protein complex between WT and V407F structures. On the other hand, the V407F mutant channel exhibited reduced fluctuations in the IDF region, with the largest difference at residue K405 (Supplementary Fig. S3A). The RMSF of K405 in the WT protein complex was 2.57 angstrom, whereas the RMSF of the same residue in the V407F mutant channel was 1.22 angstrom. The difference between the two RMSF data sets (ΔRMSF = RMSF(V407F) − RMSF(WT)) is visible in Fig. 3c. The most prominent peak appears at the residue of K405 (Supplementary Fig. S3B). The negative peak amplitude of −1.35 angstrom indicates reduced structural flexibility (i.e. stabilization) of K405 in the mutant V407F complex, when compared with the WT complex.
Previously, we identified K405 as one of the positively charged residues that constitute a putative binding site for PIP231. PIP2 is critical in mediating the mechanical coupling between Ca2+ binding to CaM and subsequent channel opening (Fig. 1a). MD simulations in the presence of PIP2 revealed the enhancement of PIP2 interaction energy in the V407F mutant structure compared to the WT structure (Supplementary Fig. S4). The median PIP2 interaction energies were −21.51 kcal/mol and −27.04 kcal/mol, in the WT and mutant structures, respectively. Hence, the Ca2+ hypersensitivity of the V407F mutant channel might be attributed to the enhanced mechanical coupling between Ca2+ binding and channel opening, as a result of K405 stabilization. We tested this hypothesis by introducing a neutralizing K405N mutation into the V407F mutant SK2-a channel. The double mutant (K405N/V407F) channel yielded a ~3.9 fold decrease in Ca2+ sensitivity (Fig. 3d), with an EC50 of 0.20 ± 0.023 μM (n = 7, P < 0.0001), compared to 0.051 ± 0.0024 μM (n = 8) for the V407F single mutant. In our previous report31, the K405N mutation in the WT SK2-a channel decreased Ca2+ sensitivity to 0.62 ± 0.046 μM (n = 6), which is a ~2 fold change from the WT. We conclude that the V407F mutant channel is more susceptible than the WT channel to the neutralizing impact of K405. The elevated Ca2+ sensitivity of the V407F mutant channel may be attributed, at least in part, to stabilizing K405 (Fig. 3b,c), a key residue for PIP2 interactions and subsequent mechanical coupling31.
In the V407F mutant structure, residues M411, V420, L463, K467 and Q470 are in the vicinity of the ectopic phenylalanine 407 (Supplementary Fig. S5A). We tested the effect of alanine substitutions of these five residues on the hypersensitivity of V407F mutant channel to Ca2+ activation using inside-out patches. Among these five residues tested, M411A mutation had the biggest impact. The double mutant channel V407F/M411A drastically decreased the Ca2+ sensitivity, with an EC50 of 0.21 ± 0.013 μM (n = 9, P < 0.0001), compared to 0.051 ± 0.0024 μM (n = 8) of the single V407F mutant (Supplementary Fig. S5B).
Next, we compared the IDF conformation in the WT and V407F mutant structures. When WT and V407F mutant protein complex structures are overlaid (Fig. 4a), V407 is 7.9 angstrom away from the IDF hydrophobic residue M411 in the WT structure, but the 407-to-411 distance is shortened to 4.7 angstrom in the V407F mutant structure. We used the MD simulations to explore changes in the dynamic interactions between amino acid residues, with particular focus on the IDF region. In the distance distribution histogram of MD simulations, the 407-to-411 median distance is 7.13 (V407-to-M411) angstroms for the WT structure (Fig. 4b). We note that M411A substitution did not change the Ca2+ sensitivity of the WT SK2-a channel (Fig. 2a,b), echoing the median 407-to-411 distance of 7.13 angstroms and the lack of prominent 407-to-411 interaction in the MD simulation of the WT structure (Fig. 4b). The distance distribution histogram also demonstrated a much shorter 407-to-411 median distance of 4.13 (F407-to-M411) angstroms for the mutant structure, indicating a strengthened hydrophobic interaction between the residues 407 and 411 (Fig. 4b). We calculated the interaction energies between the residues 407 and 411 using the Discovery Studio 2017 molecular modeling program (BIOVIA). The 407-to-411 interaction energies were −0.12 kcal/mol and −2.34 kcal/mol in the WT and mutant structures, respectively. We speculated that interruption of this 407-to-411 interaction through mutagenesis of M411 might compromise the Ca2+ hypersensitivity of the V407F mutant channel. To address this, we first introduced a phenylalanine at the location 411 in the V407F background, and resulted in the double mutant channel V407F/M411F. The double mutant did not exhibit significant change in the Ca2+ sensitivity (Fig. 4c), with an EC50 of 0.040 ± 0.0052 μM (n = 9, P = 0.89), compared to the single V407F mutant 0.051 ± 0.0024 μM (n = 8) (Fig. 4d). On the other hand, the double mutant channel V407F/M411A drastically decreased the Ca2+ sensitivity (Fig. 4d). Notably, a single M411A mutation did not change the Ca2+ sensitivity compared to the WT channel (Fig. 2b), suggesting that the side chain of M411 is required for interaction with the ectopic phenylalanine but not the original valine. Therefore, the V407F substitution results in formation of a new hydrophobic interaction with M411, which might drive stabilization of the IDF residues, especially PIP2-interacting K405 (Fig. 3c).

