
Abstract
Colorectal cancer (CRC) growth and progression is frequently driven by RAS pathway activation through upstream growth factor receptor activation or through mutational activation of KRAS or BRAF. Here we describe an additional mechanism by which the RAS pathway may be modulated in CRC. PTPRS, a receptor-type protein tyrosine phosphatase, appears to regulate RAS pathway activation through ERK. PTPRS modulates ERK phosphorylation and subsequent translocation to the nucleus. Native mutations in PTPRS, present in ~10% of CRC, may reduce its phosphatase activity while increasing ERK activation and downstream transcriptional signaling.
Similar content being viewed by others


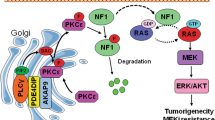
Introduction
Colorectal Cancer (CRC) is the second leading cause of deaths from cancer in the United States1. Genetic analysis of CRC has recently led to a wealth of information concerning the initiation and development of CRC that may help improve our ability to treat this disease2,3. Activation of the RAS pathway is likely a key driver of tumorigenesis as evidenced by the fact that activating mutations of KRAS or BRAF are frequently seen in many cancers (~50% of CRCs)3,4,5,6. Although most CRC tumors (80%) are thought to be initiated through dysregulation of the WNT pathway, CRC tumors with activated KRAS/BRAF/MEK/ERK appear to be associated with poor outcomes7,8 and are difficult to treat.
EGFR inhibitors have been FDA approved for the first and second line treatment of CRC in patients with wild-type KRAS/NRAS9,10,11,12,13,14,15. Despite careful selection of wild-type RAS patients, ~60% of patients still fail to respond to these therapies, suggesting that there may be RAS pathway activation secondary to mutations in genes beyond canonical RAS pathway genes. To decipher the genes with high frequency mutations that might modulate RAS pathway activation, we carried out an integrated analysis of gene expression and sequencing data on 468 CRCs that were molecularly characterized by us recently5,8 using an 18-gene “RAS pathway activation” signature score16,17. We found that mutations in PTPRS were highly concordant with the RAS pathway signature. Moreover these mutations were identified in a significant number of cases.
PTPRS (PTPσ) is a receptor-type protein tyrosine phosphatase (PTP) whose physiological role has been well-established in the nervous system and in pituitary development, as well as in spinal cord injury and repair18,19,20. PTPRS has also been shown to play a role in ulcerative colitis, intestine epithelial permeability, autophagy regulation21,22,23, and tumor suppression24,25,26. PTPRS was recently shown to dephosphorylate EGFR in the A431 and other cancer cell lines25,26,27, and a genomic analysis revealed frequent deletion of PTPRS in head and neck cancers was associated with activation of the EGFR/PI3K pathway25. Now we report a new role for PTPRS in negatively regulating the RAS pathway in CRC by a mechanism modulating ERK activation. Moreover, we show that multiple, native, missense point mutations affecting various domains in ~10% of CRC patients may affect PTPRS function, underscoring their significance.
Results
Identification of PTPRS as one of the top-ranked RAS pathway signature-associated genes
We recently evaluated a cohort of 468 CRC patient tumor samples using both global gene expression and targeted sequencing of 1321 cancer-related genes5,8. In order to identify mutated genes beyond KRAS, BRAF and NRAS that might account for expanded RAS pathway activity, we stratified these 468 CRCs using an 18-gene RAS pathway gene expression signature score that measures pathway activation via MEK functional output16. We recently adapted this signature from use in fresh frozen CRC samples to more clinically-available, archived formalin-fixed, paraffin-embedded (FFPE) tissues17 as a means to predict RAS pathway dependence regardless of RAS/RAF mutation status. In the ranking analysis (see Methods for detailed description) we evaluated both the correlation of mutant genes with the RAS pathway activation score and their mutational frequencies. When all patient samples (n = 468) were included, not surprisingly, the mutated gene most correlated with RAS pathway activation was KRAS, followed secondarily by BRAF. Upon removal of KRAS-mutated tumors (n = 278), BRAF became the No.1 gene (Fig. 1). When the influence of KRAS and BRAF was removed (n = 225), the ranking of NRAS rose from #170 to #1, and became the most correlated mutant gene, thereby validating the approach to further identify contributing mutant genes (Fig. 1). Once out of the shadow of KRAS, BRAF and NRAS (n = 209), a list of 15 top-ranked, potentially new RAS pathway activation-associated genes was identified, in which ADAMTSL3, ITGB4, APC2, GNAS, PTPRS, ATG2B showed >5% mutational frequency in the 209 remaining tumors, while TGFBR2, SLC2A4, INSRR, NLRP3, MAP3K9, MAPT, MN1, MCM3AP and PTK2B had 2.5–4.9% frequencies (see Supplementary Table 1). PTPRS was the most mutated, top-ranked gene (22/209, mutation frequency 10.5%), and it was also the only protein tyrosine phosphatase that stood out among sequenced phosphatases, upon removal of the masking effects of the RAS/RAF common drivers. Notably, the other 16 sequenced receptor type and non-receptor type PTPs including PTPRT had a much lower ranking (#223 or below). This was a surprising result given previous observations that PTPRT might be one of the most prominent phosphatases in CRC28. Interestingly, PTPRS was recently confirmed to be mutated in ~10% of CRC tumors in the database from the Dana Farber Cancer Center6. Our data show that mutations in PTPRS were equally present in CRC tumors with (25/257) and without (22/209) mutation-activated RAS or BRAF.

Identification of PTPRS by a hybrid analysis of global gene expression (Afffymetrix) and observed DNA mutations derived from targeted exome nextgen DNA sequencing of 1321 genes. 468 CRC cases were first scored for RAS pathway activity with an 18-gene RAS pathway gene expression-based activation score. PTPRS emerged as a lead candidate gene to activate RAS pathway when shadows of mutant KRAS, BRAF and NRAS were removed. See Methods for detailed description of the ranking analysis.
Inhibition of PTPRS with a peptide specific inhibitor activated ERK and AKT
To confirm a potential regulatory role of PTPRS in RAS pathway activation, we inhibited PTPRS activity in vivo in CRC cell lines containing both mutation-activated and wild-type KRAS (i.e. HCT116 (KRAS G13D), SW620 (KRAS G12V) and KM12L4A (WT KRAS)). All cell lines harbored wild-type PTPRS29. We employed a 33 amino acid peptide specific inhibitor of PTPRS (ISP) that has been shown to effectively inhibit PTPRS in neuronal cells30. Cell extracts were prepared from HCT116, SW620 and KM12L4A cells that had been treated for 24 hours with 10 μM of the ISP or a scrambled control peptide (SC). Western blot analysis was used to visualize the phosphorylation of ERK1/2, a direct indicator of RAS pathway activation. As can be seen in Fig. 2a, ISP brought about an increase in the level of ERK 1/2 phosphorylation in all three cell lines, regardless of KRAS activation. Notably, the ISP treatment did not bring about an increase in MEK1/2 phosphorylation in KM12L4A cells (WT KRAS) but caused a small (15–25%), albeit statistically significant, increase in p-MEK in HCT116 and SW620 (mutant KRAS) cells. We also found that vanadate, a pan tyrosine phosphatase inhibitor, also brought about an increase in ERK phosphorylation of similar magnitude in all cell lines (Supplementary Fig. 1), supporting inhibition of PTPRS phosphatase activity by ISP. In addition, we observed that ISP also increased AKT phosphorylation at S473 (Fig. 2a), suggesting that inhibition of PTPRS might mediate AKT activation as well.

