
Abstract
Regulation of cerebral endothelial cell function plays an essential role in changes in blood-brain barrier permeability. Proteins that are important for establishment of endothelial tight junctions have emerged as critical molecules, and PDZ domain containing-molecules are among the most important. We have discovered that the PDZ-domain containing protein periaxin (PRX) is expressed in human cerebral endothelial cells. Surprisingly, PRX protein is not detected in brain endothelium in other mammalian species, suggesting that it could confer human-specific vascular properties. In endothelial cells, PRX is predominantly localized to the nucleus and not tight junctions. Transcriptome analysis shows that PRX expression suppresses, by at least 50%, a panel of inflammatory markers, of which 70% are Type I interferon response genes; only four genes were significantly activated by PRX expression. When expressed in mouse endothelial cells, PRX strengthens barrier function, significantly increases transendothelial electrical resistance (~35%; p < 0.05), and reduces the permeability of a wide range of molecules. The PDZ domain of PRX is necessary and sufficient for its barrier enhancing properties, since a splice variant (S-PRX) that contains only the PDZ domain, also increases barrier function. PRX also attenuates the permeability enhancing effects of lipopolysaccharide. Collectively, these studies suggest that PRX could potentially regulate endothelial homeostasis in human cerebral endothelial cells by modulating inflammatory gene programs.
Similar content being viewed by others


Introduction
Human cerebral endothelial cells possess special properties that are integral for normal homeostatic brain function. In particular, endothelial cells participate in the formation of the blood-brain barrier (BBB), a key function of the cerebrovasculature that prevents transit of blood-borne substances into the central nervous system1,2,3. A defined set of endothelial proteins is critical for the establishment and maintenance of this barrier function4. One important group of proteins includes transmembrane tight junction molecules (including claudin55, occludin6, and junctional adhesion molecule7). Another key group of proteins includes cytoplasmic adaptor proteins that localize with tight junction membrane proteins. Important cerebral endothelial adaptor proteins described to date include the MAGUK class of proteins (ZO18, ZO29, and ZO310). Additional proteins in endothelial barriers include cingulin11, AF-612, and 7H613.
Each of these adapter proteins contains a homologous and evolutionarily conserved structural motif, the PDZ domain (postsynaptic density protein of 95 kilodaltons, disc large, zona occludens-1). This domain, described in several membrane associated scaffolding molecules, binds to the carboxy-terminal tail of many of the tight junction transmembrane proteins14,15,16,17. The presence of multiple PDZ domains and other protein binding motifs in these endothelial adapter proteins mediates molecular linkage between junctional membrane proteins to the underlying actin cytoskeleton networks within the cell18,19. These adaptor proteins play an important role in regulating barrier properties, including in disease states4.
The multitude of PDZ-domain containing proteins that regulate endothelial cell function suggests there may be additional regulatory proteins containing similar domains. To identify new human brain endothelial cell proteins, we screened a public database for cerebral factors with vascular expression. From this screen, we identified periaxin (PRX) as a novel human cerebral endothelial PDZ-domain protein. Unlike in humans, PRX was not expressed in the cerebral endothelium in multiple other species, even though it was present in other tissues. Expression of PRX in cerebral endothelial cells in vitro tightened the BBB and altered inflammatory responses. However, PRX was primarily present in the nucleus rather than associated with tight junctions at the cell membrane, suggesting the effects of PRX, while PDZ dependent, are mediated by regulating gene expression.
Results
Identification of a human specific PDZ-domain protein in cerebral endothelial cells
While conducting a bioinformatic study of protein expression patterns in the brain20, we identified PRX as a previously unrecognized brain endothelial protein in humans based on images from the Human Protein Atlas. To validate the expression pattern in an independent set of human samples, we immunostained for PRX in frontal cortex obtained at autopsy from 8 individuals (Fig. 1). Our staining for PRX showed strong capillary expression without neuronal or astrocyte reactivity (Fig. 1A and C). In addition, the endothelium of small white matter vessels were stained (Fig. 1B). Large arteries and veins of the leptomeninges did not react with PRX antibodies. In situ hybridization localized PRX mRNA to endothelial cells of the cortex, in agreement with immunohistochemistry results (Fig. 1D).

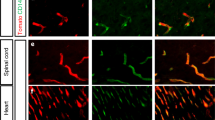
Expression of PRX in the human brain and in non-human tissues. Immunohistochemical analysis of human brain was performed with antibodies raised against human PRX. In the cerebral cortex, capillary staining (but not neurons or astrocytes) was detected in both gray (A) and white (B) matter. High power images demonstrate capillary and small vessel endothelial-predominant expression in the cortex (C). Positive vascular signals are highlighted with left pointing arrows. PRX mRNA was localized to capillaries by in situ hybridization ((D) pink granular stain shows RNA hybridization; left pointing arrows). Immunohistochemical analysis of PRX in mouse cortex (E) kidney (F,G) lung (H) and tail (I) was conducted for PRX expression using the same antibodies. Kidney and tail tissues demonstrated peripheral nerve staining while lung showed capillary expression (left pointing arrows). Right pointing arrows in (E–G) show endothelial cells that fail to stain for PRX in brain and kidney. Negative staining was observed in rat, pig, and marmoset brain (not shown). Scale bar for (A,B) shows 600 microns and for all other photographs measures 150 microns.
PRX has previously been demonstrated in Schwann cells of mouse peripheral nerves but not the central nervous system. We used the same antisera against PRX (which highlighted human endothelium) to stain brain from several non-human species: two strains of mouse, rat, pig, and marmoset. No brain PRX expression was detected in those species (Fig. 1E shows negative mouse brain staining). In mouse kidney, vascular structures, including the glomeruli, did not stain (Fig. 1F), but perivascular nerve bundles showed reactivity (Fig. 1G). The PRX antibody also strongly recognized lung capillaries (Fig. 1H). In the mouse tail, nerve bundles and endothelial cells were stained with the PRX antibody (Fig. 1I). These studies demonstrated that the polyclonal antibodies to PRX recognized both human and mouse protein but that there is human-specific expression in the brain endothelium.