V407F interacts with M411. (a) Local conformational changes in the IDF of the V407F mutant crystal structure (salmon) compared to that of the WT structure (grey). (b) In MD simulations, the distance distribution histogram shows a shorter distance between residue 407 and residue 411 in the V407F mutant protein complex compared to the WT complex. (c) M411A mutation right-shifted the Ca2+ dependent activation of the V407F mutant SK2-a channel. (d) M411A mutation significantly compromised the Ca2+ hypersensitivity of the V407F mutant SK2-a channel. Statistical analysis was performed using one-way ANOVA followed by Tukey’s post hoc tests. All data are presented in mean ± s.e.m.
The effectiveness of the equivalent valine to phenylalanine mutations in SK1 and SK3 channels
There are three mammalian SK channel subtypes expressed in the neurons (SK1, SK2, and SK3)3. IDF region amino acid sequences are completely identical in all three subtypes, including the valine corresponding to V407 (Fig. 1b). We determined if mutating these valines would change the Ca2+ sensitivity of SK1 and SK3 channels. Substitution of phenylalanine for V378 in the SK1 channel shifted the Ca2+ dose response curve to lower concentrations (Fig. 5a) and changed the EC50 value from 0.26 ± 0.017 μM (n = 6) to 0.081 ± 0.0079 μM (n = 6; P < 0.0001) (Fig. 5b). Substitution of phenylalanine for V560 in the SK3 channel has a similar effect (Fig. 5c). The EC50 values for Ca2+ activation of the WT SK3 and mutant SK3-V560F channels are 0.30 ± 0.024 μM (n = 8) and 0.080 ± 0.0095 μM (n = 8; P < 0.0001), respectively (Fig. 5d). Thus, substitution of phenylalanine at the cognate valine in all three SK channel subtypes effectively increases the Ca2+ sensitivity of these channels heterologously expressed in cell culture.

Phenylalanine substitution of the cognate valine in SK1 and SK3 channels increases Ca2+ sensitivity. (a) Dose-dependent activation of the WT and V378F mutant SK1 current by Ca2+. (b) EC50 values for the activation of the WT and mutant SK1 channels by Ca2+. (c) Dose-dependent activation of the WT and V506F mutant SK3 current by Ca2+. (d) EC50 values for the activation of the WT and mutant SK3 channels by Ca2+. Statistical analysis was performed using two-tailed t-test. All data are presented in mean ± s.e.m.
Substitution of the cognate IDF valine in C. elegans kcnl-2 ameliorates defects in a C. elegans model of SOD1 Amyotrophic Lateral Sclerosis
To address the impact of IDF substitutions and SK2 channel activation in vivo, we used a previously validated model of ALS, in which C. elegans expresses human superoxide dismutase (SOD1) in neurons39.
Animals expressing ALS mutant SOD1G85R-YFP fusion protein in all neurons (hSOD1G85R) have impaired locomotion, compared to animals expressing human wild-type SOD1-YFP (hSOD1WT) protein39. To determine the impact of SK2 channel activity on ALS-associated defects in this model, we re-created the V698F (equivalent to SK2-a V407F) mutation in the orthologous C. elegans gene, kcnl-2 (Fig. 6a), using CRISPR/Cas9-mediated homologous recombination-based genome editing. To test the impact of kcnl-2(V698F) mutation on the locomotion of hSOD1WT or hSOD1G85R animals, we used a center-out dispersal locomotion assay (Fig. 6b). Most animals expressing hSOD1WT dispersed quickly, while animals expressing mutant hSOD1G85R were defective (n = 60; P = 0.0006) (Fig. 6c). The kcnl-2(V698F) mutation significantly increased dispersal in animals expressing hSOD1G85R, compared to hSOD1G85R animals carrying the wild type kcnl-2 gene (n = 60; P = 0.0105) (Fig. 6c). Yet, kcnl-2(V698F) activity did not alter dispersal of hSOD1WT animals, consistent with a specific impact on animals expressing hSOD1G85R (Fig. 6c). Thus, kcnl-2(V698F) IDF mutation partially rescues locomotion defects in a C. elegans model of SOD1 ALS.

