
Abstract
Drought is one of the major abiotic stresses that negatively impact alfalfa growth and productivity. The role of microRNA156 (miR156) in drought has been demonstrated in plants. To date, there are no published studies investigating the role of miR156 in regulating global gene expression in alfalfa under drought. In our study, alfalfa genotypes overexpressing miR156 (miR156OE) exhibited reduced water loss, and enhanced root growth under drought. Our RNA-seq data showed that in response to drought, a total of 415 genes were upregulated and 169 genes were downregulated specifically in miR156OE genotypes. Genotypic comparison revealed that 285 genes were upregulated and 253 genes were downregulated in miR156OE genotypes relative to corresponding WT under drought. Gene Ontology enrichment analysis revealed that the number of differentially expressed genes belonging to biological process, molecular function and cell component functional groups was decreased in miR156OE genotypes under drought. Furthermore, RNA-Seq data showed downregulation of a gene encoding WD40 repeat in a miR156-specific manner. 5′ RACE experiments verified cleavage of WD40-2 transcript under drought. Moreover, alfalfa plants overexpressing WD40-2 showed drought sensitive, whereas those with silenced WD40-2 exhibited drought tolerant phenotypes. These findings suggest that miR156 improves drought tolerance in alfalfa by targeting WD40-2.
Similar content being viewed by others

Introduction
Crop losses caused by extreme environmental conditions have risen steadily over the past decades1. Climate change models predict more frequent incidence of drought and extreme temperature in the near future2,3,4,5,6,7. An increased demand for water consumption by plants suggests the need for stress resilient crop genotypes to ensure sustainable food production8.
Alfalfa (Medicago sativa) is the most widely cultivated forage legume crop in the world9. In addition to its primary use as a forage for livestock feed, alfalfa enhances soil fertility by fixing atmospheric nitrogen10. Furthermore, alfalfa is contemplated for use as a feedstock for biofuel production, a practice that would contribute to a cleaner environment11. However, drought negatively affects alfalfa growth and biomass yield12.
Genome-wide transcriptome analysis has emerged as a powerful tool to discover genes that regulate various traits in plants. A transcriptomics study of root-knot nematode resistant and susceptible alfalfa genotypes revealed several differentially expressed genes common to both genotypes, as well as genes unique to individual genotypes13. Leaf transcriptome analysis of these nematode-related genotypes also led to the identification of candidate genes related to fall dormancy14. Transcriptome analysis of Medicago truncatula has been conducted in response to salt stress15,16. Moreover, the full sequence of M. truncula genome is available to use as a reference to discover alfalfa (Medicago sativa) genes involved in various traits such as abiotic stress17. The M. truncatula genome has been used to annotate transcriptome profiles of alfalfa under salinity stress13,18,. Although alfalfa and M. trunctula share a high degree of sequence similarity, genetic diversity between the two still exists19. Hence, genetic information obtained from the M. truncatula genome may not always be applicable to alfalfa. This is apparent from the large number of unannotated genes in a recently published salt resistant study on alfalfa breeding populations20.
MicroRNAs are eukaryotic gene regulators that repress gene expression by inducing transcript cleavage or translation repression21. Several studies have been undertaken to discover the role of microRNAs in improving abiotic stresses in numerous plants22,23,24.
MicroRNA156 (miR156) and its SPL target genes play crucial roles in regulating different aspects of growth, development, and flowering in many plant species25,26,27,28,29,30,31. Apart from SPL genes, miR156 is known to regulate non-conserved WD40 genes, which function in signal transduction, transcription regulation, and apoptosis in eukaryotes32. Naya et al.32 showed that a specific isoform of miR156 cleaves non-conserved WD40 targets in M. truncatula root apices. Although WD40 regulatory genes are known mainly for their role in anthocyanin biosynthesis and trichome development in plants33,34,35,36, there are other studies that link WD40 to nodule and cell wall formation37,38, and response to hormones, light and abiotic stress39.
More recently, we showed a positive role of miR156 in drought and salinity stress responses of alfalfa12,40. Despite a series of miR156-related studies in various plant species, there has been no reported transcriptome analysis on miR156OE alfalfa roots under drought stress. A root transcriptome analysis of contrasting alfalfa genotypes under these conditions could provide an insight into the underlying molecular mechanisms that control this trait in alfalfa. Hence, we conducted RNA sequencing (RNA-Seq) analysis on two miR156OE alfalfa genotypes (A8 and A16b) generated in a previous study25, after exposure to drought. Previously, these two genotypes showed improved drought tolerance and root biomass under drought12. In the current study, these genotypes exhibited increased root length and reduced water loss. The present analysis identified differentially expressed genes in alfalfa roots and investigated the role of a miR156-targeted WD40-2 gene in modulating drought responses in alfalfa.
Results
MiR156 promotes root growth and reduces water loss under drought conditions
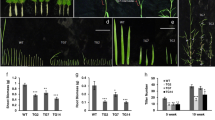
We measured root length of WT and miR156OE plants under control and drought stress. Under non-stress control conditions, A16b had longer roots than WT, whereas no statistical differences were observed between A8 and WT under non-stress conditions (Fig. 1a). Upon exposure to drought, we observed even more pronounced differences in root length between WT and miR156OE plants. Both A8 and A16b genotypes showed longer roots than WT (Fig. 1a). Drought-induced increase in root length ranged from 42% (A16b) to 130% (A8) in miR156OE plants after 13 days of drought stress, whereas only a 12% increase was recorded in WT during this period (Fig. 1a). This experiment provides evidence that miR156 overexpression enhances root growth under drought conditions.

Physiological response of WT and miR156OE genotypes to drought. (a) Root length, (b) water loss, measurements in drought stressed and well-watered control WT and miR156OE plants at day 13. An asterisk (*) shows statistical significance at p < 0.05 while double asterisk (**) indicates significance at p < 0.01 where n = 3–8 (ANOVA).
In a water loss assay, we did not observe differences between miR156OE and WT under control conditions, whereas upon withholding water for 12 days, water loss in miR156OE plants was reduced (~137%) compared to WT (Fig. 1b). This provided additional evidence that miR156 plays a role in water conservation during drought stress. In addition to the above results, A16b and A8 exhibited other drought tolerance traits which were reported in our previous study12.