To determine whether PRX is present in young humans, we stained a different cohort of human brains ranging in age from 2 months to 18 years. We found expression in cortical capillaries in all human samples, regardless of age (Fig. 2). Notably, in many but not all vessels, PRX reactivity localized heavily to endothelial cell nuclei.

Expression of PRX in capillaries in young human brain. Immunohistochemical analysis of human brain confirmed the expression of PRX over a wide span of ages. Capillary staining (left pointing arrows) similar to that seen in aging human brain was observed in brains from individuals at 3 months (A) 6 years (B) and 18 years (C) of age. Capillary cells in (A) show an abundance of staining in a nuclear pattern. Scale bar shows 150 microns for all photographs.
We also assessed the expression of PRX protein in vascular tissue by Western blotting. Crude vessel fractions were prepared from human cortical tissue. Vascular fractions showed substantially increased expression of a major band corresponding to the predicted molecular mass of full length L-PRX, the larger of two known gene products (Fig. 3). On longer exposure of the blot, a cluster of protein sizes were observed in cells transfected with L-PRX cDNA, whole brain, and brain vessel preparations. The major band from transfected cells was slightly higher than the brain bands, but all samples demonstrated clusters of high molecular weight sizes that demonstrated overlap as shown in Supplemental Fig. 1. The degree of PRX enrichment in vascular fractions of the brain was similar to that of established endothelial markers, vWF and CD31 (Fig. 3E).

Expression of PRX protein in human brain vessel fractions. Fractions of human brain from autopsy were analyzed for presence of indicated proteins. Lane 1: 293 cells transfected with L-PRX full length cDNA (encoding Genbank Protein ID AAH67266.1; purchased from OriGene); Lane 2: Human whole brain (Hwb) lysate; Lane 3: Partially purified microvessel proteins from human brain. Lane 1 was underloaded to avoid overloading PRX. (A) Western blotting of brain vessels showed expression of a protein band corresponding to the larger form of PRX (L-PRX), as seen in transfected 293 cells. As controls, the same protein lysates were probed for (B) vWF, (C) CD31, and (D) tubulin. Quantification of the degree of enrichment of vascular proteins in vascular fractions was computed by first calculating the ratio of the protein of interest to tubulin (D) and then normalizing this ratio in vessels to the ratio in human whole brain. (E) shows overall levels of protein enrichment in vessel fractions of multiple individuals (n = 3 for PRX and vWF and n = 2 for CD31; *indicates p < 0.01). Additional brain samples and higher exposure PRX blots are shown in Supplemental Fig. 1.
Subcellular expression of PRX in brain endothelial cells
Because immunohistochemical studies suggested PRX could be localized to both cell nuclei and cytoplasm in human brain, we examined the subcellular expression of PRX protein in finer detail in cultured endothelial cells. The large form of PRX is composed of an N-terminal PDZ domain, a middle repeat domain, and a C-terminal domain. To map regions of PRX that could contribute to subcellular localization, we prepared GFP fusions (Fig. 4A) to PRX corresponding to the full length PRX (L-PRX), a splice form of PRX which generates a short form (S-PRX) that contains the PDZ domain of the protein and very small C-terminal tail21, and a construct that deletes the PDZ domain from the full length protein (D-PRX, which does not exist naturally). Constructs were expressed in primary mouse cerebral endothelial cells, and GFP was examined along with the tight junction markers claudin-5 and ZO1 (Fig. 4B). GFP-L-PRX was found largely in the nuclei of mouse endothelial cells. A modest amount of L-PRX was found in the cytoplasm and very little L-PRX was located at the cell membrane where the predominance of ZO-1 protein is enriched. The small isoform of PRX (S-PRX), expressed as a fusion protein to GFP was localized outside of the nucleus of endothelial cells, with a minor component of protein seen in the nucleus. There was very little GFP-S-PRX at the plasma membrane of transfected cells. To test for nuclear localization signals in the C-terminus of PRX, we also tested the localization of a GFP fusion to PRX sequences that are not present in S-PRX. We found that this protein (GFP-D-PRX; Fig. 4B) had a similar distribution to S-PRX. Confocal Z-stack images confirm that endothelial cells expressing PRX harbor proteins that reside within the nucleus rather than on the surface (Fig. 4C). We concluded that L-PRX is mostly found in the cytosol and nucleus and that, in endothelial cells, nuclear localization cannot be accounted for by a single dominant nuclear localization sequence. Transfection of untagged PRX construct into mouse cerebral endothelial cells resulted in similar, robust localization to the nucleus in a population of cells (Supplemental Fig. 2), and HEK293 expression of recombinant PRX resulted in protein expression in nuclear fractions of cellular protein preparations (Supplemental Fig. 3).

Subcellular localization of PRX in cerebral endothelial cells. Primary mouse brain endothelial cells were transfected with GFP constructs (A). L-PRX is the full length form of PRX which includes a PDZ domain and a long, repetitive C-terminal sequence. S-PRX is short form that is composed principally of the PDZ domain and is generated naturally by alternative splicing. The D-PRX construct, which deletes the PDZ domain from L-PRX, was used to determine cell localization mediated by sequences outside of the PDZ domain. Cells were fixed and then stained for claudin-5 and ZO-1, with DAPI to highlight nuclei (B). (C) Z-stack images were processed in order to assess localization of the three PRX constructs in three dimensional space. Scale bar measures 50 microns.
Gene expression alterations triggered by endothelial PRX
Global gene expression analysis was performed to determine the impact of PRX expression in human cerebral endothelial cultures. Microarray analysis of RNA from PRX-expressing cells (relative to control cells expressing RFP) revealed significant increases in a small fraction of transcripts, shown in Table 1 (top). On the other hand, a larger number of transcripts were significantly decreased by expression of L-PRX (Table 1, bottom). A large fraction of transcripts decreased after L-PRX expression participates in inflammatory responses, and, in particular, the five most strongly suppressed genes are canonical members of the Type I interferon response pathway (Table 1). Validation experiments using independent sets of cultured human endothelial cells expressing L-PRX or RFP demonstrated strong suppression of IFIT1, IFIT3, IFIT6, IFITM1, IL1A, and MX1(Fig. 5A). TNF-alpha and IL1-beta transcript levels were not affected (Supplemental Fig. 4). On the other hand, in the same transcriptome experiment, no endothelial tight junction factors were increased by more than 1.5 fold after expression of L-PRX (Table 2).

