
Abstract
The genus Calotropis (Asclepiadaceae) is comprised of two species, C. gigantea and C. procera, which both show significant economic potential for use of their seed fibers in the textile industry, and of their bioactive compounds as new medicinal resources. The available wild-sourced germplasm contains limited genetic information that restricts further germplasm exploration for the purposes of domestication. We here developed twenty novel EST-SSR markers and applied them to assess genetic diversity, population structure and differentiation within Calotropis. The polymorphic information index of these markers ranged from 0.102 to 0.800; indicating that they are highly informative. Moderate genetic diversity was revealed in both species, with no difference between species in the amount of genetic diversity. Population structure analysis suggested five main genetic groups (K = 5) and relatively high genetic differentiation (FST = 0.528) between the two species. Mantel test analysis showed strong correlation between geographical and genetic distance in C. procera (r = 0.875, p = 0.020) while C. gigantea showed no such correlation (r = 0.390, p = 0.210). This study provides novel insights into the genetic diversity and population structure of Calotropis, which will promote further resource utilization and the development of genetic improvement strategies for Calotropis.
Similar content being viewed by others

Introduction
The genus Calotropis (Asclepiadaceae), which is native to the tropics and subtropics of Africa and Asia, has great potential for use as a fiber and medicinal plant1,2. Its long, fine seed hair (similar to that of cotton) is a high quality fiber, while the sap contains unique chemical compounds that have the diverse bioactive and pharmacological properties indicative of potential for new drug discovery3. In addition, Calotropis’ growth properties include drought hardiness, fast growth after establishment, a short reproductive cycle and adaptation to salty soils. This suggests that it is an important shrub which could serve as a key plant for ecological restoration in arid and semiarid regions4,5.
There are two closely related species of the genus; Calotropis procera (mainly distributed in the tropic and subtropics of Africa) and Calotropis gigantea (mainly found in tropic and subtropics of Asia). Due to their morphological similarity, with major differences occurring only on the floral structures, distinguishing them during the non-flowering season is difficult4. Taxonomically, C. procera is distinguished from C. gigantea by its flower characteristics such as subglobose flower buds, corona as long as gynostegium and erect corolla with five petals with dark purple tips4. Development of a genetic marker to distinguish the two species would facilitate species identification outside the flowering season.
Utilization of Calotropis products have mostly been explored from wild populations, which raises concerns regarding their sustainability and low product yield. Currently, there is a Calotropis domestication trial as a fiber source underway in Kenya (personal communication). The available wild germplasm has limited genetic information, with few molecular markers pinpointed that separate the two species. This currently limits germplasm exploration and genetic improvement of Calotropis. To date, various marker systems have been used in population genetic analysis of Calotropis. In flower polymorphic (pink and white) C. gigantea, genetic analysis using random amplified polymorphic DNA markers (RAPDs) found rich genetic diversity and high genetic similarity6. In 18 accessions of C. procera studied in Egypt, RAPDs unveiled a high polymorphism level7. Moreover, RAPDs were also used to discriminate thirteen salt tolerant plant species including C. procera according to their genetic relationships8. Analysis of three populations of C. procera on the basis of their climo-geographic adaptations using zymograms of superoxide dismutase and peroxidase indicated high genetic variation9. Further, SDS-PAGE for protein and isozyme in genetic variation analysis successfully discriminated between the genotypes of the studied C. procera populations10. The analysis of C. procera from Benin using Amplified Fragment Length Polymorphism (AFLP) uncovered high genetic variation among the sampled populations11.
However, these previous studies were based on only a few samples within a limited sampling area and mainly focused on C. procera. In addition, AFLP and RAPDs are dominant markers which cannot distinguish between heterozygotes and homozygotes, thus their use may over-estimate genetic diversity, making them less robust for use in genetic analysis12. On the other hand, isozymes are phenotypic markers whose usefulness is limted by the requirement for fresh samples, the fact that they are influenced by environmental changes and that only a few loci are analysed13. Expressed sequence tags-simple sequence repeats (EST-SSR) are more robust and efficient in population genetic analysis based on their intrinsic characteristics such as codominance, multi-allelic, highly reproducible, transferability across taxon, hypervariable and ubiquitous distribution across the genome14. In particular, EST-SSRs target protein coded are conservative and their markers are usually transferable within species, thus they are often applied to investigate the genetic diversity and population structure across related species15,16,17,18.
In our previous study19, we generated transcriptome data for C. gigantea. Using that data, in the present study, we developed 20 polymorphic and efficient EST-SSR markers. These markers were used to investigate genetic diversity and differentiation and perform population structure analysis for ten populations collected from Asia and Africa (as shown in Fig. 1). These results provide novel insights into the genetic diversity and population differentiation in Calotropis. This study not only increases our understanding of the genetics of Calotropis, but also is useful to development of feasible strategies for Calotropis genetic resource management and conservation.

