
Abstract
Microorganisms display diverse biogeographic patterns in the three-dimensional contiguous seawater. The distance-decay relationship, the change in species composition similarity between different communities over a geographic distance, is a commonly observed biogeographic pattern. To study biogeographic patterns and the corresponding driving forces, the bacterial distance-decay patterns along the horizontal and vertical dimensions in the South China Sea (SCS) were investigated through the sequencing of partial 16 S rRNA gene regions. Along the horizontal geographical distances (up to ~1000 km), no significant distance-decay pattern in community compositions was observed in any of the tested seawater layers. However, vertical depths (up to ~4 km) had strong effects on bacterial community variation, which was apparently governed by dispersal barriers due to limited water mass mixing. In addition, community variations in the vertical direction were strongly correlated with the prominent variation of environmental factors. Apparently, the changes in bacterial community compositions along vertical distances were much greater than those along horizontal distances. The results showed that the distance-decay relationship in bacterial communities at the medium spatial scale was associated with vertical depth rather than with horizontal distance, even though the horizontal distance is much larger than the vertical distance in the open SCS.
Similar content being viewed by others

Introduction
The ocean is the largest three-dimensional contiguous aquatic environment on the Earth and contains approximately 106−109 microbial taxa and 3 × 1028 microbial cells1,2. These marine microorganisms play vital roles in diverse oceanic biogeochemical processes and are recognized as indispensable agents of nutrient cycling and climate change on a global scale3,4. Hence, understanding the patterns of marine microbial distribution and the factors that drive these distribution patterns is essential for predicting the response of marine microbial ecosystems to future environmental changes2,5. In recent years, there has been extensive research regarding microbial biogeography, which studies the diversity of microorganisms over space and time6,7. Due to their small sizes, high dispersal abilities and large population sizes, microorganisms are thought to be globally distributed and primarily selected by environmental factors8. This situation is very suitable for marine microorganisms because global seawater is a dynamic and contiguous system and horizontal and vertical currents can disperse marine microorganisms to every corner of the world’s oceans, as demonstrated in the Southern Ocean9 and in deep seas10. In addition to the dispersal rate being governed by ocean currents, microbial biogeographic patterns are also dependent on selective processes6, including environmental pressures (e.g., wind, sunlight and nutrient availability)11,12 and biotic interactions (e.g., algal blooms, viral lysis and grazing)13,14. Additionally, marine microbial communities follow scale-dependent spatial biogeographic patterns2,5. However, contradictory biogeographic patterns have been observed in marine microorganisms. Some studies, such as the microbial seed bank theory15, have shown the cosmopolitanism of marine microbial species characterized by the lack of biogeographic patterns. Meanwhile, other studies have shown that marine microbial species are provincial, i.e., different environments exhibit different microbial abundances and diversities16. Several factors can lead to contradictory biogeographic patterns, including differences in spatial scales, taxonomic resolutions, sequencing platforms and regions, as well as the habitat types considered for the studies of microbial biogeographic patterns.
A commonly studied microbial biogeographic pattern is the distance-decay relationship, which describes how similarities in community compositions vary with geographical distance2,14,17. The ocean is a three-dimensional contiguous aquatic system where microbial diversity can be affected by horizontal and vertical distances. There are sinking and upwelling currents that can vertically mix seawater and microorganisms. Horizontal currents can also mix the seawater at a single depth. Therefore, there are distance-decay relationships along the vertical depth and horizontal distance in seawater. Because of the stratification of the ocean, distance-decay relationships are prominent along the vertical depth18. However, it remains unclear if there are any differences between the patterns of distance-decay relationships along the vertical depth and horizontal distance, and if there are differences, it is unclear which factors drive these differences.