Effect of PRX expression on inflammatory markers in endothelial cells. (A) Effect of PRX on inflammatory factor mRNA levels in endothelial cells. Human primary cerebral endothelial cells were infected with L-PRX viruses or RFP control viruses. After 24 hours, RNA was analyzed by quantitative RT-PCR by normalizing Ct values to 18S rRNA levels. Expression of a series of inflammatory mRNA species (green background) was significantly suppressed, as shown. Few mRNA species (red background) were upregulated. Samples were analyzed in triplicate with error bars shown as SD; all comparisons between PRX and RFP infected samples were significant with p < 0.05 (t-test). Experiments were repeated showing the same differences with three sets of cell cultures. (B) Effect of PRX on human cerebral endothelial protein expression after inflammatory challenge. We display protein array signals for human cytokines present in control (RFP-transfected cells) and L-PRX overexpressed HBMEC treated with/without IL-1β (10 ng/ml). Data represent several highly increased cytokines/chemokines in cell culture media collected at 24 hrs after introducing IL-1β. L-PRX presence in brain endothelial cells decreased expression of several proinflammatory cytokines. Values are means ± SD, n = 3; *p > 0.05, **p > 0.01, ***p > 0.001.
Secreted cytokine/chemokine expression in endothelial cells after inflammatory challenge
The potential effect of PRX on inflammatory cytokines was assessed in human cerebral endothelial cells that were challenged with IL-1b. Cells were infected with viruses expressing PRX or RFP (as a control) and after 48 hours, were exposed to IL-1b. The secreted proteins from the media were analyzed using a protein based antibody array for human cytokines. Selected cytokines were activated by IL-1b but suppressed by the expression of PRX. In total, of the 80 cytokines analyzed, 22 predominantly proinflammatory cytokines/chemokines were suppressed by PRX expression. As such, the inflammatory suppressive effect of PRX extends beyond suppression of gene expression and results in decreased protein release (Fig. 5B).
Endothelial PRX strengthens BBB function
To test if PRX has a role in barrier function, we investigated potential effects of PRX on primary mouse cerebral endothelial cell barrier function. Cells expressing L-PRX (versus control cells shown) for up to 7 days were grown in transwell chambers. Cell barrier function was assessed in permeability assays using three different size tracers (Inulin 5 kDa, Dextran 20 kDa and Dextran 40 kDa). As shown in Fig. 6, beginning at day 3 after introduction of the transgene, the expression of L-PRX significantly reduced permeability to all three tracers, significantly enhancing monolayer barrier function at days 5 and 7. This was most significant for, Dextran 40 kDa (p < 0.001) and Dextran 20 kDa (P < 0.01). In addition, transendothelial electrical resistance (TEER) progressively increased during each day of observation with significant increases in tightness at days 5 and 7 (p < 0.05). RFP lentivirus infected cells showed the same permeability profiles as uninfected cells. In sum, expression of the human protein L-PRX in mouse cerebral endothelial cells exhibits significant barrier strengthening properties.

Effect of PRX on mouse cerebral endothelial cell permeability. L-PRX was expressed in primary mouse cerebral endothelial cells using recombinant lentiviruses. Uninfected cells were used as a control. At the number of days after infection indicated on the x-axis, permeability to indicated fluorescent tracers was determined (A–C). (D) TEER was also determined at the same time points. L-PRX significantly strengthened the barrier function of endothelial cells, accelerating both the acquisition of barrier function and the final resistance of cells to tracers of a wide range of molecules. RFP lentivirus infected cells showed the same permeability profiles as uninfected cells (not shown). Data represent means +/− SD for n = 5 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 comparing control and L-PRX transfected cells.
PDZ domain is necessary and sufficient for barrier function modification
To map domains of the PRX coding sequence which mediate barrier strengthening function, we generated recombinant viruses encoding S-PRX and D-PRX (and N-terminal truncation that eliminates the PDZ domain). We compared mouse cerebral endothelial cells infected with these viruses to cells infected with full length L-PRX for permeability (Fig. 7). Cells expressing S-PRX had improved barrier function to all tracers and increased TEER compared to control cells. The enhancement of function was similar that induced by expression of full length (L-PRX). In contrast, D-PRX did not have an impact on endothelial permeability.

Mapping structural domains of PRX that mediate enhancement of endothelial permeability. The effect of three components of PRX on endothelial permeability was compared. Lentiviruses were used to express either full length L-PRX, S-PRX, or D-PRX (C-terminal PRX) (similar to the sequences shown in Fig. 4A, minus the GFP tag) and permeability was measured as in Fig. 6 for each group of infected primary mouse brain endothelial cells. Permeability to both inulin (A) and dextran (B) were measured. Enhancement of barrier function was observed in cells expressing S-PRX and full length L-PRX, but not with D-PRX. Uninfected cells served as controls. RFP lentivirus infected cells showed the same permeability profiles as uninfected cells (not shown). Data represent means +/− SD for n = 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, comparing control and S-, D- and L-PRX transfected cells. ###p < 0.001 comparing L-PRX with S- and D-PRX transfected cells.
PRX blunts inflammation driven endothelial permeability
Inflammatory mediators such as cytokines and bacterial products (e.g. lipopolysaccharide; LPS) cause increased endothelial permeability. Since PRX mediates coordinated repression of inflammatory response genes, we determined whether PRX expression could counteract inflammatory factor-mediated increased endothelial permeability. Control endothelial cells exposed to LPS demonstrated 3–7 fold increases in permeability to three tracers in transwell assays. RFP lentivirus infected cells showed the same permeability profiles as uninfected cells (not shown). L-PRX expression resulted in a marked suppression of inflammatory factor-stimulated permeability (Fig. 8). Cells expressing L-PRX resisted challenges to barrier function, although this function was not fully normalized. As before, S-PRX also led to resistance to inflammatory challenge, but D-PRX (lacking the PDZ domain) did not exhibit barrier enhancing function.