Western blot analysis for ERK and MEK activation. The indicated CRC cell lines were cultured, cells harvested, extracts prepared and western blots performed. (a) Cells (HCT116, SW620 and KM12L4A) cultured with ISP, an inhibitor of PTPRS, or a scrambled control peptide (SC). Western blots to detect ERK, tyrosine phosphorylated ERK, MEK and phosphorylated MEK, and AKT and phosphorylated AKT are shown as indicated. The quantitations were determined by normalizing the phosphorylated protein values with the total protein; then dividing the ISP values by the SC values. (b) The indicated cells had PTPRS knocked down with siRNA to PTPRS (siPTPRS) or were treated with a scrambled siRNA control (siCtl). Western blot analysis shows PTPRS, phospho-ERK, ERK, phospho-MEK, MEK, phospho-AKT, AKT, and alpha-tubulin. Knockdown of PTPRS via siRNA shows results consistent with the ISP treatments. (c) CRISPR knockouts of PTPRS in HCT116, SW620, and KM12L4A cell lines and their CRISPR control cell lines where cell extracts were used in western blot analysis for phosphorylation of ERK and MEK. This analysis shows PTPRS, ERK, phospho-ERK, MEK, phospho-MEK, AKT, phospho-AKT, and alpha tubulin in the cell line pairs (Ctl and KO) as indicated. (d) ddPCR analysis of PTPRS expression in CRISPR KO cells for HCT116, SW620, and KM12L4A. Analysis of the ddPCR result shows a near complete knockout for HCT116 and KM12L4A; SW620 shows >85% knockout. All experiments were done in triplicate. The mean and standard deviation are shown. Two-tailed, paired t test was used to determine the statistical significance for comparison as indicated.
The effect of siRNA-knock down or CRISPR PTPRS knockout on activation of ERK and AKT
In order to validate the results with ISP and to confirm the specificity of action of PTPRS, we used a functionally validated PTPRS siRNA24 to selectively silence the endogenous expression of PTPRS in HCT116, SW620 and KM12L4A CRC cell lines. Figure 2b shows the decrease in PTPRS protein expression after siRNA treatment for 48 hours as compared to the cells treated with a scrambled siRNA. In agreement with ISP inhibition (Fig. 2a), reduced PTPRS expression in these cell lines brought about increased ERK 1/2 phosphorylation (Fig. 2b). This increase in ERK phosphorylation was verified with a second siRNA to PTPRS (Supplementary Fig. 2). In these experiments, the inhibition of PTPRS expression by siRNA did not bring about an increase in MEK phosphorylation.
To further investigate the sustained effect of the loss of PTPRS activity on ERK activation in CRC cell lines and compare to ISP inhibition and siRNA knockdown of PTPRS, we applied CRISPR technology to permanently knock out expression of PTPRS in the HCT116, SW620 and KM12L4A cell lines. PTPRS was successfully knocked out in each of the cell lines as seen by the loss of PTPRS protein expression (Fig. 2c) and mRNA expression (Fig. 2d). The knockout of PTPRS was associated with an increase in ERK 1/2 tyrosine phosphorylation (Fig. 2c). PTPRS KO caused a small, albeit statistically significant, increase in p-MEK in SW620 but not in HCT116 and KM12L4A cells.
To support and validate the observations seen in Fig. 2, we overexpressed PTPRS in the two CRC cell lines (HCT116 with mutant KRAS and KM12L4A with wild-type KRAS) with CRISPR knockout of PTPRS to determine if the exogenous expression of PTPRS could reduce the phosphorylation of ERK. The exogenously expressed PTPRS seen in Fig. 3(a,b) did reduce ERK phosphorylation that appeared independent of KRAS mutational activation. Thus, the data (Figs 2 and 3) indicate that PTPRS negatively regulates ERK phosphorylation.

Transfections of PTPRS plasmid back into CRISPR KO cells decreases Phospho-ERK. HCT116 and KM12L4A CRISPR PTPRS KO cells and their CRISPR control cells were grown and transfected with full length PTPRS containing vector or empty vector alone. 48 hours after transfection cells were harvested and analyzed for ERK, AKT and MEK phosphorylation. Panel (a) Western blots are for HCT116 CRISPR PTPRS (KO) knockout cells and CRISPR controls (Ctl) as indicated. Cells not treated (NT), transfected with empty vector alone, and transfected with plasmid with PTPRS are shown. Panel (b) is the same as Panel (a) but with KM12L4A cells. The PTPRS expression is shown in two blots. The top blot is a darker exposure to elucidate the native PTPRS expression in the control cells. The second blot for PTPRS is a lighter exposure to show the PTPRS expression of the plasmid transfected cells. Quantitations were determined by normalizing the phosphorylated protein values with the total protein; then dividing the PTPRS KO by the CRISPR control values. All experiments were done in triplicate. The mean and standard deviation are shown. Two-tailed, paired t test was used to determine the statistical significance for comparison as indicated. (c) Co-immunoprecipitation of Flag-tagged PTPRS and ERK. Flag-tagged PTPRS was transfected into HCT116 and KM12L4A PTPRS KO cells. Cell lysates were then immunoprecipitated (IP) with a Flag Ab (left blots). The Flag IP was successful in pulling down the Flag-tagged PTPRS as well as pulling down ERK (lane 3 of left panel). Conversely PTPRS transfected cells were also immunoprecipitated using an ERK Ab (right panel). It was showed that the Flag-tagged PTPRS was pulled down along with ERK (lane 3 of right panel), again supporting a direct association between PTPRS and ERK.
In addition, siRNA knock-down or CRISPR KO of PTPRS in all the cell lines tested also increased p-AKT at S473, whereas transfection of PTPRS plasmid into cells completely blocked the AKT phosphorylation (Figs 2 and 3), indicating that PTPRS negatively regulates AKT activation as well.
PTPRS co-immunoprecipitated with ERK
In order to explore if PTPRS associated with ERK, we sought to determine if immunoprecipitates of PTPRS contained ERK. HCT116 and KM12L4A cells were grown and transfected with plasmid containing PTPRS constructs with Flag tag or with empty vector. Cells were grown for 48 hours and then harvested for cell extract preparation. The PTPRS was immunoprecipitated with antibody to the Flag. The immunoprecipitates were analyzed by western blotting with antibodies to PTPRS-Flag and ERK. Figure 3c shows the PTPRS immunoprecipitates contained ERK and PTPRS. Conversely, when the extracts were immunoprecipitated with ERK they were shown to contain PTPRS by western blotting (Fig. 3c). These data demonstrate that PTPRS and ERK were likely associated in these cells.
Differential effects of PTPRS KO in cells harboring wild-type vs. mutant KRAS
Since our data showed that loss of PTPRS increased the phosphorylation of ERK in tumor cells driven by both mutant KRAS as well as wild-type KRAS, we sought to further confirm the effects of PTPRS knockout (KO) on CRC cells, independent of KRAS mutation status. We compared the parental HCT116 cell line (KRAS G13D/+) with one activated KRAS allele to its isogenic, engineered derivative HCT116 (−/+) in which the activated KRAS allele was deleted, leaving the cell lines with only one wild-type KRAS allele. In each of these two cell lines, PTPRS was knocked out by CRISPR and paired with CRISPR controls. Cells from growing cultures of these paired cell lines were harvested. Extracts were then prepared and activated ERK was measured by western blots of phosphorylated ERK. Figure 4a shows that the cells without PTPRS had an increased ERK phosphorylation greater than its CRISPR control cell line. Clearly, not only did the reduction of PTPRS expression modulate the ERK activity in cells with a mutant KRAS driven ERK pathway, but an increase in ERK activation was also seen in the cells with wild-type KRAS. While mutant KRAS HCT116 (KRAS G13D/+) seemed to induce more phospho-ERK than wild-type KRAS HCT116 (−/+), the loss of PTPRS did not bring about an elevated MEK phosphorylation in either cell line.