Phenylalanine substitution of the cognate valine in C. elegans kcnl-2 ameliorates SOD1G85R ALS model locomotion defects. (a) Amino acid sequence alignment of mammalian SK2 channels and orthologous C. elegans KCNL-2 at the region connecting the CaMBD and the S6 transmembrane domain. Amino acids are numbered according to KCNL-2 isoform j. Seven amino acid residues from the IDF shown in magenta are the focus of this study. Human and rat SK2 sequences are identical in this region. (b) Diagram: to measure locomotion defects, adult animals were scored for dispersal 1 hour after they were placed at the center of a thin bacterial lawn on a 60-mm petri dish (food source). Coordinated animals disperse radially from the center of the bacterial lawn. Uncoordinated animals disperse more slowly. (c) Pan-neuronal expression of hSOD1G85R slowed dispersal, compared to hSOD1WT controls (one-tailed t-test, P = 0.0006; n = 60). Phenylalanine substitution of the cognate valine in C. elegans kcnl-2 increased dispersal in hSOD1G85R animals, compared to hSOD1G85R animals carrying the kcnl-2 wild type allele (one-tailed t-test, P = 0.0105; n = 60). Each determination is average of four trials. All data are presented in mean ± s.e.m. All animals were tested in sod-1(tm776) null background.
Discussion
The SK channels are a unique group of potassium channels with an important role in regulating membrane excitability. These channels are activated exclusively by Ca2+-bound CaM40. Therefore, understanding the Ca2+ sensitivity of the CaM/SK complex is critical8,31,35. Previously described modifications usually decrease SK channel function. For example, phosphorylation of CaM threonine79 (T79) inhibits SK channels; this led to creation of the phosphomimetic CaM-T79D substitution that negatively modulates SK2-a channel Ca2+ sensitivity8. Or, in another example, an alternative RNA splice variants of rat or chicken SK2 channels (SK2-b) reduces channel Ca2+ sensitivity35,41. Also, disturbing positively charged residues in the putative SK2 PIP2 binding site decreases Ca2+ sensitivity31,32. But, to our knowledge there are no reports of SK channels with increased Ca2+ sensitivity. For the intermediate-conductance Ca2+-activated K+ (IK) channels, there are amino acid changes that can lock the channel into a low-conducting state with a high open probability, rather than the full conducting state42. Here, we report for the first time an alteration that increases SK channel sensitivity to Ca2+. The V407F substitution of SK2-a channel will facilitate future structural studies on the modulation of SK channel activity, as well as genetic and functional studies of SK channels and their impact in neurodegenerative diseases.
The V407F substitution is located in the SK2-a channel intrinsically disordered fragment (IDF), between E404 and M412 (Fig. 1b). Previous work demonstrated that IDF residue K405 is critical for the channel-PIP2 interactions31, when PIP2 mediates the mechanical coupling between Ca2+ binding to CaM and subsequent channel opening. The newly identified V407F substitution stabilizes K405 in MD simulations (Fig. 3c), reminiscent of the impact of the small molecule NS309 on SK channel IDF conformation32. In turn, stabilization of K405 enhances the mechanical coupling to channel opening (Fig. 1a), which is the likely mechanism for the Ca2+ hypersensitivity of the V407F mutant channels. Methionine 411 in the IDF seems to form a stronger hydrophobic interaction with the V407F mutation in the mutant channel structure, than with the V407 in the WT structure (Fig. 4a,b). The mutation of M411 to a phenylalanine also modestly increases the Ca2+ sensitivity of the SK2-a channel (Fig. 2c,d). The double mutant V407F/M411F, however, does not further enhance the Ca2+ sensitivity beyond the single V407F mutant (Fig. 4c,d), suggesting that one phenylalanine mutation at the residue 407 is sufficient to facilitate the 407-to-411 hydrophobic interaction. While the present study was in the final stage of review, cryo-electron microscopy (cryo-EM) structures of a human IK (also called SK4) channel have been reported43. Using the IK channel structure at the Ca2+-bound state (PDB code: 6CNN), we were able to generate a homology model of rat SK2 channel with the V407F substitution (Supplementary Fig. S6a). In this SK2 channel homology model, the N-lobe of CaM interacts with the S4–S5 linker of the SK2 channel in the presence of Ca2+. The CaMBD of the SK2 channel also forms substantial contacts with the S4–S5 linker in the presence of Ca2+. Interestingly, both residue 407 and residue 411 are located at the interface between CaMBD and the S4–S5 linker (Supplementary Fig. S6b). A top down view of the homology model shows the location of the two residues at the CaMBD-S4-S5 linker interface even more clearly (Supplementary Fig. S6c and d). The S4–S5 linker has been identified to be crucial for the Ca2+-dependent activation of the IK channel43. If the SK2 channel utilizes the same gating mechanism as IK channels, then the V407F substitution may enhance the hydrophobic interactions between the CaMBD and the S4–S5 linker, and thus increase the Ca2+ sensitivity of the SK2 channel. However, the possible interactions between the CaMBD and the S4–S5 linker and the effect of the valine to phenylalanine substitution on the interactions still need to be tested in the context of the SK2 channel. Equivalent valine to phenylalanine changes in SK1 and SK3 channels also confers Ca2+ hypersensitivity, addressing the conserved nature of IDF interactions (Fig. 5).