PRX as an attenuator of inflammatory elevation of endothelial permeability. Murine endothelial cells were infected with viruses to express L-PRX. Afterwards, cells were challenged with LPS (5 ug/ml), which stimulates endothelial permeability. Measurements of permeability to a series of tracer proteins shown and of TEER demonstrate that L-PRX reduces LPS-stimulated permeability to proteins of a range of molecular weights. Uninfected cells served as controls. RFP lentivirus infected cells showed the same permeability profiles as uninfected cells (not shown). Data represent means +/− SD for n = 3 independent experiments. **p < 0.01, ***p < 0.001, comparing LPS-challenged to control cells. ###p < 0.001 comparing L-PRX expressing to control cells.
Discussion
A complex array of molecular components regulates cerebral endothelial properties. This study identified PRX as a new protein expressed in human cerebral endothelial cells that is capable of regulating cell barrier physiology. The core findings of our studies are that: (1) PRX is unique among vascular PDZ-domain containing proteins in that it is present in human (but not other vertebrate) brain endothelial cells and that it does not preferentially localize to tight junctions; (2) PRX sharply down-regulates gene and protein expression related to inflammation; and (3) PRX markedly enhances barrier properties of endothelial cells and suppresses inflammatory-stimulated increases in endothelial permeability.
Motivation for human cerebrovascular protein identification
Use of non-human species to model cerebrovascular disease such as stroke has largely fallen short in yielding information that has advanced therapy. The reasons for the disconnection between animal and human clinical trials have been debated with vigor, and an often overlooked consideration is that vascular protein content may have species differences. PRX was among the first of over a dozen factors discovered to be human-only cerebrovascular markers in an ongoing database screen for novel human cerebrovascular factors. This effort, which is still ongoing, involves the manual annotation of human cerebral protein expression patterns20, publically available on the Human Protein Atlas22. The identification of PRX as a functional BBB protein that is present in humans (but not in traditional experimental animals) suggests that vascular protein expression differences could account for limitations of animal models.
A small number of proteins have already been demonstrated as human-enriched or human-specific in endothelial cells23,24 and Schwann cells25. Our new characterization of PRX demonstrates, for the first time to our knowledge, that a human-specific factor modifies cerebral endothelial cell permeability. Future studies, such as the generation of mice expressing human PRX and other human-only factors, will allow critical testing of the possibility that humanizing the cerebrovascular will advance the predictive value of mouse models of stroke and other brain blood vessel disorders.
PRX expression in the nervous system
Previous work in mouse has demonstrated PRX in Schwann cells and the lens26 but not the central nervous system27. The function of PRX has been elucidated in mouse and human genetic studies which show that it plays an essential role in the establishment of functional myelin and lens fiber structure26,28. Rare cases of human PRX loss of function mutations cause recessive peripheral neuropathies, including Dejerine-Sottas syndrome29 and Charcot-Marie-Tooth disease30,31. These studies firmly establish PRX as an essential protein in the peripheral nervous system across species.
In the Human Protein Atlas, PRX antibodies stain peripheral nerves of humans; furthermore, staining of multiple organs of the mouse, in this study, confirm that PRX is expressed robustly in the peripheral nervous system. The strong localization of PRX to the same structures shows that the antibodies used in this study recognize the protein in multiple species.
We used these antibodies to demonstrate that PRX is expressed in human capillaries of the brain and that it is not expressed at detectable levels in brain of a variety of other species. Further confirmation of PRX expression in human brain vessels is provided by in situ hybridization and by Western blotting of vessel-enriched protein preparations. While the human expression of PRX in the brain has not previously been reported, our inability to find PRX in mouse brain confirms previous studies27, which employed mRNA detection in tissues. We emphasize that in addition to its absence in the mouse, PRX was not found in the brain vasculature in a variety of species, including the non-human primate, marmoset, that is sometimes used as an experimental model. Because antibodies recognize both mouse and human PRX proteins, we believe that the antibodies should recognize PRX in most other species that are more closely related to human because of substantial homology (similarity of human vs. mouse [84.1%], vs. rat [82.9%], vs. pig [86.4%], vs. marmoset [95.2%]). A potential limitation of our study is that antibodies were generated to the C-terminus of PRX that is present only in L-PRX. Thus, we cannot address the species distribution of the minor S-PRX form, though we can ascertain that L-PRX is human specific for the brain endothelium.
The subcellular distribution of PRX within cerebral endothelial cells distinguishes it from other PDZ-domain containing proteins that regulate junction function. Those proteins localize to the cell-cell junctions and are thought to play key roles in formation of the BBB by physically stabilizing these structures. In myelinated peripheral nerve, PRX is localized to appositions between myelin and the plasma membrane that define Cajal bands32. Thus, we expected to find at least a fraction of PRX associated with the plasma membrane and with the tight junction proteins claudin-5 and ZO-1. In contrast, we found no junctional PRX and a large fraction of nuclear PRX in endothelial cells, raising the possibility of a role for PRX in transcriptional processes (see below).
Potential molecular mechanisms of action of PRX
PRX effected substantial enhancement of cell barrier function, a key property of brain endothelial cells. The increase in barrier strength was consistent across a wide range of molecules of diverse molecular weights, including ions (measured by TEER). PRX is an unusual example of a single regulatory protein whose overexpression strengthens baseline endothelial barrier function and inhibits the deleterious effects of an inflammatory stimulator (i.e. LPS) on endothelial barrier function.
Specific immediate signaling pathways by which PRX acts in endothelial cell are suggested by comparisons between S-PRX, L-PRX, and D-PRX. Since the PDZ domain of PRX is both necessary and sufficient for its barrier enhancing function, proteins that bind to the PDZ domain of PRX likely initiate molecular events that improve endothelial barrier function.
Proteins sequences that bind to PDZ domains are generally located at the C-termini of target proteins. Other PDZ proteins in endothelial cells generally target tight junction proteins and are thought to directly participate in structures that strengthen barrier function. But, we identified PRX principally in the nucleus and cytoplasm and failed to find localization with either claudin-5 or ZO-1. Prior work showed expression of PRX in the nucleus and that control of subcellular localization was responsive to a nuclear export signal within S-PRX33 and by nuclear localization signals in the PDZ domain34. Our results suggest that in agreement with previous studies, nuclear localization signals within the PDZ domain influences cell distribution of PRX in cerebral endothelium.