Located in the subtropical and tropical areas adjacent to the western North Pacific Ocean, the South China Sea (SCS) is the world’s largest marginal sea, with a maximum depth of over 5000 m4. The water of the SCS has frequent and complicated horizontal and vertical currents19. The surface ocean currents are mainly driven by monsoons and changes in seasons. Winter currents flow northeast to southwest, but summer currents flow in the opposite direction. The upper layer (0–200 m) of seawater circulates cyclonically in winter but anticyclonically in summer20; this is also true of the circulation in the bottom layer of the SCS shelf 21. Meanwhile, large-scale horizontal flow is observed in the intermediate current of the SCS19. The Kuroshio branch intrudes into the SCS through the Luzon Strait in winter, but sometimes this intrusion occurs in summer, which greatly affects hydrological states and circulation structures22,23. With low temperature and high salinity, the Pacific intermediate waters near the east side of the Luzon Strait are characterized by high density24. The Pacific waters sink when they enter the SCS, and the local deep-water masses of the SCS upwell to compensate for the sinking movement25. Besides, monsoon winds cause offshore delivery of seawater and lead to the formation of upwelling and sinking currents20. In addition, submarine mountains can regulate vertical currents10,19. All these currents constantly mix the seawater in vertical and horizontal directions.
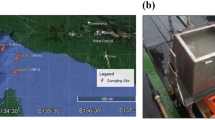
Therefore, the SCS environment is an ideal region for the study of the biogeographic patterns of marine bacterial communities along the horizontal and vertical directions. In this study, bacterial communities from different samplings at horizontal distances up to ~ 1000 km and vertical distances up to ~ 4 km were systematically investigated to study and compare the distance-decay relationship patterns along horizontal and vertical distances (Fig. 1). Furthermore, the major factors driving the biogeographic patterns were also explored.

Map locations of the sampling sites in the open South China Sea.
Results and Discussion
Bacterial taxonomic assignment and community variance
After quality control, a total of 3,128,028 high-quality sequences were retained for post-run analysis. A total of 2665 OTUs were assigned at a 3% dissimilarity threshold. The number of OTUs changed from 373 to 1310 across 24 samples, with the highest OTU numbers seen in the 200-m layer (Wilcoxon test, P < 0.05) (Supplementary Table S1). For Shannon diversity, no remarkable difference was observed between samples collected from the same layer at two different sites. However, there were clear differences in community diversities between the various vertical layers (Supplementary Fig. S1). The Shannon index of the surface water was dramatically higher than that of deep water (800-m, P = 0.026; Deep, P = 0.008) and was comparable to that of the 200-m layer.
In total, 35 bacterial phyla were found in all the samples. Proteobacteria had the largest proportion in most of the upper seawater samples. Firmicutes were abundant in the intermediate and deep layers. Actinobacteria, Cyanobacteria, Bacteroidetes and Acidobacteria were also present at a high abundance across all samples (Supplementary Fig. S2). The microbial phyla network, with 21 nodes and 72 edges, can be divided into two community groups. The degrees of Acetothermia, Acidobacteria, Thermodesulfobacteria and Gemmatimonadetes were 10, which indicated a high incidence of inter-phylum correlations (Supplementary Fig. S3a).
To identify phylogenetic differences at finer taxonomic levels, bacterial abundance variations between different layers were analyzed at the class level (Fig. 2). As the most abundant group in Proteobacteria, Gammaproteobacteria showed a decreasing trend in community abundances along the vertical depth, with the abundance changing from 29.3% in the surface zones to 13.2% in the Deep areas. This trend was also observed in Alphaproteobacteria. The distribution pattern was in agreement with previous reports that Cyanobacteria and the classes α- and γ-proteobacteria were the predominant surface-seawater bacterial groups in the SCS4. Burkholderiales and Rhodocyclales, with high abundance in Betaproteobacteria, were observed in our sampling sites, though they were predominant in freshwater areas instead of saltwater areas26. Cyanobacteria accounted for 23.7% of the bacterial communities from surface sites, and a majority of them belonged to the GpIIa family, including Prochlorococcus and Synechococcus. Furthermore, Prochlorococcus was observed in the deep seawaters, as reported previously1. Deep-sea Prochlorococcus may have evolved particular characteristics, such as the ability to take up and use dissolved organic carbon, to survive in the aphotic zone27. The class Flavobacteriia (predominantly composed of Flammeovirgaceae) had high abundance in the surface layer likely due to its important functions. The genus Zunongwangia, which is affiliated with Flammeovirgaceae, has the important ability to hydrolyze carbohydrates and proteinaceous organic nitrogen-containing compounds28. Genomic analyses of Polaribacter revealed a key function of this genus in the degradation of sulfated polysaccharides that are major components of algal cell walls29. Previous reports have demonstrated that Flavobacteriia can mediate algal organic matter degradation, supporting the growth of the SAR11 clade and marine Gammaproteobacteria12,30. This phenomenon was consistent with our network analysis at the family level, which showed that Flammeovirgaceae had wider positive correlations with members from other phyla, including Alphaproteobacteria, Gammaproteobacteria and Cyanobacteria (Supplementary Fig. S3b). Thus, some synergistic heterotrophic microorganisms in marine waters might grow partly due to Flavobacteriia-derived organic products.