Comparison of Isogenic HCT116 Cell lines with and without activated KRAS. (a) The parental CRC HCT116 (KRAS G13D/+) cell line has an activating mutation of KRAS. Here we compare this cell line to an isogenic HCT116 (−/+) cell line that has the mutated KRAS allele knocked out, leaving the cell with only one WT KRAS allele. PTPRS CRISPR KO and CRISPR control paired cell lines were made in both HCT116 parental (KRAS G13D/+) and the isogenic HCT116 (−/+) cell lines. Extracts prepared from growing cultures were used for western blot analysis of ERK, phospho-ERK, MEK, phospho-MEK, AKT andphospho-AKT. The HCT116 (−/+) is shown to have natively less phospho-ERK compared to its parental HCT116 cell line with the activated KRAS mutation. The KO of PTPRS for both cell lines shows a dramatic increase in phospho-ERK and phospho-AKT. Quantitations were determined by normalizing the phosphorylated protein values with the total protein; then dividing the PTPRS KO by the CRISPR control values. All experiments were done in triplicate. The mean and standard deviation are shown. Two-tailed, paired t test was used to determine the statistical significance for comparison as indicated. (b) Active Ras pull down for all cell lines. The mutant KRAS cells lines, HCT116 and SW620, show a constitutively active Ras. The WT KRAS PTPRS KO cell lines, KM12L4A and the isogenic HCT116 (−/+), show a reduced amount of active Ras. KM12L4A was also used for negative and positive controls incubating the samples with GDP and GTPσ; HCT116 and HCT116 (−/+) samples were incubated with GTPσ as a positive control.
RAS-GTP is a measure of RAS activity. It has been recently reported that activated ERK might have a negative inhibitory effect on RAS activity31. Interestingly, here we found that PTPRS KO cells show evidence of feedback inhibition of RAS-GTP expression in wild-type RAS cells (KM12L4A and HCT116 wild-type KRAS). This inhibition by PTPRS KO was not seen in mutant KRAS cells (SW620 and HCT116 parental) (Fig. 4b)
The effect of PTPRS KO on the expression of ERK regulated genes
In order to validate the effect of PTPRS on the modulation of ERK activation, we examined the protein expression of several ERK-regulated genes including c-MYC and DUSP632,33,34,35. In addition, we also examined the ERK specific phosphorylation of ELK1, MSK1 and p90-RSK33,34. To determine and compare the effects of the loss of PTPRS on the expression of these genes in CRC cell lines, we compared HCT116 (KRAS G13D/+) to HCT116 (−/+) in paired cell lines, with and without CRISPR KO of PTPRS. In addition, we also investigated these same ERK regulated genes in KM12L4A (wild-type KRAS) and SW620 (mutant KRAS) PTPRS CRISPR KO cell lines. As seen in Fig. 5, the cells lacking PTPRS had a marked increase in the protein expression of ERK targeted genes and in the phosphorylation of ERK-specific downstream protein targets. Notably, HCT116 (KRAS G13D/+) cells that have activated KRAS produced higher protein expression than the HCT116 cells (−/+) with wild-type KRAS. The loss of PTPRS, however, brought about increased gene expression in both cell lines regardless of KRAS mutation status. These data demonstrate that increased ERK phosphorylation was correlated with an increase in the ERK biological response of regulated gene (protein) expression and signaling.

ERK targeted gene regulation and specific phosphorylations. The CRC cell lines SW620, KM12L4A, HCT116 (WT KRAS) [HCT116 (−/+)], and HCT116 [HCT116 (KRAS G13D/+)] along with their paired CRISPR PTPRS knockout cell lines were grown and cell extracts were prepared. Western blot analysis for Phospho-p90RSK (Ser 380), DUSP6, and C-Myc were performed. The PTPRS knockout cell line in each pair shows the presence of more protein for each product tested than its parental matching cell line. This effect is consistent even between the WT KRAS and mutant KRAS HCT116 cells. Phosphorylation of Elk-1 (S383) and MSK1 (T581) are shown. Quantitations were determined by normalizing the phosphorylated protein values with the total protein; then dividing the PTPRS KO by the CRISPR control values. The mean and standard deviation are shown.
The effect of PTPRS on EGFR signaling in CRC cell lines requires wild-type RAS
The loss of PTPRS brought about elevated ERK signaling in CRC cell lines as demonstrated by the increased protein expression of ERK-regulated genes. Since PTPRS was reported to be an EGFR phosphatase in other cancer cell lines25,26,27, we wanted to determine if the effect of PTPRS KO might be through modulating EGFR activity, upstream of RAS signaling. We first examined the level of EGFR phosphorylation at Y1068 and Y1173 in CRC cell lines, with and without expressed PTPRS (CRISPR KO). Figure 6a shows that no change in p-EGFR at Y1068 and Y1173 in HCT116 parental cells, whereas SW620 lacked expression of both P-EGFR and EGFR. However, we observed that PTPRS KO caused a modest, but statistically significant, increase in the EGFR phosphorylation at Y1068 and Y1173 in HCT116 (−/+) and KM12L4A cells both of which have WT KRAS. To further confirm a role of KRAS mutation status in PTPRS KO-mediated modulation of EGFR phosphorylation response, we used isogenic cell pairs HCT116 (KRAS G13D/+) vs HCT116 (−/+) with and without PTPRS expression. Cells were starved for 24 hours and then challenged with EGF. At the indicated times after EGF addition, cells were harvested, and the amount of phosphorylated EGFR was determined by western blotting. We found that in HCT116 (KRAS G13D/+) cells, PTPRS KO had no or minimal effect on EGFR phosphorylation (Fig. 6b,d). However, wild-type KRAS HCT116 (−/+) cells lacking PTPRS had a more prolonged activation of phospho-EGFR than cells containing PTPRS (Fig. 6c,d). Similar to P-EGFR, a stronger effect on the AKT phosphorylation was also seen in wild-type KRAS HCT116 cells (Fig. 6b–d).

The activation of EGFR and ERK in PTPRS knockout cell lines and in response to EGF stimulation. (a) The CRC PTPRS CRISPR KO cell lines SW620, KM12L4A, HCT116 [mutant KRAS G13D/+], and HCT116 [WT KRAS −/+] and their CRISPR control cells were cultured, and harvested for western blotting to determine the phosphorylation of EGFR. Phosphorylation of EGFR Y1173, Y1068 and total EGFR are shown. Quantitations were done normalizing each phospho-EGFR levels against total EGFR levels; normalized phospho-EGFR values for the KO were then divided by the control values to see the fold change difference. SW620 shows no EGFR expression revealing that EGFR appears to be not the driving mechanism of the observed change in phospho-ERK. HCT116 [KRAS G13D/+] also shows no change in EGFR phosphorylation. The WT KRAS cell lines, KM12L4A and HCT116 [WT KRAS −/+] do show a change in phospho-EGFR. All experiments were done in triplicate. The mean and standard deviation are shown. Two-tailed, paired t test was used to determine the statistical significance for comparison as indicated. (b) Time course of serum starved HCT116 parental [mutant KRAS, G13D/+] PTPRS CRISPR KO and CRISPR control cells treated with EGF (50 ng/ml). At the times indicated, cells were harvested and extracts were prepared for western blotting. EGFR, EGFR phosphorylated at Y1173 andY1068, as well as ERK, phospho-ERK, AKT and phospho-AKT are shown. (c) HCT116 WT KRAS [−/+] PTPRS CRISPR KO and CRISPR control cells are shown with identical treatment as in (b). (d) This graph shows the normalized values of phospho-EGFR (left panels, average of Y1173 and Y1068), phospho-ERK (middle panels), and phospho-AKT (right panels) for the blots shown in Fig. 6b (top three panels) and Fig. 6c (bottom three panels). All experiments were done in triplicate. The mean and standard deviation are shown. Two-tailed, paired t test was used to determine the statistical significance for comparison. Significant P values (<0.05) are shown; * − near significant P values.