De novo assembly of alfalfa root transcriptome
The samples of WT and the two most prominent miR156OE genotypes A16b and A825 were selected after drought stress for next generation sequencing to identify differentially expressed genes (DEGs). A total of 755,528,618 short reads were generated from cDNA libraries of non-stressed and drought stressed roots of WT, A8 and A16b (Supplementary Table 1). Poor quality reads such as adapters, short reads and unpaired reads were filtered out and high quality reads were obtained (Supplementary file S1).
Due to the absence of alfalfa genome sequences, we performed de novo assembly on alfalfa root transcriptome using Trinity assembly program41. From de novo assembly, a total of 455,303 transcripts were obtained with an average length of 728.05 bp. We recorded a N50 value of 1072 bp (Fig. 2a, Supplementary file S2), which was higher than what was previously reported (Postnikova et al.13). The de novo assembly contained a total of 302,193 Trinity genes (Fig. 2a), of which 64,959 genes were between 200–999 bp long and constituted the majority of the genes (Fig. 2b).

Summarized statistics of de novo assembly of Medicago sativa transcriptome using the Trinity program. (a) Summary of de novo assembly of M. sativa root transcriptome, (b) assembled transcript length distribution.
Drought-related DEGs
We carried out root transcriptome analysis in drought stressed plants to identify DEGs that may contribute to drought stress responses. We compared expression between non-stressed control and drought stressed roots of WT, A8 and A16b genotypes. To that affect, we observed a reduced number of total DEGs in both miR156OE genotypes (A8, A16b) compared to WT under drought (Fig. 3a, Supplementary files S3, S4, S5). A total of 998 and 988 unique DEGs were significantly upregulated in response to drought specifically in either A16b or A8, respectively, and not in WT (Fig. 3b,c, Supplementary files S3, S4, S5), with 415 DEGs being common genes upregulated both in A16b and A8 (Fig. 3d, Supplementary files S3, S4, S5). Similarly, we found 694 DEGs that were downregulated in response to drought in A16b, 574 DEGs in A8, but none of the genes were silenced in WT (Fig. 3,f, Supplementary files S3, S4, S5). Furthermore, we observed a downregulation of 169 DEGs in response to drought in both miR156OE genotypes (Fig. 3g, Supplementary files S3, S4, S5), indicating these DEGs may be regulated by miR156 in response to drought.

Comparison of significant DEGs found in WT and two miR156OE genotypes in response to drought stress. Venn diagram shows statistically significant DEGs in (a) total, (b–d) upregulated, and (e–g) downregulated, in WT, A8 and A16b under drought stress conditions relative to corresponding well-watered control plants.
Genotypic differences in DEGs
A total of 1794 and 1894 DEGs were found in A8 and A16b, respectively, relative to WT under non-stress conditions. Furthermore, we observed contrasting effects of drought on a number of DEGs in these two miR156OE genotypes where drought increased the number of DEGs to 2249 in A16b but reduced it to 1040 in A8 (Fig. 4a; Supplementary files S6, S7, S8 and S9). Of the total DEGs in A8 under control conditions, 517 genes were upregulated and 1277 were downregulated relative to WT (Fig. 4b,c, Supplementary file S6). Number of DEGs in A8 was reduced under drought stress when 580 genes were upregulated and 460 were downregulated relative to WT (Fig. 4b,c, Supplementary file S8). On the other hand in A16b, a total of 929 genes were upregulated and 965 were downregulated under control conditions relative to the corresponding WT (Fig. 4b,c, Supplementary file S7). Under drought stress, number of DEGs was increased in A16b with 1240 genes showing upregulation and 1009 downregulation relative to the corresponding WT (Fig. 4b,c, Supplementary file 9). We detected 538 differentially expressed genes common between A8 and A16b under drought stress conditions (Fig. 4b,c), indicating these genes may be specifically regulated by miR156 under drought conditions.

Comparison of significant DEGs found in two miR156OE genotypes relative to corresponding WT. Venn diagram shows statistically significant DEGs (a) total, (b) upregulated and (c) downregulated in A8 and A16b under well-watered control (A8-C, A16b-C) or drought stress (A8-D, A16b-D) conditions relative to corresponding WT.
Gene ontology (GO) enrichment analysis
GO analysis revealed that drought reduced the number of DEGs belonging to various functional groups in miR156OE roots compared to WT (Fig. 5). In biological process, we found a reduced number of DEGs that were downregulated in miR156OE genotypes compared to WT (Fig. 5a), whereas either one or both miR156OE genotypes showed a small increase in the number of DEGs that were upregulated in carbohydrate metabolic process, response to abiotic stimulus, secondary metabolite process and response to water deprivation (Fig. 5b). Similar results were observed in the molecular function category where the number of downregulated genes was reduced in miR156OE roots compared to WT (Fig. 5c). A small increase in the number of upregulated genes was observed in one or both miR156OE genotypes in the functional sub-categories of catalytic activity, transporter activity and transcription factor activity (Fig. 5d). Consistently, a decreased number of DEGs was recorded in the cellular component category in miR156OE genotypes compared to WT (Fig. 5e), but an increased number of upregulated genes was observed in one or both miR156OE genotypes in response to drought in this category. These latter sub-categories include extracellular region, cell periphery and plasma membrane (Fig. 5f).

Distribution of gene ontology (GO) categories involved in drought stress responses among three GO domains. Left panel (a,c,e) indicates number of downregulated whereas right panel (b,d,f) shows upregulated DEGs in WT and miR156OE genotypes in response to drought stress.
RNA-Seq data validation by quantitative real time PCR
We validated RNA-Seq data by randomly selecting 27 genes that included upregulated, downregulated, and unchanged expression in response to drought stress and/or miR156. These genes were tested for expression pattern consistency using quantitative real time PCR (qRT-PCR). The expression levels of the selected genes in response to drought stress (relative to non-stress controls) was compared with RNA-Seq data and presented in Supplementary Table 2. A strong correlation existed between the two expression data sets showing Pearson correlation coefficient r = 0.82 (Supplementary file S10). Under different drought stress levels and genotypes, NGS and RT-qPCR data showed similar trends, providing strong confidence in our RNA-seq data.