Map showing geographical locations where samples were collected. The marked points are exact points of collection. ArcGIS v10.2.2 (http://www.esri.com/) was used to generate the map.
Results
Polymorphism of EST-SSR Markers
In total, 170 primer pairs were randomly selected from those designed from the transcriptome of C. gigantea in our recent study19. To inspect polymorphism of primers the screening was initially carried out using eight samples (four from C. gigantea and four from C. procera), selected at random from the sampled populations. Among the 170 selected primer pairs, 151 amplified successfully across C. gigantea and C. procera while 19 pair primers did not yield any PCR products at diverse annealing temperatures. Out of the 151 successful primer pairs, 109 yielded amplification products of the expected size, and the other 42 primer pairs generated PCR products that were larger or smaller than expected or unspecific bands. Of those successfully amplified, 20 primers (13.2%) showed polymorphisms whereas 89 were identified as monomorphic. The 20 polymorphic EST-SSR primers (see Table S1) were used for further population analysis. In total, 97 alleles were identified from C. gigantea samples, ranging from two (CG71) to eight (CG 84), with an average of 4.85 alleles per locus, whereas in C. procera samples, 84 alleles were identified with an average of 4.2 alleles per locus (Table S2). Across the20 loci, observed heterozygosity (HO) had a mean of 0.223 in C. procera and 0.194 in C. gigantea while the mean genetic diversity (HS) was 0.487 and 0.379 for C. gigantea and C. procera respectively. PIC values ranged from 0.065 to 0.769, with the mean = 0.429 in C. gigantea and 0.045 to 0.670, with the mean = 0.338 in C. procera (Table S1). From the average number of alleles and PIC values, it is evident that both C. procera and C. gigantea accessions display similar amount of genetic diversity (Table S1). Combining all populations of both species; the PIC ranged from 0.102 (CG71) to 0.800 (CG28). Inbreeding coefficient (FIS) values ranged from −0.365 to 0.879 (mean = 0.167) in C. gigantea, and from −0.597 to 0.951 (mean = 0.177) in C. procera (Table S2).
Population Genetic Diversity Analysis
The genetic diversity (HS) of the ten populations ranged from 0.157 to 0.363 with an average of 0.245. In C. gigantea the average HS was 0.249. The Nepal population showed the highest (HS = 0.363), while the Dongchuan population had the lowest genetic diversity values (HS = 0.157). In C. procera the Mali S population had the highest genetic diversity (HS = 0.339) and the lowest was the Kibwezi population (HS = 0.195) with a mean of 0.248. The average levels of observed heterozygosity (HO) across all populations was HO = 0.214 while in C. procera and C. gigantea HO was 0.223 and 0.198 respectively, while within species the level of genetic diversity (HS) was 0.249 and 0.256 for C. gigantea and C. procera, respectively. Allelic richness (AR) ranged from 2.25 (Honghe) to 3.3 (Nepal) in C. gigantea and 2.05 (Kibwezi) to 2.85 (Mali S) in C. procera. The fixation index (F) for all the ten populations ranged from 0.048 (Kibwezi) to 0.320 (Honghe). Using various genetic diversity parameters there was no noticeable difference between the two species (Table 1).
Population Genetic Structure
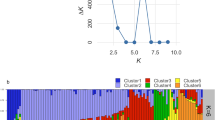
PCoA is often used to show genetic similarity among populations, with populations clustered according to their geographical location and species identity. Our PCoA results showed that the two species were clearly clustered and that the first two axes explained 64.62% of the total observed variation, suggesting that a distinct genetic structure exist between C. gigantea and C. procera (Fig. 2). STRUCTURE analysis indicated the optimal cluster number as K = 5 based on delta K (Fig. S1). Due to their biological relevance, other genetic structure K = 2, K = 3 and K = 4 are also displayed in Fig. 3. At K = 2, strong genetic structure was found among our samples that corresponded to respective species. Further at K = 5, the results revealed that Nepal, Mali S and Tanzania populations each had a unique gene pool (see Fig. 3). In addition, we performed the genetic relationship analysis among populations with neighbor joining criteria. As shown in Fig. 4, the sampled individuals were clearly grouped into two clusters (C. gigantea and C. procera), in concordance with the results of PCoA and STRUCTURE analyses.