Native PTPRS mutations found in CRC decreased PTPRS activity
A significant number of somatic mutations in PTPRS were found in our 468 tumor database, and in the Dana Farber CRC database recently published6. The landscape of these PTPRS mutations is shown in Supplementary Fig. 3. In order to determine if the mutations in PTPRS could alter its functionality, we performed a biochemical analysis using phospho-ERK and phospho-AKT as readouts. We selected 7 native PTPRS mutants in CRC for further analysis (Fig. 7a). These PTPRS mutants were selected based on their frequency and the position of the mutation within specific domains in the PTPRS protein structure. In addition to the native mutations, we also prepared three plasmids with deletion mutations to remove the immunoglobulin (Ig) domain or the phosphatase domains (D1 and/or D2) of PTPRS (Fig. 7a). The HCT116 (mutant KRAS) and KM12L4A (WT KRAS) CRISPR PTPRS KO cell lines that had highly elevated ERK and AKT phosphorylation (Fig. 2c) were used here. When full-length wild-type PTPRS was transfected back into the cells for 48 hours, the dramatically reduced phospho-ERK and phospho-AKT were observed compared to the empty vector control (Fig. 7b,c), indicating the inhibitory activity of PTPRS. The constructs with the appropriate PTPRS mutations were then transfected into HCT116 for 48 hours for western blot analysis. Results show that 6 of 10 mutations tested (R714C, R1608Q, R1384Q, -D2, -D1&D2 and –Ig) exhibited completely or considerably reduced PTPRS activity compared to wild-type PTPRS plasmid (Fig. 7b,c). For example, two PTPRS point mutations (R1608Q and R1384Q) showed a distinct reduction in ERK de-phosphorylation (i.e. phospho-ERK with the PTPRS mutations compared to wild-type PTPRS transfection). The levels of p-ERK and p-AKT seen in these mutants (lanes 7 and 12) match those of the empty vector (Control, lane 1) and the truncation mutant that removes both D1 and D2 phosphatase domains (lane 10), implying that point mutations R1608Q and R1384Q are complete, de-activating mutations. Interestingly, removal of just the D2 domain (lane 9) or of the IG domain (lane 11) appears to reduce the activity of PTPRS as measured by phospho-ERK levels, but is not completely de-activating, as is seen in lanes 7, 10, and 12. The remaining 4 mutations (T103I, S717F, V363I and R1091Q) had only minimal or modest effects on the PTPRS activity (Fig. 7b,c). Notably, similar results were also observed for phospho-AKT in HCT116 and KM12L4A cell lines.

Effects of endogenous native PTPRS mutations on ERK de-phosphorylation assay. (a) Endogenous, native mutations were selected for further study based on frequency and location. 7 plasmids containing selected point mutations were synthesized. Additionally, 3 truncated mutants were synthesized. (b,c) HCT116 (KRAS G13D/+) and KM12L4A (WT KRAS) PTPRS KO cells were transfected with the various mutant plasmids and cultured for 48 hours. Extracts were prepared and levels of ERK and AKT phosphorylation were determined with western blotting. Results show that 6 of 10 mutations we tested (R714C, R1608Q, R1384Q, -D2, -D1&D2 and –Ig) exhibited completely or considerably reduced PTPRS activity compared to WT PTPRS plasmid. Similar results were observed in both cell lines tested. The experiments were done in triplicates. Quantitations were determined by normalizing the phosphorylated protein values with the total protein. All experiments were done in triplicate. The mean and standard deviation are shown. Two-tailed, paired t test was used to determine the statistical significance for comparison between WT PTPRS (Ref) and PTPRS mutants. Significant P values (<0.05) are shown; * − near significant P values; Vector − empty vector as a control.
PTPRS expression reduced nuclear ERK staining
HCT116 PTPRS CRISPR KO and control paired cell lines were grown and then fixed and stained for total ERK. Immunofluorescent staining (Fig. 8a) showed strong ERK nuclear localization in HCT116 PTPRS KO cells (upper left). By contrast, ERK staining was present throughout the cells in HCT116 control cells (lower left). A comparison of the control cells to the KO cells showed that the PTPRS KO cells had nuclei that were enriched with total ERK (arrow in top left compared to arrow in bottom left), which correlated with the PTPRS KO cells having increased levels of phospho-ERK as shown previously in western blot analysis (Fig. 2c). The importance of ERK phosphorylation with its location is confirmed when looking at ERK in these same cells and conditions with a MEK inhibitor (PD98509, middle panels). The inhibition of MEK prevented ERK phosphorylation resulting in ERK not being translocated to the nucleus (middle images have nuclei with very low-stained signals for ERK). Staining for phospho-ERK (right most columns) showed a very dynamic difference with PTPRS KO cells showing a bright signal and control cells displaying very weak signal. The linear profile of fluorescence intensity for both DAPI and ERK/phospho-ERK confirmed the increased signal seen in the nuclei of PTPRS KO cells (Supplementary Fig. 4). The effect of the MEK inhibitor was also verified by Western blotting (Fig. 8b). Figure 8c shows multichannel blotting of cells transfected with PTPRS with a C-terminal Flag tag to illustrate the natural cleavage of PTPRS36. Full length PTPRS is 217 kDa (yellow), the N-terminal subunit containing extracellular and transmembrane domains are 140 kDa (green - PTPRS antibody) the C-terminal subunit containing phosphatase D1 and D2 domains are 78 kDa (red - Flag antibody). Notably, this cleavage was a consistent result, as seen in all cell lines used.