Identification of drought and miR156 responsive transcription factors
Using the Plant Transcription Factor Database (http://planttfdb.cbi.pku.edu.cn/), we identified a number of transcription factors that were significantly upregulated or downregulated in response to drought and miR156. Among major classes of transcription factors, number of up-regulated C2H2 was increased, whereas TCP was decreased in miR156OE genotypes under drought stress conditions (Supplementary Table 3). Genotypically, drought affected more bZIP TFs in A16b than A8. In addition, we observed a decreased number of WRKY genes that were downregulated in miR156OE genotypes under drought relative to non-stress conditions (Supplementary Table 3). Moreover, number of downregulated SBP-box transcription factor was increased in miR156OE genotypes, whereas WD40 repeat was similar under drought stress conditions (Supplementary Table 3).
MiR156 regulation of WD40 genes under drought
Our RNA-seq analysis showed that expression of WD40-2 (Medtr2g028050) was reduced in miR156OE genotypes under drought stress (Supplementary files S8, S9). To investigate whether miR156 targets the WD40-2, we identified putative miR156 recognition sites using sequence alignment, and carried out 5′-RACE experiments25 to analyze cleavage of the WD40-2 transcript. Transcript cleavage was detected outside of the predicted miR156 target sites in all 25 sequenced clones (Fig. 6a). On further investigation with qRT-PCR, we observed downregulation of WD40-2 in miR156OE genotypes, but only under drought stress, as non-stressed control plants did not show a difference in WD40-2 transcript level (Fig. 6b).

WD40-2 cleavage in root of miR156OE genotypes. (a) Validation of WD40-2 cleavages by 5′ RACE in transgenic alfalfa overexpressing miR156. The predicted miR156 target sequences are highlighted in yellow. The WD40-2 sequences are shown and cleavage sites are highlighted in green. The denominator refers to the number of clones sequenced whereas the numerator represents the number of clones cleaved. (b) qRT-PCR analysis of WD40-2 expression in roots of WT and miR156OE genotypes under well-watered control and drought stress conditions.
Misexpression of WD40-2 affects growth and drought tolerance in alfalfa
Further, we generated overexpression and RNA interference (RNAi) genotypes. This included four transgenic genotypes overexpressing WD40-2 (WD-1OE, WD-4OE, WD-6OE and WD-7OE) (Supplementary file S11), and four RNAi plants with reduced WD40-2 levels (WD-1R, WD-3R, WD-17R, WD-18R) (Supplementary file S11). These WD40-2 overexpression, RNAi and WT plants were then tested for tolerance to drought stress.
Under non-stress control conditions, we observed stunted growth of transgenic alfalfa overexpressing WD40-2, where plants looked dwarf, small in stature with only one stem compared to WT (Fig. 7a). We noted that WD-6OE and WD-7OE plants (which have the highest WD40-2 expression) exhibited the most severe phenotypes (Supplementary file S11; Fig. 7), suggesting high WD40-2 expression leads to negative effects on plant growth. On the other hand, WD40-2 RNAi genotypes looked large, green with multiple stems compared to WT (Fig. 7A). In addition, WD-3R and WD-17R with medium level of downregulation (Supplementary file S11) showed increased plant growth (Fig. 7a), indicating that reduced WD40-2 expression at moderate levels enhances alfalfa performance.

Effect of drought stress on water conservation and shoot biomass of alfalfa. WT and WD40-2 overexpression, and RNAi genotypes (a) well-watered control conditions, (b) under drought stress, (c) leaf water potential, (d) water loss, and (e) shoot biomass. Data are average of 3-5 biological replications. Single asterisk (*) shows significance of WD40-2 RNAi and overexpression plants with WT at P < 0.05 and double asterisk (**) indicates P < 0.01 (ANOVA).
We tested whether drought tolerance in alfalfa is affected by WD40-2 expression. After withholding water for 22 days, WD40-2 RNAi genotypes remained relatively green and turgid, but WT and WD40-2 overexpression transgenic plants wilted (Fig. 7b). Moreover, WD40-2 overexpression plants were dwarfed and wilted with a single stem compared to WT (Fig. 7b).
We also measured the changes in water potential and water loss with and without drought stress. We exposed wild-type, WD40-2 overexpression and RNAi transgenic plants to drought by withholding water. Without drought, water potential in overexpression genotypes (except WD-4OE) was significantly reduced relative to WT while no water potential differences were found between WT and RNAi plants (Fig. 7c). During drought treatment, overexpression genotypes exhibited significantly lower, and RNAi genotypes significantly higher water potential than WT (Fig. 7c). These data suggest that drought causes a small reduction in water potential when WD40-2 expression is reduced and vice versa.
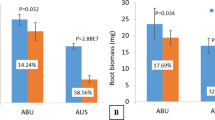
We observed no differences of water loss among these plants under control conditions, water loss in response to drought was significantly increased in two WD40-2 over-expression genotypes, and was significantly reduced in all WD40-2 RNAi genotypes compared to WT (Fig. 7d). Thus, attenuated expression of WD40-2 decreases water loss, making plants resistant to drought, suggesting that WD40-2 plays an important role in controlling water status in alfalfa.
We compared the shoot biomass accumulation in WT and transgenic plants. The WD40-2 overexpression plants showed a lower biomass than WT under non-stress. On the contrary, biomass in RNAi plants was significantly increased in two genotypes under this condition (Fig. 7e). Similarly, biomass accumulation was reduced in overexpression plants and increased in RNAi plants under drought stress (Fig. 7e). This result is consistent with the rapid wilting, lower water potential and faster water loss in overexpression plants, and higher water potential, reduced water loss and greenish plants of RNAi plants upon drought treatment.
WD40-2 affects root growth in alfalfa
We analyzed differences in root length and biomass under non-stress control and drought stress conditions. Compared to WT, roots of over-expression genotypes looked thin and had a reduced volume, with one major root and only a few adventitious roots originating from the main root, during non-stress control conditions (Fig. 8a). Under identical conditions, thick, long roots with many adventitious roots were observed in RNAi genotypes compared to WT (Fig. 8a). Similar patterns were obtained during drought stress where roots of over-expression plants looked brownish and thin, whereas thick roots with extra adventitious roots were observed in RNAi plants compared to WT (Fig. 8b). We further quantified the root data and showed that without drought, one over-expression genotype (WD-6OE) had shorter roots than WT, whereas two RNAi genotypes (WD-3R, WD-17R) exhibited significantly increased root length relative to WT (Fig. 8c). During drought stress, overexpression plants did not show difference of root length with WT. RNAi plants, however, exhibited enhanced root length in three genotypes (Fig. 8c).