Heatmap showing the phylogenetic abundances of major bacterial clades at the class level across all the samples. Color bars represent the row-scaled value, in which a blue curve illustrates the distribution density percentage.
Generally, the observed bacterial community compositions and variation trends in this study are consistent with previous studies in this area.
Bacterial distance-decay pattern along the horizontal distance and vertical depth
Based on an average Bray-Curtis similarity and Jaccard similarity between each pair of samples, the bacterial communities from the four seawater layers were not significantly correlated with horizontal geographical distance (126 km to 989 km) (by Pearson correlations (P > 0.05)) (Fig. 3 and Supplementary Fig. S4). This result was inconsistent with a previous study in the SCS that showed a biogeographic distribution of bacteria due to environmental control and dispersal limitation31. It was reported that distance-decay patterns of beta-diversity (variations in community composition) are dependent on spatial scales5. Therefore, the medium scale of 100–1000 km between the sample sites in the open SCS may be a limiting factor, leading to the absence of a distance-decay pattern. Notably, the environmental parameters (nitrate, nitrite, DOC, pH, phosphate and temperature) versus the geographic distance in each layer showed no remarkable difference (Wilcoxon test, P > 0.05) (Table 1). This showed that environmental perturbation did not drive changes in microbial communities across horizontal distances. With native SCS water masses exchanged at each water layer, there was no obvious dispersal limitation leading to reduced differences in bacterial communities19. In addition, the Kuroshio branch intruded into the north of the SCS through the Luzon Strait with large-scale surface and subsurface circulations; these horizontal current flows also decreased bacterial dispersal limitation. Average Bray-Curtis similarities of bacterial communities from Surface, 200-m, 800-m and Deep layers were 56.8%, 66.0%, 56.4% and 59.7%, respectively (Supplementary Fig. S5a). The trend was similar with Jaccard similarities (Supplementary Fig. S5b). These relatively high community similarities in the same water layers implied a weak distance-decay pattern along the horizontal direction.

Correlations between bacterial community similarities at the same depth versus geographical distance. Bacterial communities were structured on Bray-Curtis similarity. Correlations from (a) Surface, (b) 200-m, (c) 800-m and (d) Deep layers are represented as Pearson correlations.
The pronounced ocean stratification implied that vertical depth would have a strong effect on microbial community diversity and result in an obvious distance-decay relationship. Previous studies had reported a universal biogeographic pattern of variations in microbial community similarity versus geographic distance2,14,17. In this study, significant bacterial community differences between different water layers were observed along the vertical distance (0.2 km to 3.8 km), which was far shorter than the horizontal distances measured (>100 km) (Fig. 4). Besides, the average Bray-Curtis similarities of bacterial community between the surface-layer zones and the 200-m, 800-m and Deep layers were 34.8%, 30.0% and 31.0%, respectively. These low similarities that also were supported by Jaccard similarity indice hinted at obvious variations in community structures between the surface and other layers (Supplementary Figs S6 and S7). The unconstrained non-metric MDS analysis clearly showed that surface seawater and the other three water layer samples had different bacterial compositions that clustered into two larger groups (Fig. 5). This distribution pattern was in agreement with the constrained RDA result, which showed that the two larger groups were remarkably divided by the RDA 1 axis and that the intra-group members changed along the RDA 2 axis. Accordingly, a remarkable separation between the surface seawater group and the groups from the other layers was confirmed (ANOSIM, Surface vs 200 m, R = 0.98, P = 0.002; Surface vs 800 m, R = 0.97, P = 0.002; Surface vs Deep, R = 0.99, P = 0.002). Furthermore, the heterogeneity observed in the surface seawater community structures was relatively higher than that observed in the intermediate and deep layers. This high bacterial heterogeneity of surface seawater may be due to sunlight and more active conditions at the surface, as reported in other studies (Figs 5 and 6)32.