In resting neurons, the intracellular Ca2+ concentration is estimated to be ~0.1 μM2. As a result, the WT SK2-a channel (EC50 ~0.30 μM) is usually in the closed state in resting neurons, whereas the mutant V407F SK2-a channel (EC50 ~0.051 μM) could be in the activated state more than ~90% of the time, barring other regulatory events (Fig. 2c). To determine the in vivo consequences of SK2 channel activation in an intact nervous system, we turned to the model organism, C. elegans. The C. elegans genome contains four genes that encode SK family channels (kcnl-1 through kcnl-4)44, but kcnl-2 encodes the channel most similar to the mammalian SK2 channel. Previous work demonstrated that loss of C. elegans kcnl-2 channel decreases sensitivity to compounds that directly modulate SK2 channel function (apamin and riluzole) and loss of kcnl-2 function has behavioral consequences14. Loss of kcnl-2 activity exacerbates neuromuscular defects in a C. elegans model of Spinal Muscular Atrophy and kcnl-2 is required for the beneficial effects of riluzole in this model14. Riluzole increases SK2 channel activity, which may contribute to the modest benefits that riluzole provides to ALS patients.
Here, we used a C. elegans model of SOD1 ALS to examine the consequences of the cognate V698F mutation of kcnl-2 in the intact nervous system. In this C. elegans ALS model, human SOD1 containing the patient G85R mutation is expressed at relatively high levels in C. elegans neurons (hSOD1G85R). In control animals, wild type human SOD1 protein is similarly expressed at high levels (hSOD1WT). Substitution of phenylalanine for the cognate valine in the C. elegans KCNL-2 IDF region had no obvious impact on C. elegans on locomotion, but did suppress locomotion defects in animals expressing SOD1G85R (Fig. 6c). These results are consistent with previous work on the role of SK2 channels in ALS and confirm that this phenylalanine substitution at this valine impacts channel function in vivo.
Given the importance of SK2 channels in neuronal excitability, understanding their regulation is critical. Here, we focus on the IDF fragment of SK2 and discover a phenylalanine to valine amino acid substitution that can increase SK2 channel Ca2+ sensitivity in tissue culture and also increases SK2 function in vivo in an animal model of ALS. This is the first SK channel variant with increased activity due to increased Ca2+ sensitivity that has been developed. Future studies can take advantage of these results to precisely dissect how SK channel activity contributes to neuronal activity under normal conditions and to neuron survival in neurodegenerative disease.
Methods
Electrophysiology
In our studies, the effect of mutations on the Ca2+ sensitivity of SK channels was investigated as previously described29,30,31,45. Briefly, mutations were introduced to the IDF region of rat SK2-a, human SK1 or human SK3 channels using QuickChange II site-directed mutagenesis kit (Agilent). The mutant channel cDNAs, along with CaM and GFP, at a ratio of 5:2.5:1 (weight), were transfected into TsA201 cells by the calcium–phosphate method. SK currents were recorded 1–2 days after transfection, with an Axon200B amplifier (Molecular Devices) at room temperature.
pClamp 10.5 (Molecular Devices) was used for data acquisition and analysis. The resistance of the patch electrodes ranged from 3–5 MΩ. The pipette solution contained (in mM): 140 KCl, 10 Hepes (pH 7.4), 1 MgSO4. The bath solution containing (in mM): 140 KCl, 10 Hepes (pH 7.2), 1 EGTA, 0.1 Dibromo-BAPTA, and 1 HEDTA was mixed with Ca2+ to obtain the desired free Ca2+ concentrations, calculated using the software by Chris Patton of Stanford University (http://www.stanford.edu/~cpatton/maxc.html). The Ca2+ concentrations were verified using Fluo-4 and standard Ca2+ buffers (Thermo Fisher Scientific).