Effect of drought stress on root growth and root biomass of alfalfa. WT and WD40-2 overexpression, and RNAi genotypes (a) well-watered control, (b) drought stress, (c) root length and (d) root biomass. Data are average of 3-5 biological replications. Single asterisk (*) shows significance of WD40-2 RNAi and overexpression plants with WT at P < 0.05 and double asterisk (**) indicates P < 0.01 (ANOVA).
We further determined root biomass accumulation in WT and transgenic plants with increased or reduced WD40-2 expression. All four overexpression genotypes showed significant reduction in root biomass than WT under control conditions whereas two RNAi genotypes (WD-3R, WD-17R) gained significantly higher root biomass than WT under this condition (Fig. 8d). Moreover, all over-expression genotypes accumulated lower biomass, while two RNAi genotypes built up more biomass than WT also under drought stress (Fig. 8d). Our root biomass data are consistent with root phenotype in over-expression and RNAi genotypes with and without stress (Fig. 8a,b).
Discussion
Numerous studies have shown that miR156 affects biomass production and grain yield in plants42,43,44,45. Our group previously generated six alfalfa genotypes (A16, A8a, A8, A11, A17 and A11a) overexpressing miR15625. These alfalfa genotypes showed enhanced aerial biomass, root length, forage yield, quality and delayed flowering under non-stress conditions25. The increase in root length caused by miR156 suggested to us that miRNA may also be involved in controlling drought stress in alfalfa. Our current study revealed that two of the miR156OE genotypes A16b and A8 with moderate miR156 expression, also showed enhanced root length and, additionally showed reduced water loss under drought conditions. Previously, these two genotypes displayed the most pronounced phenotypes, such as increased root biomass, proline accumulation, relative water content and survival after drought stress12. We therefore chose them for RNA-Seq analysis. Transcriptomic analysis revealed that miR156 can affect: 1) differentially expressed genes commonly found in both genotypes 2), drought-specific genes commonly expressed in both genotypes and 3) genes expressed in an individual miR156OE genotype relative to non-stress WT control. Our study has found DEGs that are specifically found in miR156OE genotypes but not in WT under well-watered control (Supplementary file S13) and drought stress conditions (Supplementary file S14). These genes may provide an interesting insight on what processes, mechanisms and pathways are affected by miR156, and open an array for further studies.
Overall, GO-enrichment analysis revealed that miR156OE genotypes had a reduced number of genes that are downregulated under drought conditions in three functional categories; biological process, molecular function and cell component. The GO terms such as effect of ROS metabolic process, response to oxidative process, peroxidase activity and protein kinase activity are closely related to drought tolerant phenotypes46. Reduced number of downregulated genes in these categories may show that miR156 alleviates drought-related suppression of these genes for a better response to stress. On the other hand, we noted an increased number of upregulated genes in A16b in some functional categories such as carbohydrate metabolic process, secondary metabolic process, catalytic activity, cell periphery and plasma membrane. Higher levels of starch content in A16b compared to control and A8 plants25, may justify increased number of genes at least in one category “carbohydrate metabolic process” in A16b plants. In addition, increased number of upregulated genes in both genotypes (A16b, A8) belonging to the ‘response to abiotic stress stimuli’, may suggest a pronounced role for miR156 in drought response of alfalfa. Our group has previously shown that expression of several genes positively involved in abiotic stress responses was consistently upregulated in A16b and A8 under drought12, which supports the outcome of our current study.
Our results indicated many genotypic differences in DEGs in response to drought. During drought conditions, WT contained >400 DEGs compared to A16b and A8, which showed a similar number of DEGs under drought stress. Relative to the corresponding WT, numbers of DEGs in A16b were twice of those found in A8 under drought stress. This may indicate a stronger response by A16b to drought stress presumably due to different miR156 levels. A strong response of A16b was also observed in various GO categories such as “transport”, “carbohydrate metabolic process” “catalytic activity” “extracellular region” and “cell periphery”. These results showed that different levels of miR156 can affect similar genes and GO categories, as well as expression of some unique non-overlapping genes in each genotype47. For example, CATALASE and ZEAXANTHIN EPOXIDASE were upregulated in A16b, but not in A8, in response to drought. On the contrary, GLUTATHIONE SYNTHETASE expression was induced only in A8, but not in A16b under drought conditions12. These uniquely expressed genes in an individual genotype can bring about different degrees of phenotypic change in plants13,47.
Transcription factors (TFs) play a crucial role in regulating gene expression under abiotic stress in plants. The RNA-seq study revealed that expression of many TFs was altered in miR156OE genotypes under drought stress. Major TFs affected by drought stress in miR156OE genotypes belonged to bHLH, HD-ZIP, TCP, C2H2 and WRKY family. Previous studies have shown positive roles for these TFs in drought responses of various plant species48,49,50,51,52, indicating that miR156 improves drought responses in alfalfa by modulating expression of at least some members of these TF families. The SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) are known targets of miR15625,47, and the high number of SPLs that were downregulated in miR156OE genotypes under drought may indicates that at least some SPLs may act as negative regulators of drought responses. This is also evident from our previous study, which showed an improved drought tolerance in alfalfa genotypes with reduced expression of SPL1312. Moreover, we observed altered expression of WD40 TFs in miR156 genotypes under drought stress. Given that WD40 genes could be potential target of miR15632, we hypothesized that miR156 downregulates WD40-2, and decided to conduct functional characterization experiments with this non-SPL gene.
Our group has identified seven SPL genes that are targets of miR156 in alfalfa25,47. Given the diversity of important transcription factors, genes and physiological traits affected by miR156 in alfalfa, it is critical to identify and characterize downstream target genes, not only SPLs but also other genes and transcription factors such as WD40. Genome wide analysis of the WD40 repeat proteins has been conducted in Arabidopsis and cucumber53, but their role has not been elucidated in alfalfa. In the present study, we focused on identifying non-SPL targets of miR156 and identified a gene encoding a putative WD40/repeat protein (WD40-2) that also regulates drought responses. Some studies have shown involvement of WD40 in anthocyanin biosynthesis33,34,35,36,54, nodule development38, cell wall biosynthesis37, as well as hormone, light and stress responses39.