In the absence of colocalization with claudin-5 or ZO-1, available evidence suggests an alternative role for the PDZ domain of PRX which may affect nuclear function in addition to (or instead of) tight junction complexes. Gene expression studies of endothelial cells demonstrate that intermediate events stimulated by PRX predominantly reflect suppression of genes; genes suppressed more than 2-fold vastly exceeded those activated more than 2-fold. Furthermore, the most strongly suppressed genes were factors that have been implicated in response to inflammation and infection. Inflammation and infection, in turn, are generally associated with increases in cerebral vascular permeability. In accordance, inflammatory stimulation of levels of a wide array of proteins was attenuated by PRX expression in endothelial cells. The link between PRX-induced gene suppression and PRX-mediated permeability suppression may thus conceivably be functionally linked. Our findings are also congruent with the known effects of corticosteroids, first line therapy for reducing BBB permeability caused by vasogenic brain edema35,36 and potent global inhibitors of inflammatory gene expression.
Surprisingly, we did not find evidence for notable transcriptional upregulation of tight junction genes in PRX expression studies (Table 2). Among proteins thought to play a role in endothelial permeability, we did not find more than 1.5-fold increases in expression of any gene encoding tight junction proteins, and a similar number of tight junction genes were up- and down-regulated. In aggregate, these studies imply that PRX potentially mediates tight junction tightening via post-transcriptional effects, which may include protein phosphorylation37 and other post-translational controls of tight junction proteins4.
PRX in normal physiology and disease
Brain viability clearly does not require endothelial expression of PRX, since all non-human species examined do not express this protein. In addition, loss of function PRX mutations in humans cause a demyelinating peripheral neuropathy, but there are no symptoms that can be attributed to impaired cerebral endothelial function reported in affected individuals. At first approximation, this may suggest that the impact of PRX on the brain endothelium is minimal. However, it is known that some components of endothelial tight junctions are frequently not required for in vivo function of the blood brain barrier (for example ZO-338). It remains possible that PRX plays a homeostatic role under perturbed states (e.g. during inflammatory challenges and ischemia).
An important impetus for studying proteins that modify the BBB is to identify potential control points that can be leveraged to modulate cerebral endothelial function in disease. In brain conditions that require medications to cross the BBB, impairing barrier function could be beneficial. In other conditions, such as brain injury due to trauma, stroke or dementia, tightening a dysfunctional BBB is advantageous. To control BBB function, identification of a protein that is not absolutely required for BBB function but whose levels can increase or decrease BBB function would be attractive. As a protein that is clearly not essential for BBB function in other species, but that increase barrier function of endothelial cells, PRX may be positioned favorably as a target for development of approaches to control the human BBB during pathological states. From a practical perspective, PRX, if delivered in an active state, could offer substantial advantages over current medications for treating increased brain vascular permeability. Corticosteroids, while effective, affect many other cells and thus induce significant side effects. PRX and pathways stimulated by this protein may offer alternative targets for BBB modification that act on the same pathways as endothelial cell glucocorticoid receptors.
Methods
Animal studies
This study used vertebrate animals. All experiment that involved animals were performed in accordance with NIH guidelines using protocols approved by the Institutional Animal Use and Care Committee (IACUC) at the University of Michigan and the VA Ann Arbor Healthcare System.
Immunohistochemistry
Formalin fixed frontal lobes sections were obtained from the Alzheimer’s Disease Center at the University of Michigan and the Brain Bank of the National Institute for Developmental and Childhood Disorders at the University of Maryland39. Five micron sections were analyzed using chromagenic immunohistochemical staining using a rabbit polyclonal antibody to PRX (Sigma, St. Louis, MO) after citrate-induced antigen retrieval. Staining was followed by hematoxylin counterstaining. Human tissue antigen integrity was ascertained by staining with the mouse monoclonal antibody BRIC231 (anti-H; Santa Cruz). Integrity of mouse tissue antigens was evident in most tissue blocks because PRX is uniformly expressed in peripheral nerve bundles in all organs.
In situ hybridization
Localization of PRX in human brain was performed using a chromogenic detection method (Advanced Cell Diagnostics)40. Hybridization of antisense nucleic acid probes against PRX was followed by multiple non-enzymatic amplification steps, and, finally, probe detection was accomplished with an alkaline phosphatase conjugated terminal probe. After colorimetric detection of the in situ signal, we counterstained with hematoxylin. Brain blocks that resulted in signal with ubiquitous positive control probes were selected for this study.
DNA constructs and virus production
Full length, human PRX cDNA was generated by PCR from a commercially obtained clone (Origene) and cloned into the NotI and XbaI sites of the pLENTI-EV vector for expression studies; the long form in this study is designated L-PRX. D-PRX was generated by PCR using primers to delete sequence encoding the first 199 amino acids of the N-terminus of PRX, replacing this with a Kozak sequence to initiate translation. S-PRX was generating using PCR from the original template and inserted into the NheI and BamHI sites of pLENTI-EV; a unique sequence encoding the short C-terminal tail of S-PRX and stop codon were engineering into the PCR primer. Corresponding GFP fusion clones were generated by inserting these PRX fragments into pEGFP-C3 (Clontech), resulting in a fusion of EGFP at the N-terminus of the PRX sequences. These clones were inserted into XhoI (Klenow blunted) and EcoRI sites of the vector and NcoI (Klenow blunted) and EcoRI sites in the inserts. As a control for viral infection, lentiviruses encoding RFP were also generated.