PTPRS affects the localization of ERK. (a) Immunofluorescent staining shows ERK nuclear localization in HCT116 PTPRS KO cells (upper left) and general whole cell staining in HCT116 control cells (lower left). A comparison of the control cells to the KO shows that the PTPRS KO cells have nuclei that are enriched with total ERK (arrow in top left compared to arrow in bottom left). The ERK phosphorylation in its location is confirmed with a MEK inhibitor (PD98509, middle panels). The inhibition of MEK prevent ERK phosphorylation resulting in ERK not being translocated to the nucleus (middle images have nuclei with very low-stained signals for ERK). Staining for phospho-ERK (right panels) shows a very dynamic difference with the PTPRS KO cells showing a bright signal and the control cells having very weak signal. Supplementary Fig. 4 shows the linear profile of fluorescence intensity for both DAPI and ERK/phospho-ERK. These measurements confirm the increased signal seen in the nuclei of PTPRS KO cells. (b) Western blot for the cells in Fig. 8a. The left two lanes are untreated HCT116 cells and right two are treated with 5 μM of MEK inhibitor PD98509 for 24 hours. The upper blot confirms the PTPRS knockout in the KO cells. The lower blots for phospho-ERK and total ERK confirm the increased phospho-ERK for the PTPRS KO in the left two lanes. The right most lanes confirm that the MEKi prevented the phosphorylation of ERK. (c) Multichannel blot of cells transfected with PTPRS with a C-terminal Flag tag. This blot uses both a PTPRS (rabbit green) and Flag (mouse red) antibody. Here the cleavage of PTPRS is illustrated. Full length PTPRS is 217 kDa (yellow), the N-terminal subunit containing extracellular and transmembrane domains are 140 kDa (green) the C-terminal Subunit containing phosphatase D1 and D2 domains are 78 kDa (red). This cleavage was a consistent result, and seen in all cell lines used. (d) Assessment of PTPRS transfection on ERK localization. HCT116 and KM12L4A PTPRS KO cells were transfected with a RFP C-terminal tagged PTPRS. Here we examined the localization of PTPRS (red) and total ERK (green) as well as their co-localization (orange). The PTPRS transfected cells (red) show a critically reduced level of ERK in their nuclei (white arrows) when compared to the cells not over expressing PTPRS (red arrows), which have bright green nuclei (ERK). (e) Western blotting corresponding to the cells used in 8d and 8f. The left three lanes show the PTPRS KO compared to the control cells and PTPRS KO cells transfected with PTPRS. The third lane shows that PTPRS transfected back into PTPRS KO cells reduces the increased phospho-ERK back to levels equivalent to the control cell line. The right three lanes show the ISP inhibited the transfected PTPRS activity allowing for increased phospho-ERK. (f) The nuclear reduction of ERK as a result of PTPRS transfection is reversed when PTPRS is inhibited by the ISP. HCT116 and KM12L4A PTPRS KO cells were transfected with the RFP tagged PTPRS and then treated with the PTPRS inhibitor ISP. The reduction in nuclear ERK (8d) is completely reversed (8f) when PTPRS is inhibited. Both the cells overexpressing PTPRS (white arrows) and non-transfected cells (red arrows) show bright ERK signal in their nuclei. Supplementary Fig. 5 shows the DAPI stains for these images. (g) Duo-Link In Situ staining for PTPRS and ERK co-localization. HCT116 PTPRS KO cells were transfected with a C-terminal FLAG tagged PTPRS or control empty vector. The cells were then labeled with a FLAG mouse Ab and an ERK rabbit Ab. The red dots indicate a successful duolink reaction, which requires both antibodies to be in close proximity. The PTPRS transfected cells show an ample amount of red signal (left), and the empty vector cells do not show a significant amount of signal (right). These data suggest a direct association between PTPRS and ERK.
An effect of PTPRS transfection on ERK localization was further assessed. HCT116 and KM12L4A PTPRS KO cells were transfected with a RFP C-terminal tagged PTPRS (Fig. 8d). For PTPRS, we see two localizations: (1) the cell membrane and (2) surrounding the nucleus. The perinuclear PTPRS is likely the cleaved form of the protein that includes the C-terminal D1 and D2 phosphatase domains. The PTPRS transfected cells (red) showed a critically-reduced level of ERK in their nuclei (white arrows) when compared to the cells not over expressing PTPRS (red arrows), which have bright green nuclei (ERK). In addition, the nuclear reduction of ERK as a result of PTPRS transfection was verified by addition of the PTPRS inhibitor ISP and Western blotting (Fig. 8e,f).
We have previously shown that PTPRS and ERK co-immunoprecipitated with each other (Fig. 3c). Moreover, we used DuoLink (22,23) to further confirm that ERK and PTPRS are proximally-associated in cells (Fig. 8g). These data suggest a direct association between PTPRS and ERK.
Discussion
Receptor type and non-receptor type protein tyrosine phosphatases (PTPs) are thought to be important in regulating the RAS/ERK pathway, although their functional role in cancer is much less understood than their counterpart protein tyrosine kinases (PTKs)37,38,39. For example, receptor-type PTPs PTPRE (PTPε) and PTPRJ (DEP-1) were shown to inhibit ERK activation in vitro using NIH3T3, HEK293 and/or HeLa model cell lines40,41. Recently, PTPN11 (SHP2), a non-receptor type PTP, was reported to play an oncogene-like role in laryngeal cancer, hepatocellular carcinoma and glioblastoma, and the mechanism appeared to involve dephosphorylating RAS to activate the RAS/ERK pathway42,43,44. Moreover, while genetic and epigenetic alterations in a number of PTPs have been observed in CRC3,5,6,28,38,45,46, a role for these PTPs in regulating RAS/ERK pathway is not yet known.
We identified PTPRS mutations as significantly associated with RAS pathway activation (Fig. 1), suggesting a regulatory role for PTPRS in RAS/ERK signaling in CRC. PTPRS was frequently mutated in our CRC dataset (46/468, 9.8%). This is in close agreement with the somatic mutation rate reported for PTPRS by DFCI (57/619, 9.2%)6 (Supplementary Fig. 3). Since PTPRS was reported to have a tumor suppressor-like role24,25,26, we postulated that the somatic mutations of PTPRS, if functional, might be inactivating mutations, which could mediate RAS/ERK pathway activation, which is a driver of tumorigenesis3,4,5,6. In support of this notion, our biochemical analyses using a specific peptide inhibitor, siRNA and CRISPR knockout demonstrated that inhibition or loss of PTPRS resulted in elevated ERK phosphorylation in both mutant KRAS and wild-type KRAS CRC cell lines (Fig. 2). The increase in ERK phosphorylation was associated with an increase in ERK-stimulated gene expression (DUSP6, CMYC) and ERK-specific phosphorylation of p90RSK, ELK1 and MSK132,33,34,35 (Fig. 5). The role of PTPRS in regulating ERK activation was also confirmed by using a PTPRS expression plasmid, which reduced ERK phosphorylation (Fig. 3), indicating that PTPRS is a negative regulator of ERK activation.
PTPRS was reported to be an EGFR phosphatase in A431 epidermoid carcinoma cells and head and neck cancers25,27. We observed that loss of PTPRS (knockout) caused a modest but statistically significant increase in phospho-EGFR at Y1068 and Y1173 in wild-type KRAS CRC cell lines yet had no effect in mutant KRAS cell lines (Fig. 6a). Using HCT116 parental (KRAS G13D/+) and the isogenic HCT116 (KRAS −/+) cell line, we demonstrated that loss of PTPRS had no or minimal effect on EGFR phosphorylation in mutant KRAS HCT116 cells following EGF stimulation whereas wild-type KRAS HCT116 (−/+) cells lacking PTPRS had a more prolonged activation of phospho-EGFR compared to the control cells containing PTPRS (Fig. 6c,d). These data indicate that PTPRS may be involved in negative regulation of EGFR signaling in the absence of oncogenic activation of KRAS in CRC. Since we consistently observed a significant increase in ERK phosphorylation by inhibition/loss of PTPRS in both mutant and WT KRAS cell lines, the regulation of EGFR signaling appears to be not necessary for PTPRS’s role in moderating ERK activation. Activated ERK not only mediates RAS pathway downstream signaling that regulates various cellular process but can also mediate feedback regulation of RAS pathway32,33,34,35,47,48. We also observed PTPRS KO-mediated feedback inhibition of RAS-GTP expression in association with ERK activation in WT KRAS but not mutant KRAS cell lines (Fig. 4b). This suggests that mutation-activated RAS might block feedback regulation of RAS pathway activation by PTPRS.