In addition to common miR156 targeted SPL genes in alfalfa25,47, WD40-2 represents another family of genes targeted by miR156. Contrary to some SPL genes in Arabidopsis and alfalfa25,55, we did not detect cleavage sites of WD40-2 within the predicted miR156 target region, but sites were instead located upstream of the predicted target region. This discrepancy could be due to an RNA-induced silencing complex affecting the transcript during miRNA-related cleavage56. Our results however are consistent with some previous studies that have also shown variation in cleavage sites, for example, a cleavage site of rice SPL14 was found away from the miR156 target site44. In addition, our group has recently shown similar results where target sites of SPL2, SPL3, SPL4 and SPL9 were detected beyond the target sites47.
Two recent studies showed a positive role for WD40 gene in drought response in Arabidopsis57, as well as salt and osmotic stress responses in wheat58. However, Arabidopsis and wheat WD40 genes phylogenetically differ from alfalfa WD40-2 (Supplementary file S12), indicating functional differences potentially among various species. Alfalfa plants overexpressing WD40-2 exhibited stunted growth under control and drought stress conditions. In addition, low water potential, shoot, root biomass, and root length coupled with increased water loss further elucidates its role as a negative regulator. Furthermore, RNAi genotypes of WD40-2 seem to alleviate the negative effects of drought, and show high water potential, increased shoot and root biomass along with root length, and reduced water loss under drought stress conditions. This provides evidence of WD40-2 role as a negative regulator of drought response in alfalfa. In contrast, WD40 has been shown to positively modulate osmotic stress responses in Arabidopsis and wheat57,58. Studies have shown that different genes belonging to the same family could have opposite roles in plants. For example, seven Arabidopsis PP2Cs are negative regulators59,60,61,62,63,64,65, and one is a positive regulator of ABA signaling66. Recently, our group has discovered that one gene (SPL13) belonging to the miR156 network acts as a negative regulator of drought responses in alfalfa12. These observations support our current results which show that WD40-2 negatively regulates drought responses in alfalfa through a relationship with SPL13.
In conclusion, we present here the first report on the effect of miR156 overexpression on global gene expression in alfalfa under drought stress. We identified a non-SPL gene (WD40-2) that is targeted by miR156. Functional assessment of WD40-2 indicated that overexpressing and RNAi plants exhibited opposite effects, indicating its role as a negative regulator of drought responses in alfalfa. However, the possibility exists that the alfalfa drought tolerance phenotype is in part regulated by additional drought-responsive genes identified in our RNA-seq study, and these may directly or indirectly be regulated by miR156. For example, SPL13 is directly regulated by miR15625, and downregulation of SPL13 contributes to drought tolerance in alfalfa12. We have proposed a model that shows how miR156 regulates drought responses in alfalfa by targeting WD40-2, and affecting other physiological traits (Supplementary file S15). The model proposes that drought stimulates miR156 expression, which in turn cleaves WD40-2 transcript in alfalfa. Downregulation of WD40-2 results in improved drought tolerance whereas overexpression negatively affects drought responses. This model suggests that WD40-2 is a negative regulator of drought tolerance in alfalfa. Nonetheless, it is important to undertake functional and molecular studies also for at least some of DEGs under drought conditions. Similarly, a recent study shows that miR156 improves salinity stress tolerance in alfalfa40. Thus, a detailed analysis of the regulation of miR156 and downstream targeted drought-related genes remains an important study topic, as it will be of key interest to see whether miR156 and WD40 gene family also play roles in improving tolerance to other stresses and whether they function similarly in other plant species.
Methods
Plant material, growth conditions and drought experiments
Alfalfa plants overexpressing microRNA156 (miR156OE) were developed previously by our group25 for use in this study. Rooted stem cuttings were made from two of these genotypes (A8 and A16b) and a wild type control genotype (WT) that was generated through plant tissue culture. Also, rooted stem cuttings were made from four WD40-2 overexpression genotypes (WD40-1OE, WD40-4OE, WD40-6OE, WD40-7OE) and four RNAi genotypes (WD40-1R, WD40-3R, WD40-17R, WD40-18R). Stem cuttings were then transferred to pots (8 × 6′′) containing equal amount of homogenized PRO-MIX® BX soil. Plants were grown on a greenhouse bench under a 16-hour light/ 8-hour dark regime, and the soil was watered twice a week. Drought experiments were initiated on two-month-old plants as described in our previous study12. Briefly, at the start of each experiment, 50% soil moisture was established in each pot using a Fieldscout soil sensor (Spectrum Technologies Inc. Aurora, IL, USA). After commencing the drought experiment by withholding all water, soil pots were rotated randomly every day on the greenhouse bench to minimize environmental variation. Physiological data and root samples for RNA extraction were collected when WT plants showed stress symptoms i.e. wilting, drooping and brownish leaves, and soil moisture had dropped below 5% i.e. 13 days after imposing drought stress for miR156OE genotypes (A8, A16b), and 22 days for WD40-2 overexpression and RNAi genotypes.
Cloning medicago sativa WD40-2
Overexpression and RNAi constructs were constructed for the alfalfa homolog WD40-2 gene (MsWD40-2) using the Gateway system (Thermo Fisher Scientific, Mississauga ON). For overexpression, the full-length homologue of the Medtr2g028050 gene (a putative M. truncatula transducin/WD40 repeat gene) was amplified from Medicago sativa cDNA using primers with AttB sites, AttB1-WD40-2-cDNA and AttB2-WD40-2-cDNA (Supplementary Table 4), and cloned into the pDONR/Zeo entry vector. For RNAi, a 239 bp putative WD40-2 fragment was amplified from Medicago sativa cDNA using additional primers with AttB sites, B1-WD2-RNAi and B2-WD2-RNAi (Supplementary Table 4), and cloned into the pDONR/Zeo entry vector. After PCR screening and validation by sequencing, LR reactions were performed for the overexpression and RNAi constructs to recombine the putative WD40-2 fragments into the pMDC83 (overexpression) and pHELLSGATE12 (RNAi) vectors. Subsequently, overexpression and RNAi constructs were used to transform Agrobacterium tumefaciens strain EHA105, which was then used to transform alfalfa as described in our previous study25. QRT-PCR was used to detect transcript levels of WD40-2 gene in WD40-2 overexpressing and RNAi genotypes using primers WD2-qPCR-F and WD2-qPCR-R (Supplementary Table 4).