Western blotting
Whole brain and vessel-enriched fractions were prepared as follows41: All procedures were performed on ice or at 4 °C. Frozen human brain tissue from autopsies (10 g) was disrupted in 15 ml B1 buffer (HBSS with HEPES (10 mol/L)) using a type A glass dounce homogenizer. The brain lysates were centrifuged at 2000g for 10 min, and the cloudy supernatant decanted. The pellet was resuspended in 20 mL of B1 with 18% dextran, and centrifuged at 4400 g for 15 min. The myelin-rich layer and dextran supernatant were removed, and the microvessel pellet was resuspended in 6 ml B3 buffer (B1 supplemented with 1% bovine serum albumin). The microvessels were then passed through 70 μM nylon mesh cell strainers (BD Falcon). Microvessels in the filtrate were washed with B3 and collected in 20 ml B3, centrifuged at 2000g for 5 min, and the pellet was resuspended in 1 mL of B3 and transferred into a microcentrifuge tube, pelleted, and resuspended in RIPA buffer.
Standard electroblotting was performed as before42. Fractions were electrophoretically separated on SDS polyacrylamide gels, electrotransferred to nitrocellulose, and probed using PRX or vascular control antibodies (Sigma, St. Louis, MO) at 1:1000 dilution. Labeled secondary antibodies were detected using a Licor Odyssey infrared scanner. Expression levels were normalized to tubulin content assessed on either the same filter or a parallel Western blot.
RNA quantification
Quantitative reverse transcriptase PCR was performed and analyzed as before43. Cells were scraped off plates and then lysed in lysis buffer per manufacturer’s protocol. RNA was purified using an RNeasy spin column kit (QIAGEN). We converted RNA by reverse transcription and then cDNA was quantified by real time PCR, using HPRT, as a control to assess target gene regulation.
Human brain microvascular endothelial cell culture
Primary Human Brain Microvascular Endothelial Cells (ACBRI 376) were purchased from Cell Systems Corporation (Kirkland, WA 98034, U.S.A). Cells were cultured in CSC Complete Medium which includes 10% fetal bovine serum. The day before recombinant lentiviral infection, cells were split to 48-well-plates coated with CSC Attachment Factor. On the day of infection, 50 μl lentivirus stock was diluted into 0.5 ml CSC complete media; 1 μl of 4 mg/mL polybrene was added to the diluted virus to a final concentration of 8 μg/mL; this mixture was then used to replace the cell media. The viral media was replaced with CSC Complete Medium after 1 day of infection. After an additional day of culture, cells were harvested for RT-PCR. As a control for gene expression studies, we analyzed human brain endothelial cells that were infected with lentivirus encoding RFP.
Microarray analysis
Transcriptome analysis was performed using Affymetrix Human Gene ST 2.1 strips that were processed in the University of Michigan Microarray Facility. Labeled cDNA was prepared using a WT Pico Kit (Affymetrix). Probesets that yielded differences of more that 2-fold between PRX and control groups were considered potentially differentially expressed. Two independent replicates were performed, and the 66 genes that overlapped between two experiments were presented in the final results. A subset of these genes were assessed by quantitative RT-PCR to validate in independent cell groups, performed in triplicate, whether expression was altered by PRX expression.
Protein array analysis
A human cytokine antibody array kit (RayBiotech Inc) was used to simultaneously detect and quantify 80 cytokines/chemokines in cell culture media samples. Briefly, human brain endothelial cells with or without overexpressed PRX, were exposed to Il-1b (10 ng/ml, Preprotech) for 24 hrs, and cell culture media was sampled. The antibody array was performed according to the manufacturer’s instructions. Densitometric analysis was performed using ImageJ analysis software using a protein array analyzer plugin. The relative level of cytokines was evaluated using software provided by the manufacturer.
Mouse brain microvascular endothelial cell culture
Mouse brain microvascular endothelial cells (mBMEC) were prepared using a modified protocol already described44,45,46. Briefly, brains were collected from four to six-week-old C57BL/6 mice, minced in Hanks balanced salt solution (HBSS; Life Science Technology) and homogenized gently in a Dounce type homogenizer. Myelin was removed by 18% Dextran suspension (Dextran 60–90,000; USB, Cleveland, OH) and centrifuging at 8000 rpm for 10 min at 4 °C. Red blood cells were removed by centrifuging isolated microvessels in a Percoll gradient (Thermo Fisher Scientific) at 2700 rpm for 11 min. The isolated microvessels were digested in HBSS solution containing 1 μg/ml collagenase/dispase (Roche, Indianapolis, IN), 10 U/ml DNAase I (Sigma-Aldrich, St Louis, MO) and 1 µg/ml Na-p-tosyl-L-lysine chloromethyl ketone (TLCK) for 20 min at 37 °C and precipitated with CD31 coated magnet beads (Dynabeads, Life Science technology). These vessels were further cultured in Dulbecco’s Modified Eagles medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% inactivated fetal bovine serum (FBS), 2.5 µg/ml heparin (Sigma-Aldrich, St Louis, MO), 20 mM HEPES, 2 mM glutamine, 1X antibiotic/antimycotic (all from Invitrogen, Carlsbad, CA), endothelial cell growth supplement (BD Bioscience, San Jose, CA) and grown in 6 well plates coated with collagen type IV (BD Bioscience, San Jose, CA). This protocol typically produces primary endothelial cell cultures that are approximately 99% pure (as determined by immunocytochemistry with an anti-PECAM-1 antibody; BD Bioscience, San Jose, CA).
Mouse brain endothelial cell lines (mBECL) were purchased from Angio-Proteomie (Boston MA) and cultured in medium (DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine and 1X antibiotic/antimycotic; Invitrogen, Carlsbad, CA) at 37 °C in a humidified incubator (10%CO2/90% air).
Expression of PRX protein in mouse endothelial cells was performed by infection with recombinant lentiviruses, as in the prior section using polybrene. We used two controls for functional studies of PRX lentivirus infected mouse endothelial cells: (1) cells were infected with RFP lentivirus at the same multiplicity of infection and (2) cells were treated with polybrene without virus. In all control studies, the RFP virus had no effect compared to non-infected mouse endothelial cells. Therefore, to simplify the presentation of data, we only show uninfected cells as controls.
Permeability coefficient (PC) assessments
The in vitro permeability of mBMEC monolayers was measured as described in Kazakoff et al.47, and modified in our laboratory44,45,46. The cells were seeded on the collagen IV coat insert in a Transwell dual chamber system at a density of 10,000 cells per square cm which allows the formation of cell barriers 1–7 day after initial plating.