MEK is the only known ERK kinase34,35,47,48. Except for a slight increase in p-MEK in SW620 cell line, the inhibition/loss of PTPRS in all other cell lines (regardless of RAS mutation status) did not alter MEK phosphorylation (Figs 2 and 4). This suggests that ERK activation observed in all cell lines tested was not mediated by MEK. We found that PTPRS and ERK co-immunoprecipitated and co-localized (Figs 3c and 8g), suggesting a direct interaction between PTPRS and ERK. Using a p-ERK Y204-specific antibody, we observed significantly-increased tyrosine-specific phosphorylation in ERK1/2 induced by PTPRS KO in all cell lines (Figs 2c and 4a).
Activation of ERK is required for its entry into the nucleus and its nuclear activities47,49,50. We found that the loss of PTPRS results in enriched nuclear localization of ERK, whereas the ectopic expression of PTPRS retains ERK primarily in the cytoplasm (Fig. 8). Thus, PTPRS may inhibit ERK nuclear localization by negative regulation of ERK activation. Alternatively, it is also possible that PTPRS may directly associate with ERK in the cytoplasm by a mechanism involving scaffold protein complexes that are thought to block ERK translocation to the nucleus47. PTPRS is a membrane receptor PTP that was reported to be proteolytically cleaved into two subunits (the E subunit containing the N-terminal extracellular domain and the P subunit containing the C-terminal phosphatase domains36. In the western blot analysis, we saw both subunits as well as the full-length protein (Fig. 8c). Thus, PTPRS might be associated with ERK on the plasma membrane. Whether PTPRS might be associated with the ERK P-subunit in the cytosol is not yet clear.
Deletion of PTPRS was reported to be associated with abnormal activation of PI3K/AKT signaling in head and neck cancers25. We also found that PTPRS inhibition/KO in CRC cell lines, regardless of RAS mutation status consistently increased AKT phosphorylation at S473, indicating activation of AKT (Figs 2 and 4). There exists a crosstalk between RAS signaling and PI3K/AKT signaling47,51,52,53. However, whether increased phosphorylation of ERK and AKT by PTPRS KO were inter-dependent is not known yet. MEK has been suggested as a focal point for cross-cascade regulation51. However, inhibition/loss of PTPRS Increased p-ERK and p-AKT without moderating p-MEK in most of cell lines tested. Thus, MEK likely did not play a role here.
Finally, it was important to assess the functional effects of a variety of the observed, native PTPRS mutations. We sought to test the most commonly observed somatic alterations (Fig. 7). Surprisingly, when compared to wild-type PTPRS, 4/7 tested native variants had a measurable deleterious effect on PTPRS function as measured by ERK activation (Fig. 7). Moreover, we were able to demonstrate a functional role for the Ig-SET domain and for the D1 + D2 domains via deletion constructs. These data suggest that a substantial percentage of the ~10% SNVs observed in CRC may have a deleterious functional effect.
RAS and its downstream effectors have been targeted to develop therapeutic inhibitors in various cancers with frequently mutated KRAS or BRAF54,55,56. Although efforts to directly target RAS have not been successful to date, selective BRAF and/or MEK inhibitors have shown clinical efficacy in BRAF-mutant melanoma56. More recently, specific inhibitors of ERK have been also developed, which appear to hold promise to overcome acquired resistance to MEK inhibitors56,57,58,59,60. It is noteworthy that the combination of MEKi with the PI3K/AKT/mTOR inhibitors has been conducted in preclinical studies and in clinical trials61. Our study revealed that inactivation of PTPRS enhanced the activation of ERK and AKT, which may facilitate the development of effective targeted therapies against ERK and/or AKT in colorectal cancer.
Methods
Cell Culture
HCT116 (KRAS G13D), SW620 (KRAS G12V) and KM12L4A (WT KRAS) CRC cell lines were obtained from ATCC and tested monthly for mycoplasma contamination with Sigma LookOut® Mycoplasma qPCR Detection Kit (Cat No. MP0040A-1KT). The parental HCT116 CRC cell line (KRAS G13D/+) and the engineered HCT116-KRAS (−/+) cell line, were obtained from Horizon Discovery (Cat.No.HD104-008). Cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS and 1% penicillin and streptomycin.
Immunoblotting and Active Ras Assay
Cells were lysed in 1x RIPA buffer (9806 Cell Signaling) containing 10 mM PMSF, Protease Inhibitor Cocktail (M250 Amresco), Phosphatase Inhibitor Cocktail 2 (P5726 Milipore), and Phosphatase Inhibitor Cocktail 3 (P0044 Milipore) followed by immunoblotting using LI-COR Odyssey® CLx Imaging System. Antibodies were typically duplexed using rabbit antibodies for phosphorylated antibodies and mouse antibodies for total protein. Li-Cor secondary antibodies, Goat anti-Rabbit IRDye 680RD and Goat anti-Mouse IRDye 800CW, were used with the duplexed primary antibodies.
All primary antibodies were rabbit unless specified and were sourced as follows: PTPRS (mouse Cat.No.ab55640 Abcam); PTPRS (goat AF3430 R&D Systems); alpha-Tubulin (mouse sc-8035 Santa Cruz). All other antibodies were obtained from Cell Signaling: phospho-Erk1/2 T202/Y204 (Cat.No.4370); phospho-Erk1Y204/Erk2 Y187 (mouse D1H6G); Erk1/2 (mouse 4696); phospho-MEK1/2 S217/221 (9154); MEK (mouse 4694); phospho-EGFR Y1173 (4407); EGFR (mouse 2239); Elk-1(rabbit Ab 9182), p-Elk1 (Ser383 mouse Ab 9186), MSK1 (rabbit Ab 3489), p-MSK1 (Thr 581 rabbit Ab 9595) and Erk Rabbit Ab 4695).
Active Ras assay was performed using the Active Ras Pull Down and Detection kit from Thermo Fisher (Cat.No.16117).
RT-PCR and ddPCR
Total RNA was isolated using Autrum Total RNA Mini Kit (Cat.No.7326820 Bio Rad) followed by reverse transcription reactions with SuperScript III First-Strand Synthesis System (18080051 Thermo Fisher). ddPCR was performed using the QX200 droplet generator and reader system (Bio Rad) with ddPCR Supermix for Probes (186-3026 Bio Rad).
Samples were run in triplicate using 20 μL of final reaction mix with probes and 30 ng of cDNA per reaction. They were thermocycled on a C100 Touch Thermo Cycler using the recommended program cycle. Individual FAM probes were obtained from Bio Rad unless specified otherwise: PTPRS (dHsaCPE5055124) and the reference gene B2M HEX Probe (dHsaCPE5053101).
Intracellular Sigma Peptide (ISP)
The Intracellular Sigma Peptide (ISP) for inhibiting PTPRS activities and the scrambled ISP were designed and reported (19). These two peptides were synthesized by GenScript at >75% purity. Both peptide sequences contain TAT domain to enable membrane penetration. (1) H-ISP (GRKKRRQRRRCDMAEHTERLKANDSLKLSQEYESI) – targeted specifically against a highly conserved 24-amino-acid intracellular wedge domain of human PTPRS and (2) Scrambled ISP (GRKKRRQRRRCIREDDSLMLYALAQEKKESNMHES) – the sequence except TAT domain is scrambled. 10 μM of ISP (the scrambled ISP as a control) was used to inhibit PTPRS in CRC cell lines.
siRNA Transfection
Two PTPRS-specific siRNAs were obtained from Qiagen: PTPRS_5 siRNA (SI02759288 Qiagen, target sequence: CAGGACATTCTCTCTGCACAA); PTPRS_7 siRNA (SI03056284 Qiagen, target sequence: ATGGCGTGCCCGAATACCCAA).