Measurement of root length, water loss and water potential
For root length measurements, plants were removed from the pots and roots were fully cleaned of soil. Root length was then recorded by taking measurements with a scale from root neck to root tip of control and drought stressed alfalfa plants of WT and miR156OE and WD40-2 overexpressing genotypes as well as WD40-2 RNAi genotypes. To conduct a water loss assay, water was withheld on two-month-old plants growing in soil under greenhouse conditions for 12 days (WT and miR156OE genotypes) and 22 days (WT, WD40-2 overexpressing, and RNAi genotypes), and water loss was measured as described previously12,67. After the water loss experiment, root and shoot dry weight was obtained by incubating samples at 65 °C for 5 days. Water potential was also measured on drought stressed and non-stressed controls plants of WT, WD40-2 overexpression and RNAi genotypes using a Portable Plant Water Status Console (Soilmoisture Equipment Corp. Santa Barbara, CA, USA).
Next generation sequencing (RNA-seq)
About 5 cm of drought stressed and control root tips (13 days after withholding water) were harvested from WT and miR156OE genotypes (A8, A16b), and immediately frozen in liquid nitrogen. High quality total RNA was extracted from roots following activated charcoal protocol68. The integrity of RNA samples was confirmed on an Agilent Bioanalyzer 2100 RNA Nano chip (Agilent Technologies). An RNA library was constructed and sequenced on an Illumina Hi-Seq. 2500 using paired-end 101 bp reads at the Centre for Applied Genomics (Sick Kids Hospital, Toronto, Canada) as a fee-for-service contract. Six biological replicates were sequenced for each WT and A16b (three control and three drought stressed) while seven biological replicates were sequenced for A8 (three control and four drought stressed).
Analysis of differential gene expression
Raw Illumina pair-end reads were trimmed using Trimmomatic69 to obtain high quality reads (Q > 30). These high-quality reads were used to identify differentially expressed genes (DEGs). We also used the M. trancatula genome as a reference for alignment of RNA-Seq reads using Tophat (v2.0.10). Tophat output was then used for differential expression analysis using Cufflinks software70. Subsequently, differentially expressed genes were annotated and assigned to three major functional categories (biological process, molecular function, cell component) using the GO Term enrichment tool from PlantRegMap and the M. truncatula database at P ≤ 0.0171. Differentially expressed genes were also screened for transcription factor families using the Plant Transcription Factor Database - PlantTFDB; http://planttfdb.cbi.pku.edu.cn/71,72. Venn diagrams were generated using the Venny tool73.
De novo assembly of transcriptome
Transcriptome de novo assembly of Medicago sativa was performed directly on the set of sequenced reads using the Trinity platform41. A pair end assembly was performed on each alfalfa genotype (WT, A8 and A16b). Parameters used for assembling the transcriptome are described in Supplementary file S1.
Validation of RNA-Seq by quantitative real-time PCR
For qRT-PCR validation, RNA was treated with TURBO DNase (Ambion, Austin, TX). A total of 1 µg RNA was used to synthesize cDNA using an iScript cDNA synthesis kit (Bio-Rad Laboratories, Mississauga ON). qRT-PCR amplification was conducted using a C1000 Touch™ Thermocycler Real-Time PCR System (Bio-Rad, Canada) using SsoFast SYBR Green Master Mix (Bio-Rad Laboratories, Mississauga, ON). Alfalfa homologues for two well-known housekeeping genes, ubiquitin (Medtr3g112230) and elongation factor (Medtr1g101870) with little variation of expression in our RNA-seq study, were used as reference genes for qRT-PCR reactions. Gene-specific primers and primers for reference genes are listed in Supplementary Table 4.
Detection of cleavage sites in WD40-2
Cleavage sites in alfalfa WD40-2 genes were detected using 5′ rapid amplification of cDNA end (5′-RACE) as described by25. The experiment was conducted using a First Choice_RLM-RACE Kit (Ambion, Burlington, ON, Canada) according to the manufacturer’s instructions. PCR products from Inner 5′ RLM-RACE PCR were purified using a gel purification kit (Qiagen,Toronto, ON, Canada) and cloned into a pJET1.2/blunt cloning vector (Fermentas, Ottawa, ON, Canada). At least 25 clones were subjected to sequencing using a pJET1.2/blunt sequencing primer.
Statistical analysis
GraphPad Prism software (https://www.graphpad.com/scientific-software/prism/) was used to statistically analyze the data. For comparisons between two groups the Student t-test was used whereas for means of more than two, an ANOVA was used followed by Tukey’s test for multiple comparisons.
References
Boyer, J. S. et al. The U.S. drought of 2012 in perspective: A call to action. Global Food Security 2, 139–143 (2013).
Bita, C. E. & Gerats, T. Plant tolerance to high temperature in a changing environment: Scientific fundamentals and production of heat stress-tolerant crops. Front. Plant Sci. 4, 273 (2013).
Gourdji, S. M., Sibley, A. M. & Lobell, D. B. Global crop exposure to critical high temperatures in the reproductive period: Historical trends and future projections. Environ. Res. Lett. 8 (2013).
Pryor, S. C., Barthelmie, R. J. & Schoof, J. T. High-resolution projections of climate-related risks for the Midwestern USA. Climate Res. 56, 61–79 (2013).
Schindler, D. W. Lakes as sentinels and integrators for the effects of climate change on watersheds, airsheds, and landscapes. Limnol. Oceanogr. 54, 2349–2358 (2009).
Schindler, D. W. & Donahue, W. F. An impending water crisis in Canada’s western prairie provinces. Proc. Natl. Acad. Sci. USA 103, 7210–7216 (2006).
Stocker, T. F. et al. Climate change 2013 the physical science basis: Working Group I contribution to the fifth assessment report of the intergovernmental panel on climate change (2013).
Lobell, D. B. et al. Greater sensitivity to drought accompanies maize yield increase in the U.S. Midwest. Science 344, 516–519 (2014).