Comparison of bacterial community similarities at different depths. The average Bray-Curtis similarity value was calculated based on the similarity values between each pair of samples from different depths.

Nonmetric multidimensional scaling (NMDS) ordination of bacterial communities. The ordination was built based on the rank order of bacterial Bray-Curtis similarity. The four colored groups represent the four seawater layers: a surface group, a 200-m group and an 800-m-deep mixed group.

Redundancy analysis demonstrating the relationship between environmental variables and seawater samples. The environmental factors and various sampling depths are indicated by red arrows and different shapes, respectively.
The bacterial communities of the 200-m layer were also significantly different from those of the 800-m (ANOSIM, R = 0.53, P = 0.002) and Deep (R = 0.92, P = 0.002) layers. However, the bacterial communities of the 800-m and Deep layers had a high similarity value of 62.6% and barely showed a measurable difference (ANOSIM, R = 0.14, P = 0.134). This result showed that the bacterial community compositions changed drastically from the surface to the 800-m layer and then changed slowly as the depth increased. Thus, vertical bacterial distance-decay patterns in the seawater system showed a stepwise change instead of a smooth gradient. Meanwhile, no clear difference in bacterial communities was found within the medium-scale horizontal distances sampled.
Environmental factors driving the bacterial distance-decay pattern
It has been reported that most bacteria and archaea are limited to unique habits, mainly due to different environmental driving forces33,34. In this study, the environmental factors at each water layer were not significantly correlated with horizontal geographic distance, and no co-variance was found (Mantel test, P > 0.05). However, significant differences of environmental variables were observed along the vertical depths. The temperature of all samples, ranging from 2 to 30 °C, showed an obvious increasing trend from deep regions to surface layers; a similar trend was seen for the levels of the organic nutrient DOC. In contrast, the levels of the inorganic nutrients measured, namely, PO43−, NO2−, NO3− and DIC, were significantly lower in surface layers than in deep waters. The pH value was significantly higher in the upper zones than in the deep regions (Wilcoxon test, P < 0.05), but there was no pH difference between the 800-m layer and the Deep layer (Table 1).
To find the determinant environmental factor influencing bacterial composition patterns, RDA was performed across all the samples. The synergetic driving force of all the environmental variables provided a high explanatory power (approximately 77%) for the change of bacterial assemblages, suggesting effective impacts on variability in community patterns across seawater depths. After the removal of colinearity effects with Monte Carlo permutation tests, the four environmental variables were seen to significantly contribute to the relationships between major community distributions and the corresponding environmental factors (P < 0.05). The DOC content had the highest RDA explanatory power (44.6%) on the main phyla from different depths, followed by DO (F = 10.7, P = 0.002), salinity (F = 10.2, P = 0.002) and depth (F = 3.4, P = 0.002). The variable that best correlated with microbial community structures was DOC (r = 0.581, P < 0.001) and the top pair of environmental factors was DOC and salinity (r = 0.583, P < 0.001), as determined by BIOENV and BVSTEP analyses. A large DOC pool was reported to be present in the ocean, and the available compounds from this pool shape microbial community composition35,36. Therefore, it is reasonable that DOC content was a determinant environmental factor affecting bacterial community structures and their vertical distance-decay patterns in our study. Furthermore, clear differences in all the tested environment variables between different layers (except for salinity between surface zones and other layers, and DO and pH between the 800-m and deep layers) implied the formation of specific habitats, facilitating the vertical variations of bacterial communities (Wilcoxon test, P < 0.05). However, for the same horizontal seawater layer, the environmental variables were not significantly different between any two sites. The environmental factor analyses can partially explain why bacterial communities showed obvious discrepancies along vertical depths but no prominent community variance along the horizontal distances.