Scrambled siRNAs from Qiagen (SI03650325) and Origene (SR30004) were used as controls. Transfections were performed at 20–30% cell confluency using the RNAiMAX Lipofectamine (Life Tech) according to the provided protocol using 30 nM of siRNA.
Plasmid Transfection
The PTPRS expression vector pRK-PTPRS was kindly provided by Dr. Jeff MacKeigan (Laboratory of Systems Biology, Van Andel Research Institute, Grand Rapids, MI) (23). Cells were grown to ~50% confluency and were then treated with Lipofectamine 3000 (Cat.No.11668-019 Thermo Fisher). The PTPRS expression vectors containing various site and deletion mutations were also customarily ordered from GeneCopoeia.
CRISPR knockout of PTPRS
The CRISPR kit for PTPRS was purchased from Origene (Cat.No.KN211163) and used according to the product protocol. Cells were transfected using Lipofectamine3000. The gRNA sequence KN211163G1, PTPRS gRNA vector 1 in pCas-Guide vector, (target sequence: CTTGTGGTCCTGCTCGTTGG) proved the most effective at knocking out (KO) PTPRS expression and was thus used to create the HCT116, HT29 and SW620 PTPRS KO cell lines. CRISPR cells were then grown for 7 passages and selected using puromycin (Life Technologies). Numerous colonies were isolated and tested for absence of PTPRS via Western blot and mRNA analysis.
EGF Stimulation
PTPRS CRISPR KO and control cells of HCT116 parental and HCT116-KRAS (−/+) cell lines were plated with approximately 300,000 cells in 6-well plates with serum free RPMI. Cells were starved for 24 hours then treated with 125 ng of EGF (Cat.No.PHG0313 Thermo Fisher) in 2.5 mL serum free media (50 ng/ml). Cells were harvested at the 5, 15, and 30 minutes times points after EGF stimulation; an untreated 0 time point was used as a control.
MEK Inhibitor Treatment
HCT116 PTPRS KO and control cells were plated in 6-well plates. After 24 hours the cells were treated with 5 μM of MEK inhibitors PD98509 (Cat.No.P215 Sigma). Cells treated for 24 hours were then either harvested for western blot analysis or used for immunofluorescent staining.
Immunostaining
Immunostained slides were analyzed with a Leica DMi8. The Cherry C-terminal tagged PTPRS was obtained from GeneCopoeia. Duo Link (Sigma DU092008) assays were performed per manufacturer’s instructions using Mouse Ab DU092004
Statistical Analysis
We previously analyzed 468 stages I-IV colorectal tumors with (affymetrix) global gene expression analysis data from the surgical specimen and targeted gene sequencing of 1321 cancer-related genes5,8. Here we further used this well-curated clinico-genomics/expression database of CRC patient samples to carry out mutation ranking analysis using SAS 9.4 (Cary, NC). We first stratified the 468 CRCs by an 18-gene RAS pathway gene expression signature score16. The arithmetic mean expression of the 18 signature genes of a tumor sample is designated as its 18-gene RAS pathway score. A mutated gene list was constructed by ranking the RAS signature scores of tumors with and without a mutation in the given gene (out of 1321) using the p-value coming from one-sided Wilcoxon rank sum test with normal scores, where the mutated tumors give rise to higher RAS signature scores. For cell line studies, experiments were done in triplicates, and mean and standard deviation were calculated as indicated. Two-tailed, paired t test was used to determine the statistical significance of comparison as needed.
Availability of materials and data
The materials and datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2016. CA Cancer J Clin, https://doi.org/10.3322/caac.21332 (2016).
Fearon, E. R. Molecular genetics of colorectal cancer. Annual review of pathology 6, 479–507, https://doi.org/10.1146/annurev-pathol-011110-130235 (2011).
Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337, https://doi.org/10.1038/nature11252 (2012).
Schubbert, S., Shannon, K. & Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nature reviews. Cancer 7, 295–308, https://doi.org/10.1038/nrc2109 (2007).
Schell, M. J. et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nat Commun 7, 11743, https://doi.org/10.1038/ncomms11743 (2016).
Giannakis, M. et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep, https://doi.org/10.1016/j.celrep.2016.03.075 (2016).
Popovici, V. et al. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol 30, 1288–1295, https://doi.org/10.1200/JCO.2011.39.5814 (2012).
Schell, M. J. et al. A Composite Gene Expression Signature Optimizes Prediction of Colorectal Cancer Metastasis and Outcome. Clin Cancer Res 22, 734–745, https://doi.org/10.1158/1078-0432.CCR-15-0143 (2016).
De Roock, W. et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The lancet oncology 11, 753–762, https://doi.org/10.1016/S1470-2045(10)70130-3 (2010).
Laurent-Puig, P. et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27, 5924–5930, https://doi.org/10.1200/JCO.2008.21.6796 (2009).
Douillard, J. Y. et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. The New England journal of medicine 369, 1023–1034, https://doi.org/10.1056/NEJMoa1305275 (2013).
Bardelli, A. & Siena, S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 28, 1254–1261, https://doi.org/10.1200/JCO.2009.24.6116 (2010).
Mahipal, A. & Grothey, A. Role of Biologics in First-Line Treatment of Colorectal Cancer. J Oncol Pract 12, 1219–1228, https://doi.org/10.1200/JOP.2016.018382 (2016).
Yang, M. & Yeatman, T. J. Molecular stratification of colorectal cancer populations and its use in directing precision medicine. Expert Review of Precision Medicine and Drug Development 2, 205–215, https://doi.org/10.1080/23808993.2017.1362316 (2017).
Allegra, C. J. et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 27, 2091–2096, https://doi.org/10.1200/JCO.2009.21.9170 (2009).
Dry, J. R. et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res 70, 2264–2273, https://doi.org/10.1158/0008-5472.CAN-09-1577 (2010).
Omolo, B. et al. Adaptation of a RAS pathway activation signature from FF to FFPE tissues in colorectal cancer. BMC Med Genomics 9, 65, https://doi.org/10.1186/s12920-016-0225-2 (2016).
Elchebly, M. et al. Neuroendocrine dysplasia in mice lacking protein tyrosine phosphatase sigma. Nat Genet 21, 330–333, https://doi.org/10.1038/6859 (1999).
Wallace, M. J. et al. Neuronal defects and posterior pituitary hypoplasia in mice lacking the receptor tyrosine phosphatase PTPsigma. Nat Genet 21, 334–338, https://doi.org/10.1038/6866 (1999).
Shen, Y. et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 326, 592–596, https://doi.org/10.1126/science.1178310 (2009).
Muise, A. M. et al. Protein-tyrosine phosphatase sigma is associated with ulcerative colitis. Curr Biol 17, 1212–1218, https://doi.org/10.1016/j.cub.2007.06.013 (2007).
Martin, K. R. et al. Identification of PTPsigma as an autophagic phosphatase. J Cell Sci 124, 812–819, https://doi.org/10.1242/jcs.080341 (2011).
Murchie, R., Guo, C. H., Persaud, A., Muise, A. & Rotin, D. Protein tyrosine phosphatase sigma targets apical junction complex proteins in the intestine and regulates epithelial permeability. Proc Natl Acad Sci USA 111, 693–698, https://doi.org/10.1073/pnas.1315017111 (2014).
MacKeigan, J. P., Murphy, L. O. & Blenis, J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol 7, 591–600, https://doi.org/10.1038/ncb1258 (2005).
Morris, L. G. et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc Natl Acad Sci USA 108, 19024–19029, https://doi.org/10.1073/pnas.1111963108 (2011).