The permeability coefficient (PC; cm/min) of monolayers was calculated for the tracers FITC-inulin (5 kDa) or dextran-Texas red 20 kDa and 40 kDa (1 μg/ml, Sigma-Aldrich) at different time points 1–7 days after initial platting. FITC-inulin and dextran-Texas Red concentrations in lower and upper chambers were evaluated on a fluorescent reader (Infinity FL200) using following formula:
where C(B) and C(B)T are the concentrations (ng/ml) of tracer in the basal (receiving) chamber at the start and at the end of the time interval, respectively, and V(B) is the volume of the basal chamber (in ml). C(A) and C(A)T are, respectively, the concentrations of tracer in the apical (donor) chamber at the start and at the end of the time interval and (C(A) + C(A)T)/2 is the average concentration over the time interval. T is the duration of the time interval (in hours), while A is the area of the filter (in square cm). All samples were read on fluorescent reader (Bio-Tek Instruments, Inc., Winooski, VT U.S.A.; emission 485 and excitation 540 nm). The concentration of FITC-albumin in samples was calculated from a standard curve derived using known concentrations of tracer. Both uninfected cells and RFP lentivirus infected controls were studied and these groups showed identical results. Only data on uninfected controls are shown.
Transendothelial electrical resistance (TEER) measurements
TEERs were measured in Endohm™ TEER measurement chambers equipped with an EVOM resistance meter (World Precision Instruments, Sarasota, FL). All experiments were carried out in triplicate in three independent experiments. Both uninfected cells and RFP lentivirus infected controls were studied and these groups showed identical results. Only data on uninfected controls are shown.
Cell transfection and immunofluorescence
Mouse brain endothelial cell lines (mBECL) were grown to ~70% confluence and then transfected with GFP fusion clones L-PRX, S-PRX and D-PRX using Lipofectamine (Life Technologies) in Opti-MEM medium (Invitrogen) for 6 hrs according to the manufacturer’s instructions. After 24 hrs the samples examined for PRX expression and localization.
For immunofluorescence staining, cell samples were preincubated in blocking solution containing 5% normal horse serum and 0.05% Triton 100X (Sigma Aldrich, MO) in PBS for 30 min and then incubated overnight at 4 °C with the following antibodies: anti-claudin-5 and anti-ZO-1–Alexa Flour 594 conjugated (Life Science Technology). Reactions were visualized by Texas red anti-mouse antibodies. All samples were viewed on a confocal laser scanning microscope (Nikon).
Statistics
Unpaired Student’s t-test and one-way analysis of variance (ANOVA), with Bonferroni and Tukey’s post hoc tests were used to test group level differences (Prism analysis software). A probability value <0.05 was regarded as statistically significant.
References
Hawkins, B. T. & Davis, T. P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacological reviews 57, 173–185 (2005).
Obermeier, B., Daneman, R. & Ransohoff, R. M. Development, maintenance and disruption of the blood-brain barrier. Nature medicine 19, 1584–1596 (2013).
Zlokovic, B. V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201 (2008).
Stamatovic, S. M., Johnson, A. M., Keep, R. F. & Andjelkovic, A. V. Junctional proteins of the blood-brain barrier: New insights into function and dysfunction. Tissue barriers 4, e1154641 (2016).
Krause, G. et al. Structure and function of claudins. Biochimica et biophysica acta 1778, 631–645 (2008).
Furuse, M. et al. Occludin: a novel integral membrane protein localizing at tight junctions. The Journal of cell biology 123, 1777–1788 (1993).
Martin-Padura, I. et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. The Journal of cell biology 142, 117–127 (1998).
Stevenson, B. R., Siliciano, J. D., Mooseker, M. S. & Goodenough, D. A. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. The Journal of cell biology 103, 755–766 (1986).
Jesaitis, L. A. & Goodenough, D. A. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. The Journal of cell biology 124, 949–961 (1994).
Haskins, J., Gu, L., Wittchen, E. S., Hibbard, J. & Stevenson, B. R. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. The Journal of cell biology 141, 199–208 (1998).
Citi, S., Sabanay, H., Jakes, R., Geiger, B. & Kendrick-Jones, J. Cingulin, a new peripheral component of tight junctions. Nature 333, 272–276 (1988).
Yamamoto, T. et al. The Ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junctions in epithelial cells. The Journal of cell biology 139, 785–795 (1997).
Zhong, Y. et al. Monoclonal antibody 7H6 reacts with a novel tight junction-associated protein distinct from ZO-1, cingulin and ZO-2. The Journal of cell biology 120, 477–483 (1993).
Beatch, M., Jesaitis, L. A., Gallin, W. J., Goodenough, D. A. & Stevenson, B. R. The tight junction protein ZO-2 contains three PDZ (PSD-95/Discs-Large/ZO-1) domains and an alternatively spliced region. The Journal of biological chemistry 271, 25723–25726 (1996).
Gonzalez-Mariscal, L., Betanzos, A. & Avila-Flores, A. MAGUK proteins: structure and role in the tight junction. Seminars in cell & developmental biology 11, 315–324 (2000).
Itoh, M. et al. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. The Journal of cell biology 147, 1351–1363 (1999).
Van Itallie, C. M., Tietgens, A. J., Krystofiak, E., Kachar, B. & Anderson, J. M. A complex of ZO-1 and the BAR-domain protein TOCA-1 regulates actin assembly at the tight junction. Molecular biology of the cell 26, 2769–2787 (2015).
Fanning, A. S. et al. The unique-5 and -6 motifs of ZO-1 regulate tight junction strand localization and scaffolding properties. Molecular biology of the cell 18, 721–731 (2007).
Utepbergenov, D. I., Fanning, A. S. & Anderson, J. M. Dimerization of the scaffolding protein ZO-1 through the second PDZ domain. The Journal of biological chemistry 281, 24671–24677 (2006).
Lee, S. J. et al. Large-scale identification of human cerebrovascular proteins: Inter-tissue and intracerebral vascular protein diversity. Plos One 12(11), e0188540 (2017).