Currents were recorded using an inside-out patch configuration. The intracellular face was initially exposed to a zero-Ca2+ bath solution, and subsequently to bath solutions with a series of Ca2+ concentrations. Currents were recorded by repetitive 1-s-voltage ramps from −100 mV to +100 mV from a holding potential of 0 mV. One minute after switching of bath solutions, ten sweeps with a 1-s interval were recorded. The integrity of the patch was examined by switching the bath solution back to the zero-Ca2+ buffer. Data from patches, which did not show significant changes in the seal resistance after solution changes, were used for further analysis. To construct the dose-dependent potentiation of channel activities, the current amplitudes at −90 mV in response to various concentrations of Ca2+ were normalized to that obtained at maximal concentration of Ca2+. The normalized currents were plotted as a function of the concentrations of Ca2+. EC50 values and Hill coefficients were determined by fitting the data points to a standard dose–response curve (Y = 100/(1 + (X/EC50) − Hill)). All data are presented in mean ± s.e.m. The data analysis was performed using pClamp 10.5 (Molecular Devices) in a blinded fashion. One-way ANOVA and Tukey’s post hoc tests were used for data comparison of three or more groups. The Student’s t-test was used for data comparison if there were only two groups.
Protein crystallization and structure determination
The protein complex consisting of CaM and the SK2-a channel fragment (R395 - Q486 from rat SK2-a channel with a point mutation V407F) was purified as described in our previous papers29,30,31,45. Briefly, rat CaM was introduced into the pET28b(+) vector (Novagen) and expressed in Rosetta-2 E. Coli. cells (Novagen). The CaM protein was purified using a low substitution phenyl sepharose column (GE Healthcare). The mutant SK2-a channel fragment was also introduced into the pET28b(+) vector (Novagen) and the codons have been optimized to improve expression of this His-tagged protein in E. Coli. This His-tagged protein was expressed and purified using a Ni-NTA column. The purified mutant SK2-a channel fragment was mixed slowly with the purified CaM in the presence of Ca2+ to form a complex, followed by purification with a gel filtration column (GE Healthcare) pre-equilibrated in a solution with 10 mM Tris-HCl, 50 mM NaCl, and 10 mM CaCl2 (pH 7.5). The purified protein complex was concentrated to about 1 mM and then set up for crystallization. Protein crystals of the protein complex were grown in sitting drops by vapor diffusion at 20 °C. The complex (1 mM) was mixed in a 1:1 ratio with the reservoir solution, which consists of 1.4 M Li2SO4, 0.6 M (NH4)2SO4, 0.1 M sodium citrate, pH 5.6. Monoclinic crystals usually grew within 3 weeks. These preformed protein crystals were then incubated with SK channel modulator NS309 at its saturating concentrations for ~5 months, due to the poor water solubility of NS309. The soaked protein crystals were flash-cooled in liquid nitrogen for data collection, after a brief transfer to a suitable cryoprotectant (25% glycerol with the reservoir solution saturated with NS309). X-ray diffraction data were collected from single crystals on the X-ray diffraction system (D8 Venture Diffraction System, Bruker AXS Inc.) at our home institute and processed using PROTEUM2 (Bruker AXS Inc.). Initial phases were determined by molecular replacement (MR) using PHASER from the CCP4 suite. Our previously determined CaM/CaMBD complex structure (4J9Y) was used as a starting model to phase diffraction data. Solvent molecules were removed from the starting molecule before rigid body refinement. The crystallographic model was further constructed through iterative rounds of manual model-building using Coot and crystallographic refinement using REFMAC and PHENIX. The crystallographic statistics for data collection and model refinement are summarized in Supplementary Table S1. Structure graphics were created using PyMol (Schrödinger, LLC).
Molecular Dynamics (MD) simulations
The structures of the WT or V407F protein complex were immersed in 0.15 M KCL solvation. The optimized potentials for liquid simulations (OPLS3) force field were used for the complexes, and the simple point charge (SPC) model was used for water molecules. All of the MD simulations were accomplished using the Desmond module of the Schrödinger software 2017–2 (Schrödinger, LLC). After being relaxed by Schrodinger Maestro’s default relaxation protocol which includes two stages of minimization (restrained and unrestrained) followed by four stages of MD runs with gradually diminishing restraints, the complexes were subjected to 100-ns MD simulations (NPT, constant Number, Pressure and Temperature ensemble) without any restraint at a constant temperature of 298 K. In total, 2000 snapshot structures were collected during the MD simulations. The root mean square deviation (RMSD), and the root mean square fluctuation (RMSF) of atomic positions in the trajectories were analyzed using the built-in utility of GROMACS program. RMSF calculations were performed using protein heavy atoms (including side chains) of the associated residues. The minimum distances between atoms of residues 407 and 411 in the trajectories were also analyzed using the built-in utility of GROMACS program.
The MD simulations in the presence of PIP2 were performed as previously described31,32. The interaction energies with PIP2 were acquired from MM-GBSA calculations using the thermal_mmgbsa.py script. The latter half of the trajectory was used in the MMGBSA calculations. Of the last thousand snap shots, every 10th snap shot was used as input for the input complexes. The VSGB solvent model was used for solvation model. OPLS3 was used to parameterize the protein and the ligand.