Wang, Z. C. et al. Protein tyrosine phosphatase receptor S acts as a metastatic suppressor in hepatocellular carcinoma by control of epithermal growth factor receptor-induced epithelial-mesenchymal transition. Hepatology 62, 1201–1214, https://doi.org/10.1002/hep.27911 (2015).
Suarez Pestana, E. et al. The transmembrane protein tyrosine phosphatase RPTPsigma modulates signaling of the epidermal growth factor receptor in A431 cells. Oncogene 18, 4069–4079, https://doi.org/10.1038/sj.onc.1202794 (1999).
Wang, Z. et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 304, 1164–1166, https://doi.org/10.1126/science.1096096 (2004).
Reinhold, W. C. et al. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res 72, 3499–3511, https://doi.org/10.1158/0008-5472.CAN-12-1370 (2012).
Lang, B. T. et al. Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature 518, 404–408, https://doi.org/10.1038/nature13974 (2015).
Yao, Z. et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238, https://doi.org/10.1038/nature23291 (2017).
Lake, D., Correa, S. A. & Muller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci, https://doi.org/10.1007/s00018-016-2297-8 (2016).
Lu, Z. & Xu, S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life 58, 621–631, https://doi.org/10.1080/15216540600957438 (2006).
Morrison, D. K. MAP kinase pathways. Cold Spring Harb Perspect Biol 4, https://doi.org/10.1101/cshperspect.a011254 (2012).
Bermudez, O., Pages, G. & Gimond, C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am J Physiol Cell Physiol 299, C189–202, https://doi.org/10.1152/ajpcell.00347.2009 (2010).
Aicher, B., Lerch, M. M., Muller, T., Schilling, J. & Ullrich, A. Cellular redistribution of protein tyrosine phosphatases LAR and PTPsigma by inducible proteolytic processing. J Cell Biol 138, 681–696 (1997).
Julien, S. G., Dube, N., Hardy, S. & Tremblay, M. L. Inside the human cancer tyrosine phosphatome. Nature reviews. Cancer 11, 35–49, https://doi.org/10.1038/nrc2980 (2011).
Hendriks, W. J. et al. Protein tyrosine phosphatases in health and disease. FEBS J 280, 708–730, https://doi.org/10.1111/febs.12000 (2013).
Tonks, N. K. Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J 280, 346–378, https://doi.org/10.1111/febs.12077 (2013).
Toledano-Katchalski, H. et al. Protein tyrosine phosphatase epsilon inhibits signaling by mitogen-activated protein kinases. Mol Cancer Res 1, 541–550 (2003).
Sacco, F. et al. Tumor suppressor density-enhanced phosphatase-1 (DEP-1) inhibits the RAS pathway by direct dephosphorylation of ERK1/2 kinases. J Biol Chem 284, 22048–22058, https://doi.org/10.1074/jbc.M109.002758 (2009).
Gu, J. et al. SHP2 promotes laryngeal cancer growth through the Ras/Raf/Mek/Erk pathway and serves as a prognostic indicator for laryngeal cancer. Int J Oncol 44, 481–490, https://doi.org/10.3892/ijo.2013.2191 (2014).
Deng, R. et al. Shp2 SUMOylation promotes ERK activation and hepatocellular carcinoma development. Oncotarget 6, 9355–9369, https://doi.org/10.18632/oncotarget.3323 (2015).
Bunda, S. et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun 6, 8859, https://doi.org/10.1038/ncomms9859 (2015).
Korff, S. et al. Frameshift mutations in coding repeats of protein tyrosine phosphatase genes in colorectal tumors with microsatellite instability. BMC Cancer 8, 329, https://doi.org/10.1186/1471-2407-8-329 (2008).
Laczmanska, I. et al. Protein tyrosine phosphatase receptor-like genes are frequently hypermethylated in sporadic colorectal cancer. J Hum Genet 58, 11–15, https://doi.org/10.1038/jhg.2012.119 (2013).
Wortzel, I. & Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2, 195–209, https://doi.org/10.1177/1947601911407328 (2011).
Steelman, L. S. et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 3, 192–222, https://doi.org/10.18632/aging.100296 (2011).
Caunt, C. J. & McArdle, C. A. ERK phosphorylation and nuclear accumulation: insights from single-cell imaging. Biochem Soc Trans 40, 224–229, https://doi.org/10.1042/BST20110662 (2012).
Lidke, D. S. et al. ERK nuclear translocation is dimerization-independent but controlled by the rate of phosphorylation. The Journal of biological chemistry 285, 3092–3102, https://doi.org/10.1074/jbc.M109.064972 (2010).
Frost, J. A. et al. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J 16, 6426–6438, https://doi.org/10.1093/emboj/16.21.6426 (1997).
Hu, Q., Klippel, A., Muslin, A. J., Fantl, W. J. & Williams, L. T. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 268, 100–102 (1995).
Wennstrom, S. & Downward, J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol 19, 4279–4288 (1999).
Roberts, P. J. & Der, C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310, https://doi.org/10.1038/sj.onc.1210422 (2007).
Singh, H., Longo, D. L. & Chabner, B. A. Improving Prospects for Targeting RAS. J Clin Oncol 33, 3650–3659, https://doi.org/10.1200/JCO.2015.62.1052 (2015).
Caunt, C. J., Sale, M. J., Smith, P. D. & Cook, S. J. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nature reviews. Cancer 15, 577–592, https://doi.org/10.1038/nrc4000 (2015).
Hatzivassiliou, G. et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther 11, 1143–1154, https://doi.org/10.1158/1535-7163.MCT-11-1010 (2012).
Morris, E. J. et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 3, 742–750, https://doi.org/10.1158/2159-8290.CD-13-0070 (2013).
Carlino, M. S. et al. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Molecular oncology 8, 544–554, https://doi.org/10.1016/j.molonc.2014.01.003 (2014).
Liu, B. et al. Computational design, chemical synthesis, and biological evaluation of a novel ERK inhibitor (BL-EI001) with apoptosis-inducing mechanisms in breast cancer. Oncotarget 6, 6762–6775, https://doi.org/10.18632/oncotarget.3105 (2015).
Chappell, W. H. et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2, 135–164, https://doi.org/10.18632/oncotarget.240 (2011).
Acknowledgements
This work was supported by National Institutes of Health grant U01CA157960 (to T.J.Y.).
Author information
Authors and Affiliations
Contributions
T.J.Y. was responsible for oversight of research and entire Ms production. Specifically he was responsible for (1) ensuring that original data upon which the submission is based is preserved and retrievable for reanalysis; (2) approving data presentation as representative of the original data; and (3) foreseeing and minimizing obstacles to the sharing of data, materials, algorithms or reagents described in the work. T.D., H.W., L.M. performed the experiments to produce the data in the Ms. M.Y. and W.J.P. helped oversee the experimental work and contributed to the writing of the Ms. M.J.S. performed statistical analyses.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davis, T.B., Yang, M., Schell, M.J. et al. PTPRS Regulates Colorectal Cancer RAS Pathway Activity by Inactivating Erk and Preventing Its Nuclear Translocation. Sci Rep 8, 9296 (2018). https://doi.org/10.1038/s41598-018-27584-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27584-x
This article is cited by
-
An integrative gene expression signature analysis identifies CMS4 KRAS-mutated colorectal cancers sensitive to combined MEK and SRC targeted therapy
BMC Cancer (2022)
-
Innovative dual system approach for selective eradication of cancer cells using viral-based delivery of natural bacterial toxin–antitoxin system
Oncogene (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