Graham, P. H. & Vance, C. P. Legumes: Importance and constraints to greater use. Plant Physiol. 131, 872–877 (2003).
Lei, Y., Hannoufa, A. & Yu, P. The use of gene modification and advanced molecular structure analyses towards improving alfalfa forage. Int. J. Mol. Sci. 18, 298 (2017).
Bouton, J. The economic benefits of forage improvement in the United States. Euphytica 154, 263–270 (2007).
Arshad, M., Feyissa, B. A., Amyot, L., Aung, B. & Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. 258, 122–136 (2017).
Postnikova, O. A., Shao, J. & Nemchinov, L. G. Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol. 54, 1041–1055 (2013).
Zhang, S. et al. De novo characterization of fall dormant and nondormant alfalfa (Medicago sativa L) leaf transcriptome and identification of candidate genes related to fall dormancy. PLoS One 10, e0122170 (2015).
Li, D., Su, Z., Dong, J. & Wang, T. An expression database for roots of the model legume Medicago truncatula under salt stress. BMC Genomics 10, 517 (2009).
Zahaf, O. et al. Comparative transcriptomic analysis of salt adaptation in roots of contrasting Medicago truncatula genotypes. Mol. Plant 5, 1068–1081 (2012).
Branca, A. et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proc. Natl. Acad. Sci. USA 108, 864–870 (2011).
Jin, H. et al. Screening of genes induced by salt stress from Alfalfa. Mol. Biol. Rep. 37, 745–753 (2010).
Yang, S. et al. Alfalfa benefits from Medicago truncatula: The RCT1 gene from M. truncatula confers broad-spectrum resistance to anthracnose in alfalfa. Proc. Natl. Acad. Sci. USA 105, 12164–12169 (2008).
Gruber, M. Y. et al. Transcript analysis in two alfalfa salt tolerance selected breeding populations relative to a non-tolerant population. Genome 999, 1–24 (2016).
Jones-Rhoades, M. W., Bartel, D. P. & Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53 (2006).
Eldem, V. et al. Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PLoS One 7, 12 (2012).
Xie, F. et al. High-throughput deep sequencing shows that microRNAs play important roles in switchgrass responses to drought and salinity stress. Plant Biotechnol. J. 12, 354–366 (2014).
Zhao, B. et al. Identification of drought-induced microRNAs in rice. Biochem. Biophys. Res. Commun. 354, 585–590 (2007).
Aung, B. et al. MicroRNA156 as a promising tool for alfalfa improvement. Plant Biotechnol. J. 13, 779–790 (2015).
Gandikota, M. et al. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 49, 683–693 (2007).
Kim, J. J. et al. The microRNA156-SQUAMOSA promoter binding protein-like3 module regulates ambient temperature-responsive flowering via flowering locus in Arabidopsis. Plant Physiol. 159, 461–478 (2012).
Manning, K. et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952 (2006).
Silva, G. F. F. E. et al. MicroRNA156-targeted SPL/SBP box transcription factors regulate tomato ovary and fruit development. Plant J. 78, 604–618 (2014).
Wang, H. & Wang, H. The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol. Plant 8, 677–688 (2015).
Xie, K., Wu, C. & Xiong, L. Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 142, 280–293 (2006).
Naya, L. et al. Cleavage of a non-conserved target by a specific miR156 isoform in root apexes of Medicago truncatula. Plant Signal. Behav. 5, 328–331 (2010).
Ben-Simhon, Z. et al. A pomegranate (Punica granatum L.) WD40-repeat gene is a functional homologue of Arabidopsis TTG1 and is involved in the regulation of anthocyanin biosynthesis during pomegranate fruit development. Planta 234, 865–881 (2011).
Pang, Y. et al. A WD40 repeat protein from Medicago truncatula is necessary for tissue-specific anthocyanin and proanthocyanidin biosynthesis but not for trichome development. Plant Physiol. 151, 1114–1129 (2009).
Walker, A. R. et al. The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in arabidopsis, encodes a WD40 repeat protein. Plant Cell 11, 1337–1349 (1999).
Xie, Y., Tan, H., Ma, Z. & Huang, J. DELLA Proteins Promote Anthocyanin Biosynthesis via Sequestering MYBL2 and JAZ Suppressors of the MYB/bHLH/WD40 Complex in Arabidopsis thaliana. Mol. Plant 9, 711–721 (2016).
Guerriero, G., Hausman, J. F. & Ezcurra, I. WD40-repeat proteins in plant cell wall formation: Current evidence and research prospects. Front. Plant Sci. 6, 1112 (2015).
Yano, K. et al. CERBERUS, a novel U-box protein containing WD-40 repeats, is required for formation of the infection thread and nodule development in the legume-Rhizobium symbiosis. Plant J. 60, 168–180 (2009).
Chuang, H. W., Feng, J. H., Feng, Y. L. & Wei, M. J. An Arabidopsis WDR protein coordinates cellular networks involved in light, stress response and hormone signals. Plant Sci. 241, 23–31 (2015).
Arshad, M., Gruber, M. Y., Wall, K. & Hannoufa, A. An insight into microRNA156 role in salinity stress responses of alfalfa. Front, Plant Sci. 8, 356 (2017).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Chuck, G. S. et al. Overexpression of the maize Corngrass1 microRNA prevents flowering, improves digestibility, and increases starch content of switchgrass. Proc. Natl. Acad. Sci. USA 108, 17550–17555 (2011).
Fu, C. et al. Overexpression of miR156 in switchgrass (Panicum virgatum L.) results in various morphological alterations and leads to improved biomass production. Plant Biotechnol. J. 10, 443–452 (2012).
Jiao, Y. et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544 (2010).
Wang, S. et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 44, 950–954 (2012).
Farooq, M., Wahid, A., Kobayashi, N., Fujita, D. & Basra, S. M. A. Plant drought stress: Effects, mechanisms and management. Sustainable Agric. 153–188 (2009).
Gao, R., Austin, R. S., Amyot, L. & Hannoufa, A. Comparative transcriptome investigation of global gene expression changes caused by miR156 overexpression in Medicago sativa. BMC Genomics 17, 1 (2016).
Dong, Y. et al. A novel bHLH transcription factor PebHLH35 from Populus euphratica confers drought tolerance through regulating stomatal development, photosynthesis and growth in Arabidopsis. Biochem. Biophys. Res. Commun. 450, 453–458 (2014).