Scatter plot of two axes from a PCoA of 286 Genus Calotropis, explaining 64.62% of the total observed variation.

Bayesian STRUCTURE bar plot based on probabilities for 286 individuals of 10 populations of Calotropis. Black lines separate populations.

Radial Neighbour joining (NJ) tree showing relationships among populations of Calotropis. Bootstrap numbers (>60) were denoted on the lines.
Intraspecies population structure analysis gave the optimum K value as K = 2 and K = 3 using Delta K and L (K) approaches respectively. Since delta K may erroneously result into K = 2 (Figs S2 and S3) both K Values were considered. In C. procera at K = 2, the populations grouped into West Africa (Mali K and Mali S) and East Africa populations (Baringo, Tharaka, Kibwezi, Tanzania). At K = 3 Baringo population separates from the rest of the East Africa populations, with Tanzania and Mali S populations showing admixtures (see Fig. 5). In C. gigantea populations at K = 2, the Dongchuan population clustered with the Honghe population while the Hainan population grouped with the Nepal population, however at K = 3 the Nepal population and the Hainan population each formed a separate group (Fig. 5). These results were consistent with the analyses from both PCoA and NJ (Fig. 6).

Principal coordinate analysis (PCoA) and Neighbour-joining (NJ) tree showing the relationships among populations of C. procera and C. gigantea. (a) PCoA for six populations of C. procera. (b) NJ for six populations of C. procera. (c) PCoA for four populations of C. gigantea. (d) NJ for four populations of C. gigantea. In each case, the colours correspond to the populations in NJ and PCoA.

Structure bar plots showing the assignment of individuals into distinct genetic clusters. (a) C. procera (b) C. gigantea.
Genetic Differentiation
Patterns of genetic divergence by AMOVA analysis showed that 28.32% of total variation was accounted for by interspecific differences between C. procera and C. gigantea, with a FST value of 0.528. When AMOVA analysis was assessed within each species, C. procera had most genetic variation (55.35%) partitioned within individuals while in C. gigantea among-population divergence was highest (57.01%). The genetic differentiation (FST) for C. procera and C. gigantea were 0.366 and 0.57, respectively (Table 2). Among the sampled populations interspecies pairwise FST values were lowest between the Tharaka and Kibwezi populations both within C. procera (FST = 0.200) and the highest differentiation (FST = 0.491) was between the Baringo (C. procera) and Hainan (C. gigantea) populations (Table S3). Intraspecies pairwise FST values in C. procera showed that the most differentiated populations were between Baringo and Mali K (FST = 0.362), while in C. gigantea the Dongchuan and Hainan populations were the most differentiated (FST = 0.451) (Table S3). At the loci level the genetic differentiation (FST) ranged from 0.036 (CG71) to 0.707 (CG83) with a mean of 0.405 in C. gigantea whereas in C. procera the FST ranged from 0.024 (CG35) to 0.764 (CG83) with a mean of 0.291 (Table S2).
Mantel test analysis is often used to examine the correlation between geographic and genetic distance18. Our Mantel test analysis showed a significant correlation between geographic and genetic distance among C. procera populations (r = 0.875, p = 0.020) (Fig. 7a), whereas no such correlation was found within C. gigantea populations (r = 0.39, p = 0.21) (Fig. 7b).