Effect of currents on bacterial biogeographic patterns
In addition to the major influence of environment variables on the variations of microbial community compositions, ocean currents also may affect microbial biogeographic patterns4,20,37. It was reported that water masses in open areas of the SCS were divided into various water masses on a large scale along the vertical direction26. This distribution pattern was also found in our study. Based on the differences in temperature and salinity, a total of 24 samples were classified into the following groups: surface group, 200-m group, 800-m group and deep group (Supplementary Fig. S8). Furthermore, the LT2 surface seawater had a markedly high salinity compared to the salinity of the seawater from the other sites. Due to the southwest monsoon, local water masses from the SCS had an eastward flow in Luzon Strait, which weakened the invasion of the Kuroshio current23. Additionally, Kuroshio waters (high temperature and salinity) were not seen to cross further west than 119.5 °E and enter the SCS hinterland in the spring22,26. Thus, it was reasonable that the site LT2 (121.1 °E) was clearly affected by the Kuroshio waters.
Each stratification area produced homogeneous water masses harboring analogous physicochemical properties, such as temperature, salinity and nutrients18, which was also observed in the samples of this study (Table 1 and Supplementary Fig. S8). Water mass differences at different pelagic depths appeared to form a dispersal barrier that led to the creation of different environmental conditions and different sets of taxa13,37 even though there exist low-flow sinking and upwelling currents that can mix the vertical seawater body19. It is reported that the distance-decay relationship will be strengthened with some dispersal limitation6. Therefore, there are great variations between the bacterial communities found at different depths. Relative to the deeper regions, the surface water in the SCS frequently showed variable physical oceanographic processes, including monsoon-driven circulation and large-scale offshore transportation of nutrition20,25. These active processes in surface seawaters influenced horizontal microorganism production and movement without notable dispersal limitation. This result can further explain why no distance-decay pattern was observed along horizontal distances.
This study illustrates in detail the bacterial biogeographic patterns in the open SCS along the horizontal and vertical directions. Commonly, spatially environmental heterogeneity34,38 and dispersal limitation39 were used to interpret the observed microbial beta-diversity. The bacterial community compositions showed no detectable difference along the medium-scale horizontal geographical distances sampled in this study, showing that there is no dispersal limitation at up to ~1000 km horizontal distances in the contiguous open oceanic ecosystems. Nevertheless, bacterial community structures from different seawater layers exhibited obvious differences. Specifically, surface seawater bacteria displayed less intra-group similarities and higher inter-community diversity than deep sea bacteria. The vertical biogeographic pattern can be explained by distinct environment variables (especially DOC) and oceanographic stratification. This showed that dynamic currents and water stratification rather than simple geographic distance should be considered to interpret the bacterial biogeographical pattern in the three-dimensional contiguous seawater system. Overall, the results showed that vertical depth is the main determinant of bacterial community differentiation in the open SCS water system even though the horizontal distances were much greater than vertical depth, which will shed new lights on the understanding of microbial beta-diversity in the marine water system.
Methods
Location description and sample collection
Sample collection sites were located in the open SCS, far from coasts and river estuaries (Fig. 1). A total of 24 samples were collected from six sites (B3, D3, G1, F1, LT2 and X4) during a Dong Fang Hong 2 cruise in the spring of 2014. Seawater samples from each site were collected from layers at four depths, namely, 1–2 m, 200 m, 800 m and 2000–3800 m. For the purposes of this study, the 1–2 m and 2000–3800 m layers were named Surface and Deep, respectively. At each site, seawater from different depths was collected by a Sealogger conductivity-temperature-depth (CTD) oceanic rosette sampler (SBE 25, Sea-Bird Co., USA). After prefiltration through a 20-µm-pore-size nylon net membrane (Millipore Co., USA) to eliminate large particles, phytoplankton and zooplankton, five- to ten-liter seawater samples were filtered on board through 47-mm-diameter polycarbonate membranes with 0.22-µm pores (Millipore Co., USA). Filtered membranes were stored in sterile tubes (Corning Inc., USA) and kept in liquid nitrogen and then frozen at −80 °C (Thermo Scientific, USA) in the lab until nucleic acid extraction. Seawater physicochemical properties, such as temperature, salinity and water depth, were recorded by the onboard CTD Sealogger. To measure environmental parameters, water samples were filtered through 0.45-µm-pore-size cellulose acetate membranes (Millipore Co., USA). Nutrient contents (NO2−, NO3− and PO43−) were measured by spectrophotometric and colorimetric analyses, while dissolved organic carbon (DOC) and dissolved inorganic carbon (DIC) were calculated by a TOC analyzer (Thermo Flash 2000 Elemental Analyzer, USA).