The Discovery Studio 2017 molecular modeling program (BIOVIA) was used to conduct energy minimization and calculate the interaction energy between the residues 407 and 411 from the WT and mutant crystal structures. The structures were subjected to energy minimization using Smart Minimizer algorithm (200 steps), and Generalized Born (GB) implicit solvent model using the CHARMM force field. Interaction energies between the residue pairs were calculated using a distance dependent dielectric constant (ε = 2r) implicit solvent model.
Homology modeling
The full-length sequence of rat KCNN2 was retrieved from Universal Protein Resource ID Q9H2S1. The protein data bank structure ID 6CNN, a crystal structure of the human KCNN4 in complex with calmodulin, was selected as the template to build KCNN243. Template biological assemblies were imported as heteromers into Maestro included within the Schrodinger 2018–1 software package. Structures were prepared for homology modeling using the protein preparation wizard included within the modeling suite. This corrected the model for missing loops, side chains, and bond orders. This tool was used to assign pronation states to amino acid residues and optimize hydrogen bond placement.
Homology modeling was performed using Prime program included in the Schrodinger 2018–1 software package. Sequence alignment to the template assemble was done using the ClustalW alignment method. Four calmodulin molecules were included in the KCNN2 model. Homology building was done in a series of steps where the backbone and side chain atom coordinates of conserved residues were copied over to the new model, followed by optimization of side chains, minimization of non-template residues, and building insertions and deletions in the alignment. Non-template loops are refined using the ultra-extended loop sampling method. The model is then checked for missing atoms and minimized.
Construction of guide RNA plasmid
Embryonic U6 promoter-driven kcnl-2 guide RNA was amplified from the pU6::klp-12::sgRNA46 vector template using kcnl-2 targeting primers: kcnl-2_guide_f and kcnl-2_guide_r. The resulting linear DNA was circularized to generate the final construct pHA0815 pU6::kcnl-2::guide.
Strain construction
kcnl-1 V698F point mutation, the cognate of SK2-a V407F, was introduced into the endogenous kcnl-2 gene using CRISPR/Cas9-mediated homologous recombination (HR) with pha-1 co-conversion47. In brief, co-editing of a temperature-sensitive, lethal mutation at the unlinked pha-1 gene facilitates identification of animals with CRISPR/Cas9-mediated genomic editing events in kcnl-2. The guide RNA construct pHA0815 pU6::kcnl-2::guide was injected at 50 ng/μl into temperature-sensitive pha-1(e2123)III animals raised at 15 degrees with 50 ng/μl of Peft-3::Cas9 plasmid46, 50 ng/μl of pha-1 sgRNA + Cas9 pJW1285 plasmid47,10 μM of 200mer sense pha-1(+) rescue oligonucleotide47 and 10 μM of the single-stranded oligonucleotide template (ssODN) kcnl-2_ssODN. The kcnl-2_ssODN contained synonymous mutations to prevent further genomic editing and to generate an ApaLI restriction site for PCR genotyping. Injected animals were transferred to a restrictive temperature at 25 degrees; all surviving progeny were screened for potential HR events by PCR genotyping with primers kcnl-2_scr_f and kcnl-2_scr_r, followed by ApaLI restriction digestion. Editing events were verified by sequencing and the strain was backcrossed four times to wild type N2 before analysis. The pha-1 locus was sequenced in backcrossed lines to verify the presence of the wild-type pha-1(+) allele. The kcnl-2(rt462[V698F]) allele was crossed into animals expressing human wild-type SOD1-YFP (hSOD1WT) or human ALS-associated SOD1G85R-YFP (hSOD1G85R) under the pan-neuronal snb-1 promoter39 in sod-1(tm776) background.
Dispersal Assay
Dispersal was scored on 60-mm diameter petri dishes freshly spread with 200 μl of thin OP50 bacterial lawn (food source) on 10 ml of Nematode Growth Medium (NGM). Day 1 adult animals were placed at the center of the bacterial lawn and percentage of animals escaping the 15-mm diameter central ring is reported. One-way ANOVA and Sidak’s tests were used for data comparison. All experiments were carried out in accordance with relevant guidelines and regulations. When appropriate, protocols were approved by institutional committees.
Primers
kcnl-2_guide_f: GAATTGACAAGTTTTAGAGCTAGAAATAGC, kcnl-2_guide_r: CAACTTTCTCAAACATTTAGATTTGCAATTC, kcnl-2_ssODN: CTACCTGCTTAGTAAGTTGAGTGTCTTGCATGAAATTGTGGAAATGCTTCTCTGCGCGTGTCAATTCCAACTTTCTCGCAATAACCGCGACAACCATCGAAG, kcnl-2_scr_f: CCATTTGGCTTGTTGCTATC, kcnl-2_scr_r: AATTACCTTTGCCACGTCTC.