Zhao, Y. et al. A novel maize homeodomain–leucine zipper (HD-Zip) I gene, Zmhdz10, positively regulates drought and salt tolerance in both rice and Arabidopsis. Plant Cell Physiol. 55, 1142–1156 (2014).
Wei, W. et al. Identification and transcript analysis of the TCP transcription factors in the diploid woodland strawberry Fragaria vesca. Front. Plant Sci. 7, 1937 (2016).
Zhang, X. et al. OsMSR15 encoding a rice C2H2-type zinc finger protein confers enhanced drought tolerance in transgenic Arabidopsis. J. Plant Biol. 59, 271–281 (2016).
Ding, W. et al. Wheat WRKY type transcription factor gene TaWRKY1 is essential in mediating drought tolerance associated with an ABA-dependent Pathway. Plant Mol. Biol. Rep. 34, 1111–1126 (2016).
Li, Q. et al. Genome-wide analysis of the WD-repeat protein family in cucumber and Arabidopsis. Mol. Genet. Genomics 289, 103–124 (2014).
Gou, J. Y., Felippes, F. F., Liu, C. J., Weigel, D. & Wang, J. W. Negative Regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23, 1512–1522 (2011).
Wu, G. & Poethig, R. S. Temporal regulation of shoot development in Arabidopsis thaliana by miRr156 and its target SPL3. Development 133, 3539–3547 (2006).
Park, J. H. & Shin, C. MicroRNA-directed cleavage of targets: Mechanism and experimental approaches. BMB Rep. 47, 417–423 (2014).
Liu, W. C. et al. WD40-REPEAT 5a functions in drought stress tolerance by regulating nitric oxide accumulation in Arabidopsis. Plant Cell Environ. 40, 543–552 (2016).
Kong, D., Li, M., Dong, Z., Ji, H. & Li, X. Identification of TaWD40D, a wheat WD40 repeat-containing protein that is associated with plant tolerance to abiotic stresses. Plant Cell Rep. 34, 395–410 (2014).
Kuhn, J. M., Boisson-Dernier, A., Dizon, M. B., Maktabi, M. H. & Schroeder, J. I. The protein phosphatase AtPP2CA negatively regulates abscisic acid signal transduction in Arabidopsis, and effects of abh1 on AtPP2CA mRNA. Plant Physiol. 140, 127–139 (2006).
Merlot, S., Gosti, F., Guerrier, D., Vavasseur, A. & Giraudat, J. The ABI1 and ABI2 protein phosphatases 2C act in a negative feedback regulatory loop of the abscisic acid signalling pathway. Plant J. 25, 295–303 (2001).
Nishimura, N. et al. ABA-hypersensitive germination1 encodes a protein phosphatase 2C, an essential component of abscisic acid signaling in Arabidopsis seed. Plant J. 50, 935-949 (2007).
Rubio, S. et al. Triple loss of function of protein phosphatases type 2C leads to partial constitutive response to endogenous abscisic acid. Plant Physiol. 150, 1345–1355 (2009).
Saez, A. et al. Gain-of-function and loss-of-function phenotypes of the protein phosphatase 2C HAB1 reveal its role as a negative regulator of abscisic acid signalling. Plant J. 37, 354–369 (2004).
Saez, A. et al. Enhancement of abscisic acid sensitivity and reduction of water consumption in Arabidopsis by combined inactivation of the protein phosphatases type 2C ABI1 and HAB1. Plant Physiol. 141, 1389–1399 (2006).
Zhang, K. & Gan, S. S. An abscisic acid-AtNAP transcription factor SAG113 protein phosphatase 2C regulatory chain for controlling dehydration in senescing Arabidopsis leaves. Plant Physiol. 158, 961–969 (2012).
Reyes, D. et al. Overexpression of a protein phosphatase 2C from beech seeds in Arabidopsis shows phenotypes related to abscisic acid responses and gibberellin biosynthesis. Plant Physiol. 141, 1414–1424 (2006).
Arshad, M. & Mattsson, J. A putative poplar PP2C-encoding gene negatively regulates drought and abscisic acid responses in transgenic Arabidopsis thaliana. Trees Struct. Func. 28, 531–543 (2014).
Rajakani, R., Narnoliya, L., Sangwan, N. S., Sangwan, R. S. & Gupta, V. Activated charcoal-mediated RNA extraction method for Azadirachta indica and plants highly rich in polyphenolics, polysaccharides and other complex secondary compounds. BMC Res. Notes 6 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, 1040–1045 (2016).
Jin, J., Zhang, H., Kong, L., Gao, G. & Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, 1182–1187 (2014).
Oliveros, J. & Venny, C. V. An interactive tool for comparing lists with Venn diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html (2007).
Acknowledgements
This research was funded by a grant from Agriculture and Agri-Food Canada to AH. We thank Mr. Delwin Epp and Ms. Min Yu for generating transgenic alfalfa plants. MA was the recipient of a NSERC Visiting Fellowship to Canadian Government Laboratories.
Author information
Authors and Affiliations
Contributions
M.A. conducted experiments, analyzed data and wrote the manuscript. M.Y.G. analyzed data and edited the manuscript. A.H. conceived of the project, designed experiments, analyzed data and edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arshad, M., Gruber, M.Y. & Hannoufa, A. Transcriptome analysis of microRNA156 overexpression alfalfa roots under drought stress. Sci Rep 8, 9363 (2018). https://doi.org/10.1038/s41598-018-27088-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-27088-8
This article is cited by
-
Harnessing phytohormone dynamics to fortify crop resilience against drought: a comprehensive review
Vegetos (2024)
-
Epigenomics in stress tolerance of plants under the climate change
Molecular Biology Reports (2023)
-
The Arrangement of MicroRNAs in the Regulation of Drought Stress Response in Plants: A Systematic Review
Plant Molecular Biology Reporter (2023)
-
Comparative transcriptomic analysis and antioxidant defense mechanisms in clusterbean (Cyamopsis tetragonoloba (L.) Taub.) genotypes with contrasting drought tolerance
Functional & Integrative Genomics (2022)
-
Label-free quantitative proteomic analysis of alfalfa in response to microRNA156 under high temperature
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