DNA extraction and Illumina sequencing
Total genomic DNA were extracted with a PowerWater® DNA Isolation Kit (MOBIO, USA) from the membranes. Barcoded sequencing primers 338 F (5’-ACTCCTACGGGAGGCAGCA-3’)/806 R(5’-GGACTACHVGGGTWTCTAAT-3’) were used to amplify the V3 to V4 regions of bacterial 16 S ribosomal RNA (rRNA) gene. A 20-µL PCR, containing 4 µL of 5 × FastPfu Buffer, 2 µL of 2.5 mM deoxynucleoside triphosphate (dNTP) mix, 0.8 µL of each primer (5 µM), 0.4 µL of TransStart Fastpfu DNA Polymerase (TransGen AP221–02), 1 µL of target DNA and 11 µL of double-distilled H2O, was performed in a PCR thermal cycler (ABI GenAmp® 9700). The amplification reaction was performed in triplicate as follows: initial denaturation at 95 °C for 3 min; 27 cycles of denaturation for 30 s at 94 °C, primer annealing at 55 °C for 30 s, and chain extension for 45 s at 72 °C; and a final extension at 72 °C for 10 min. Pooled reaction mixtures were then sequenced on the Illumina Miseq PE300 sequencing platform (Majorbio Bio-Pharm Technology Co. Ltd, China) to generate large short-read libraries.
Sequence analysis, OTU clustering and taxonomy assignment
The obtained sequences were merged and trimmed by the sequencing company. All the sequence analysis was processed by Usearch 8.0.161. The sequence reads were filtered with a maximum expected error threshold to remove unqualified portions of the sequences. After standard quality control, each trimmed sequence was approximately 450-base-pairs long. The remaining reads of the 24 seawater samples were then pooled, dereplicated, and finally assigned to each OTU using a 97% identity. Then, each sample read was mapped to representative OTU sequences of using a Perl script. For equalizing the sampling efforts of statistical comparisons, the different sample reads were matched with different assignment rates, ranging from 85% to 95%. For taxonomical assignment, the representative OTU sequences were compared with a 16 S rRNA gene database at an 80% confidence threshold by RDP Naive Bayesian rRNA Classifier (RDP Release 11.1 http://rdp.cme.msu.edu/)40.
Statistical analysis
The alpha-diversity indices and Bray-Curtis similarities between samples were computed using Primer 6 (Plymouth Marine Laboratory, UK). Jaccard similarity index, bacterial community compositions and relative abundance comparisons were analyzed by R software. One-way analysis of community similarity (ANOSIM) of different layers and non-metric multidimensional scaling (NMDS) were performed in Primer 6. A standard Mantel test at each sampling layer was run in R to evaluate the correlation between horizontal geographic distance and bacterial community compositions (based on the abundances of the top 50 OTUs). Redundancy analysis (RDA) was implemented with Monte Carlo permutation tests to analyze variations in the bacterial assemblages under the constraint of environmental factors by Canoco (Version 5, Microcomputer Power). Significant environmental parameters without multicolinearity effects (variance inflation factor < 20) were obtained to explain community variability31. BIOENV and BVSTEP analyses were then performed in order to statistically determine correlations between community compositions and environmental variables.
Co-occurrence analysis
Potential interaction modes were investigated through modeling the community structure networks. Positive and negative correlations in the abundances of microbial taxa were assumed to indicate co-occurrence and co-exclusion, respectively. Correlation network patterns were determined in R software with ‘Hmisc’ and ‘reshape2’ libraries and visualized in the interactive platform Gephi41. This platform is based on a matrix checkerboard where microbial communities interact with each other. For valid network analysis, the Spearman’s rank correlations at the phylum level were both >|0.6| and statistically significant (P < 0.01)42. At the family level, a Spearman’s correlation index of >|0.8| was calculated. To reduce network complexity, the network patterns at different taxonomic levels were constructed with >50 members across all samples4. In the co-occurrence networks, each node represents a single taxon (a phylum or family), and the edge between two nodes represents a positive or negative interaction between those two taxa. The degree refers to the number of edges connected to a node in a network.
The merged sequences of all 24 samples were deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive database under project no. PRJNA 389159.
References
Flombaum, P. et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. USA 110, 9824–9829 (2013).