Strains
HA2614: kcnl-2(+)I; sod-1(tm776)II; pha-1(+)III; [hSOD1WT::YFP]
HA3351: kcnl-2(rt462[V698F])I; sod-1(tm776)II; pha-1(+)III; [hSOD1WT::YFP]
HA2612: kcnl-2(+)I; sod-1(tm776)II; pha-1(+)III; [hSOD1G85R::YFP]
HA3397: kcnl-2(rt462[V698F])I; sod-1(tm776)II; pha-1(+)III; [hSOD1G85R::YFP].
Experimental Design and Statistical Analysis
One-way ANOVA and post hoc tests were used for data comparison of three or more groups. The Student’s t-test was used for data comparison if there were only two groups.
Data availability
The structure coordinates have been deposited in the Protein Data Bank under accession codes 6ALE and 6CZQ.
References
Berridge, M. J., Bootman, M. D. & Roderick, H. L. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4, 517–29 (2003).
Clapham, D. E. Calcium Signaling. Cell 131, 1047–1058 (2007).
Adelman, J. P., Maylie, J. & Sah, P. Small-conductance Ca2+-activated K+ channels: Form and function. Annu Rev Physiol 74, 245–269 (2012).
Pedarzani, P. et al. Control of Electrical Activity in Central Neurons by Modulating the Gating of Small Conductance Ca2+-activated K+ Channels. J. Biol. Chem. 276, 9762–9769 (2001).
Stocker, M. Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5, 758–770 (2004).
Giessel, A. J. & Sabatini, B. L. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron 68, 936–47 (2010).
Maingret, F. et al. Neurotransmitter Modulation of Small-Conductance Ca2+-Activated K+ Channels by Regulation of Ca2+ Gating. Neuron 59, 439–449 (2008).
Bildl, W. et al. Protein kinase CK2 Is coassembled with small conductance Ca2+-activated K+ channels and regulates channel gating. Neuron 43, 847–858 (2004).
Devor, D. C., Singh, A. K., Frizzell, R. A. & Bridges, R. J. Modulation of Cl- secretion by benzimidazolones. I. Direct activation of a Ca(2+)-dependent K+ channel. Am J Physiol 271, L775–84 (1996).
Pedarzani, P. et al. Specific enhancement of SK channel activity selectively potentiates the afterhyperpolarizing current I(AHP) and modulates the firing properties of hippocampal pyramidal neurons. J Biol Chem 280, 41404–11 (2005).
Strobaek, D. et al. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim Biophys Acta 1665, 1–5 (2004).
Nishimura, A. L. et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75, 822–31 (2004).
Dimitriadi, M. et al. Conserved genes act as modifiers of invertebrate SMN loss of function defects. Plos Genet 6, e1001172 (2010).
Dimitriadi, M. et al. The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J Neurosci 33, 6557–62 (2013).
Luna-Cancalon, K. et al. Alterations in cerebellar physiology are associated with a stiff-legged gait in Atcay (ji-hes) mice. Neurobiol Dis 67, 140–8 (2014).
Kasumu, A. W. et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol 19, 1340–53 (2012).
Walter, J. T., Alvina, K., Womack, M. D., Chevez, C. & Khodakhah, K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci 9, 389–97 (2006).
Shakkottai, V. G. et al. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 31, 13002–14 (2011).
Deardorff, A. S. et al. Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar -motoneurons. J Physiol 591, 875–97 (2013).
Li, X. & Bennett, D. J. Apamin-sensitive calcium-activated potassium currents (SK) are activated by persistent calcium currents in rat motoneurons. J Neurophysiol 97, 3314–30 (2007).
Viana, F., Bayliss, D. A. & Berger, A. J. Multiple potassium conductances and their role in action potential repolarization and repetitive firing behavior of neonatal rat hypoglossal motoneurons. J Neurophysiol 69, 2150–63 (1993).
Zhang, L. & Krnjevic, K. Apamin depresses selectively the after-hyperpolarization of cat spinal motoneurons. Neurosci Lett 74, 58–62 (1987).
Alvina, K. & Khodakhah, K. The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J Neurosci 30, 7258–68 (2010).
Alvina, K. & Khodakhah, K. KCa channels as therapeutic targets in episodic ataxia type-2. J Neurosci 30, 7249–57 (2010).
Egorova, P. A., Zakharova, O. A., Vlasova, O. L. & Bezprozvanny, I. B. In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J Neurophysiol 115, 2840–51 (2016).
Ristori, G. et al. Riluzole in cerebellar ataxia: A randomized, double-blind, placebo-controlled pilot trial. Neurology 74, 839–845 (2010).