Dytrych, L., Sherman, D. L., Gillespie, C. S. & Brophy, P. J. Two PDZ domain proteins encoded by the murine periaxin gene are the result of alternative intron retention and are differentially targeted in Schwann cells. The Journal of biological chemistry 273, 5794–5800 (1998).
Uhlen, M. et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Molecular & cellular proteomics: MCP 4, 1920–1932.
Uchida, Y. & Terasaki, T. Quantitative targeted absolute proteomics-based ADME research as a new path to drug discovery and development: methodology, advantages, strategy, and prospects. Journal of pharmaceutical sciences 100, 3547–3559 (2011).
Uchida, Y. et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem 117, 333–345 (2011).
Alanne, M. H. et al. Tight junction proteins in human Schwann cell autotypic junctions. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society 57, 523–529 (2009).
Maddala, R. et al. Periaxin is required for hexagonal geometry and membrane organization of mature lens fibers. Developmental biology 357, 179–190 (2011).
Gillespie, C. S., Sherman, D. L., Blair, G. E. & Brophy, P. J. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron 12, 497–508 (1994).
Gillespie, C. S. et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron 26, 523–531 (2000).
Boerkoel, C. F. et al. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. American journal of human genetics 68, 325–333 (2001).
Boerkoel, C. F. et al. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Annals of neurology 51, 190–201 (2002).
Guilbot, A. et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Human molecular genetics 10, 415–421 (2001).
Sherman, D. L., Wu, L. M., Grove, M., Gillespie, C. S. & Brophy, P. J. Drp2 and periaxin form Cajal bands with dystroglycan but have distinct roles in Schwann cell growth. The Journal of neuroscience: the official journal of the Society for Neuroscience 32, 9419–9428 (2012).
Shi, Y., Zhang, L. & Yang, T. Nuclear export of L-periaxin, mediated by its nuclear export signal in the PDZ domain. Plos one 9, e91953 (2014).
Sherman, D. L. & Brophy, P. J. A tripartite nuclear localization signal in the PDZ-domain protein L-periaxin. The Journal of biological chemistry 275, 4537–4540 (2000).
Galicich, J. H., French, L. A. & Melby, J. C. Use of dexamethasone in treatment of cerebral edema associated with brain tumors. The Journal-lancet 81, 46–53 (1961).
Ingraham, F. D., Matson, D. D. & Mc, L. R. Cortisone and ACTH as an adjunct to the surgery of craniopharyngiomas. The New England journal of medicine 246, 568–571 (1952).
Tsukamoto, T. & Nigam, S. K. Role of tyrosine phosphorylation in the reassembly of occludin and other tight junction proteins. The American journal of physiology 276, F737–750 (1999).
Xu, J. et al. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Molecular and cellular biology 28, 1669–1678 (2008).
Dong, H., Blaivas, M. & Wang, M. M. Bidirectional encroachment of collagen into the tunica media in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Brain Res 1456, 64–71 (2012).
Zhang, X., Lee, S. J., Young, M. F. & Wang, M. M. The small leucine-rich proteoglycan BGN accumulates in CADASIL and binds to NOTCH3. Translational stroke research 6, 148–155 (2015).
Roberts, L. M. et al. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience 155, 423–438 (2008).
Lee, S. J., Meng, H., Elmadhoun, O., Blaivas, M. & Wang, M. M. Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy Affecting an African American Man: Identification of a Novel 15-Base Pair NOTCH3 Duplication. Arch Neurol 68, 1584–1586 (2011).
Meng, H., Zhang, X., Lee, S. J. & Wang, M. M. Von Willebrand factor inhibits mature smooth muscle gene expression through impairment of Notch signaling. Plos One 8 (2013).
Dimitrijevic, O. B., Stamatovic, S. M., Keep, R. F. & Andjelkovic, A. V. Effects of the chemokine CCL2 on blood-brain barrier permeability during ischemia-reperfusion injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 26, 797–810 (2006).
Stamatovic, S. M., Keep, R. F., Wang, M. M., Jankovic, I. & Andjelkovic, A. V. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. The Journal of biological chemistry 284, 19053–19066 (2009).
Stamatovic, S. M., Sladojevic, N., Keep, R. F. & Andjelkovic, A. V. Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells. Molecular and cellular biology 32, 3414–3427 (2012).
Kazakoff, P. W., McGuire, T. R., Hoie, E. B., Cano, M. & Iversen, P. L. An in vitro model for endothelial permeability: assessment of monolayer integrity. In vitro cellular & developmental biology. Animal 31, 846–852 (1995).
Acknowledgements
We thank the Alzheimer’s Disease Center, Cancer Histology Core, and Vector Core at the University of Michigan for tissue and services performed. Jonathan Markowitz contributed to the protein screening project that identified PRX as a human endothelial brain protein. The following funding sources are gratefully acknowledged: V.A. Merit Awards to M.M.W. (BX0003824), and NIH grants R25HL108842 (supporting S.M.; awarded to Jimo Borjigin), NS093399 (to R.F.K.), and NS062853 (to A.V.A.).
Author information
Authors and Affiliations
Contributions
M.M.W. conceived the idea for the project, analyzed results, and wrote the paper. X.Z., S.J.L., S.M., A.M.J., S.M.S. performed experiments and analyzed results. R.F.K. analyzed results and wrote the paper. A.V.A. performed experiments, analyzed results, and wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, M.M., Zhang, X., Lee, S.J. et al. Expression of periaxin (PRX) specifically in the human cerebrovascular system: PDZ domain-mediated strengthening of endothelial barrier function. Sci Rep 8, 10042 (2018). https://doi.org/10.1038/s41598-018-28190-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-28190-7
This article is cited by
-
Single cell transcriptomic analysis of murine lung development on hyperoxia-induced damage
Nature Communications (2021)
-
Modeling blood–brain barrier pathology in cerebrovascular disease in vitro: current and future paradigms
Fluids and Barriers of the CNS (2020)
-
Transcriptomic comparison of human and mouse brain microvessels
Scientific Reports (2020)
-
The year in review: progress in brain barriers and brain fluid research in 2018
Fluids and Barriers of the CNS (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