Sunagawa, S. et al. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Hutchins, D. A. & Fu, F. Microorganisms and ocean global change. Nat. Microbial. 2, 17058, https://doi.org/10.1038/nmicrobiol.2017.58 (2017).
Liu, J., Yang, H., Zhao, M. & Zhang, X. H. Spatial distribution patterns of benthic microbial communities along the Pearl Estuary, China. Syst. Appl. Microbiol. 37, 578–589 (2014).
Martiny, J. B., Eisen, J. A., Penn, K., Allison, S. D. & Horner-Devine, M. C. Drivers of bacterial beta-diversity depend on spatial scale. Proc. Natl. Acad. Sci. USA 108, 7850–7854 (2011).
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C. & Martiny, J. B. H. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Micro. 10, 497–506 (2012).
Hellweger, F. L., van Sebille, E. & Fredrick, N. D. Biogeographic patterns in ocean microbes emerge in a neutral agent-based model. Science 345, 1346–1349 (2014).
Barreto, D. P., Conrad, R., Klose, M., Claus, P. & Enrich-Prast, A. Distance-Decay and Taxa-Area Relationships for Bacteria, Archaea and Methanogenic Archaea in a Tropical Lake Sediment. PLoS ONE 9, e110128, https://doi.org/10.1371/journal.pone.0110128 (2014).
Wilkins, D., van Sebille, E., Rintoul, S. R., Lauro, F. M. & Cavicchioli, R. Advection shapes Southern Ocean microbial assemblages independent of distance and environment effects. Nat. Commun. 4, 2457, https://doi.org/10.1038/ncomms3457 (2013).
Salazar, G. et al. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 10, 596–608 (2016).
Bryant, J. A. et al. Wind and sunlight shape microbial diversity in surface waters of the North Pacific Subtropical Gyre. ISME J. 10, 1308–1322 (2016).
Bunse, C. & Pinhassi, J. Marine Bacterioplankton Seasonal Succession Dynamics. Trends Microbiol. 25, 494–505 (2017).
Brown, M. V., Ostrowski, M., Grzymski, J. J. & Lauro, F. M. A trait based perspective on the biogeography of common and abundant marine bacterioplankton clades. Mar. Genomics 15, 17–28 (2014).
Milici, M. et al. Bacterioplankton Biogeography of the Atlantic Ocean: A Case Study of the Distance-Decay Relationship. Front. Microbiol. 7, 590, https://doi.org/10.3389/fmicb.2016.00590 (2016).
Lennon, J. T. & Jones, S. E. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Micro. 9, 119–130 (2011).
Wang, K. et al. Bacterial biogeography in the coastal waters of northern Zhejiang, East China Sea is highly controlled by spatially structured environmental gradients. Environ. Microbiol. 17, 3898–3913 (2015).
Nemergut, D. R. et al. Patterns and Processes of Microbial Community Assembly. Microbiol. Mol. Biol. Rev. 77, 342–356 (2013).
Galand, P. E., Potvin, M., Casamayor, E. O. & Lovejoy, C. Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME J. 4, 564–576 (2010).
Liu, Z. & Gan, J. Three-dimensional pathways of water masses in the South China Sea: A modeling study. J. Geophys. Res. Oceans 122, 6039–6054 (2017).
Wang, X., Liu, Z. & Peng, S. Impact of tidal mixing on water mass transformation and circulation in the South China Sea. J. Phys. Oceanogr. 47, 419–432 (2017).
Lan, J., Wang, Y., Cui, F. & Zhang, N. Seasonal variation in the South China Sea deep circulation. J. Geophys. Res. Oceans 120, 1682–1690 (2015).
Nan, F., Xue, H. & Yu, F. Kuroshio intrusion into the South China Sea: A review. Prog. Oceanogr. 137, 314–333 (2015).
Wu, C. R., Wang, Y. L., Lin, Y. F. & Chao, S. Y. Intrusion of the Kuroshio into the South and East China Seas. Sci. Rep. 7, 7895, https://doi.org/10.1038/s41598-017-08206-4 (2017).
Wong, G. T. F., Ku, T. L., Mulholland, M., Tseng, C. M. & Wang, D. P. The SouthEast Asian Time-series Study (SEATS) and the biogeochemistry of the South China Sea-An overview. Deep-Sea Res. II 54, 1434–1447 (2007).