Romano, S. et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 14, 985–91 (2015).
Cao, Y. J., Dreixler, J. C., Couey, J. J. & Houamed, K. M. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol 449, 47–54 (2002).
Zhang, M., Pascal, J. M., Schumann, M., Armen, R. S. & Zhang, J. F. Identification of the functional binding pocket for compounds targeting small-conductance Ca(2)(+)-activated potassium channels. Nat Commun 3, 1021 (2012).
Zhang, M., Pascal, J. M. & Zhang, J. F. Unstructured to structured transition of an intrinsically disordered protein peptide in coupling Ca2+-sensing and SK channel activation. Proc Natl Acad Sci USA 110, 4828–33 (2013).
Zhang, M. et al. Selective phosphorylation modulates the PIP2 sensitivity of the CaM-SK channel complex. Nat Chem Biol 10, 753–9 (2014).
Zhang, M., Meng, X. Y., Zhang, J. F., Cui, M. & Logothetis, D. E. Molecular overlap in the regulation of SK channels by small molecules and phosphoinositides. Sci Adv 1, e1500008 (2015).
Shmukler, B. E. et al. Structure and complex transcription pattern of the mouse SK1 KCa channel gene, KCNN1. Biochim Biophys Acta 1518, 36–46 (2001).
Li, W., Halling, D. B., Hall, A. W. & Aldrich, R. W. EF hands at the N-lobe of calmodulin are required for both SK channel gating and stable SK-calmodulin interaction. J. Gen. Physiol. 134, 281–293 (2009).
Zhang, M. et al. Structural basis for calmodulin as a dynamic calcium sensor. Structure 20, 911–23 (2012).
Babu, M. M., van der Lee, R., de Groot, N. S. & Gsponer, J. Intrinsically disordered proteins: regulation and disease. Current Opinion in Structural Biology 21, 432–440 (2011).
Dyson, H. J. & Wright, P. E. Intrinsically unstructured proteins and their functions. Nature Rev. Mol. Cell Biol. 6, 197–208 (2005).
Wright, P. E. & Dyson, H. J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 (1999).
Wang, J. et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. Plos Genet 5, e1000350 (2009).
Xia, X. M. et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507 (1998).
Scholl, E. S., Pirone, A., Cox, D. H., Duncan, R. K. & Jacob, M. H. Alternative splice isoforms of small conductance calcium-activated SK2 channels differ in molecular interactions and surface levels. Channels (Austin) 8, 62–75 (2014).
Garneau, L. et al. Hydrophobic interactions as key determinants to the KCa3.1 channel closed configuration. An analysis of KCa3.1 mutants constitutively active in zero Ca2+. J Biol Chem 284, 389–403 (2009).
Lee, C. H. & MacKinnon, R. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 360, 508–513 (2018).
Chotoo, C. K., Silverman, G. A., Devor, D. C. & Luke, C. J. A small conductance calcium-activated K+ channel in C. elegans, KCNL-2, plays a role in the regulation of the rate of egg-laying. Plos One 8, e75869 (2013).
Nam, Y. W. et al. Structural insights into the potency of SK channel positive modulators. Sci Rep 7, 17178 (2017).
Friedland, A. E. et al. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 10, 741–3 (2013).
Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 199, 363–77 (2015).
Acknowledgements
We thank Drs John Adelman and Heike Wulff for providing the cDNAs of SK1 and SK3 channels. We thank Drs Diomedes Logothetis, Jifang Zhang for helpful comments on the manuscript. We are grateful to Kristy Harada, Michael Phan, Melody Ra, Aryan S. Shirazi, Sadaf Toutouni, Adam Viegas and Benji Whitmore for technical assistance. We thank Drs M. Benning and I. Maslennikov for help with X-ray crystallography data collection. The computations were supported by the ITS (Information Technology Services) Research Computing at Northeastern University. C. elegans work was supported by ALS FindingACure and the ALS Association (A.C.H.). M.Z. was supported by a grant 1R21NS101182-01A1 from NIH, a Scientist Development Grant 13SDG16150007 from American Heart Association and a YI-SCA grant from National Ataxia Foundation.
Author information
Authors and Affiliations
Contributions
Y.W.N., R.O. and M.Z. undertook mutagenesis, electrophysiology and X-ray crystallography studies. D.G. and M.C. performed computational work. S.N.B. and A.C.H. undertook C. elegans studies. Y.W.N., A.C.H., S.N.B. and M.Z. prepared the manuscript and the figures.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nam, YW., Baskoylu, S.N., Gazgalis, D. et al. A V-to-F substitution in SK2 channels causes Ca2+ hypersensitivity and improves locomotion in a C. elegans ALS model. Sci Rep 8, 10749 (2018). https://doi.org/10.1038/s41598-018-28783-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-28783-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