Qu, T., Girton, J. B. & Whitehead, J. A. Deepwater overflow through Luzon Strait. J. Geophys. Res. Oceans 111, C01002, https://doi.org/10.1029/2005JC003139 (2006).
Liu, J. et al. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: the potential impact of hypoxia and nutrients. Front. Microbiol. 6, 64, https://doi.org/10.3389/fmicb.2015.00064 (2015).
Jiao, N. et al. Presence of Prochlorococcus in the aphotic waters of the western Pacific Ocean. Biogeosciences 11, 2391–2400 (2014).
Qin, Q. L. et al. The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genomics 11, 247, https://doi.org/10.1186/1471-2164-11-247 (2010).
Gomez-Pereira, P. R. et al. Genomic content of uncultured Bacteroidetes from contrasting oceanic provinces in the North Atlantic Ocean. Environ. Microbiol. 14, 52–66 (2012).
Williams, T. J. et al. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15, 1302–1317 (2013).
Zhang, Y., Zhao, Z., Dai, M., Jiao, N. & Herndl, G. J. Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea. Mol. Ecol. 23, 2260–2274 (2014).
Ghiglione, J. F. et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. USA 109, 17633–17638 (2012).
Meng, L., Liu, H., Bao, M. & Sun, P. Microbial community structure shifts are associated with temperature, dispersants and nutrients in crude oil-contaminated seawaters. Mar. Pollut. Bull. 111, 203–212 (2016).
Zeglin, L. H. Stream microbial diversity in response to environmental changes: review and synthesis of existing research. Front. Microbiol. 6, 454, https://doi.org/10.3389/fmicb.2015.00454 (2015).
Jiao, N. & Zheng, Q. The microbial carbon pump: from genes to ecosystems. Appl. Environ. Microbiol. 77, 7439–7444 (2011).
Wang, P. et al. Relationship between dissolved organic carbon and bacterial community in the coastal waters of Incheon, Korea. Oceanol. Hydrobiol. Studies 46, 50–61 (2017).
Hernando‐Morales, V., Ameneiro, J. & Teira, E. Water mass mixing shapes bacterial biogeography in a highly hydrodynamic region of the Southern Ocean. Environ. Microbiol. 19, 1017–1029 (2016).
Yu, S. X., Pang, Y. L., Wang, Y. C., Li, J. L. & Qin, S. Distribution of bacterial communities along the spatial and environmental gradients from Bohai Sea to northern Yellow Sea. PeerJ 6, e4272, https://doi.org/10.7717/peerj.4272 (2018).
Villarino, E. et al. Large-scale ocean connectivity and planktonic body size. Nat. Commun. 9, 142, https://doi.org/10.1038/s41467-017-02535-8 (2018).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Bastian, M., Heymann, S. & Jacomy, M. Gephi: an open source software for exploring and manipulating networks. International AAAI Conference on Weblogs and Social Media (2009).
Barberan, A., Bates, S. T., Casamayor, E. O. & Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6, 343–351 (2012).
Acknowledgements
We thank Professor Minhan Dai and Weifang Chen for providing environmental parameters data. This work was supported by the National Science Foundation of China (grants 31290230, 31290231, 91328208, 31370056 and 41376153), AoShan Talents Cultivation Program of Qingdao National Laboratory for Marine Science and Technology (2017ASTCP-OS14), Taishan Scholars Program of Shandong Province (2009TS079), and Young Scholars Program of Shandong University (2016WLJH36 and 2016WLJH41).
Author information
Authors and Affiliations
Contributions
Y.L. and L.L.S. performed the laboratory work. M.L.S. and H.N.S. collected seawater samples. Y.L. wrote the manuscript. Q.L.Q., X.L.C., X.Y.Z. and B.B.X. helped to revise the manuscript. Y.Z.Z. and Q.L.Q. designed the study. All authors discussed the results and participated in manuscript revision.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Sun, LL., Sun, ML. et al. Vertical and horizontal biogeographic patterns and major factors affecting bacterial communities in the open South China Sea. Sci Rep 8, 8800 (2018). https://doi.org/10.1038/s41598-018-27191-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27191-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