Correlation of geographic distance (in kilometers) and genetic distance (pairwise FST) among 286 individuals of 10 populations of Calotropis, including regression line (Mantel test, R2 = 0.31, P = 0.001 at 1000 randomization).
Discussion
Development of EST-SSR
This study represents the first attempt to develop and utilize EST-SSR markers to examine genetic diversity in the genus Calotropis. EST-SSRs have been found to be important molecular markers for detecting genetic diversity, structure, and demography as well for applied and experimental research on plant populations20. These markers show high transferability to closely related species of same genus or even family since they are associated with transcribed genes conserved among homologous genes21. Here, 151 primer pairs (88.8%) were successfully amplified PCR fragments from 170 pairs that had been designed from unigenes generated from transcriptome data. 19 primer pairs did not yield any PCR product. This could have been a result of insertions, lack of specificity, assembly errors, chimeric primers and the presence of large introns22. The polymorphism rate of Calotropis EST-SSR primers was 13.5%, which is comparable to that reported from other plants such as Neotropteri nidus (11%)22 and sesame 11.9%23.
The informativeness level of markers based on PIC is usually defined as low (PIC < 0.25), medium (0.5 > PIC > 0.25) or high (PIC > 0.5)24. Based on this criteria, the EST-SSR markers developed for Calotropis have a moderate level of polymorphism, with seven having high polymorphism, 12 having medium polymorphism and only one having a low PIC value (Table S1). The slight difference in PIC between C. procera and C. gigantea may have simple been due to bias introduce by deriving the markers from C. gigantea. This is congruent with what was observed in Tilia platyphyllos and Tilia cordata, in which the former (from which the markers were designed) had a slightly higher genetic diversity17. Overall, the developed EST-SSR markers showed sufficient polymorphism levels across the loci to perform further analyses of genetic diversity, differentiation and population genetic structure in Calotropis. This could contribute to future Calotropis genetics and breeding research, in areas such as identification of elite germplasm and marker-assisted selection.
Genetic diversity analysis
It is crucial to assess genetic diversity in order to ensure that the most diverse populations are selected to widen the genetic base of germplasm. Based on the developed EST-SSR markers the current study assessed genetic diversity in both C. procera and C. gigantea. Genetic variation was lower than in previous genetic studies of Calotropis populations9,10,11. However, these studies are not directly comparable since different marker systems could result in slight differences in the results obtained.
Generally, species with a wide distribution, wind dispersal and outcrossing show high genetic diversity25, however, based on EST-SSR markers, Calotropis displays a moderate genetic diversity (HS = 0.245). This level is comparable to the genetic diversity in Blighia sapida, a woody perennial species widespread in tropics and subtropics which had an average genetic diversity of HS = 0.2926. It seems that the genetic diversity of Calotropis plants is lower than that of most outcrossing species25. The population genetic diversity did not differ between C. procera (HS = 0.248) and C. gigantea (HS = 0.249), despite the fact that C. procera was distributed widely in Afica and C. gigantea was fragmentally distributed in Asia. Generally, in fragmented populations genetic variability is expected to be low27, however, this might not be universal, and in C. gigantea there was no evidence of low variability. The European Beech has fragmented populations that nonetheless show no evidence of loss of genetic variability28. Within C. procera, the Mali S populations had the highest genetic diversity while in Nepal, C. gigantea had the highest genetic diversity (Table 1). These two populations could be an important source of germplasm for incorporating in future breeding programs.
Population genetic structure
A plant population’s genetic structure is determined by the interaction of processes such as geneflow, mutation, selection and mating strategy29. The PCoA, STRUCTURE and NJ results clearly demonstrated genetic differentiation between C. procera and C. gigantea. Intraspecies structure analysis within C. procera and C. gigantea suggested that the most likely number of populations was K = 3, which was clustered primarily according to geographical region. Within C. procera the Baringo population split from the rest of East Africa population; this could be explained by limited geneflow between the populations as a result of extensive distance between them. Within C. gigantea, distinct groups were present as shown by both PCoA and NJ. Overall, the Nepal, Mali S and Tanzania populations had unique populations with admixed genotypes likely to harbor novel and potentially beneficial alleles. Thus these populations should be prioritized as source of germplasm for breeding and planting during domestication. Both interspecies and intraspecies analysis showed that a strong genetic structure existed in Calotropis, thus efforts into collection of germplasm should focus on sampling the maximum number of populations, to maintain high levels of genetic diversity30.
Genetic differentiation
Although C. procera and C. gigantea are morphologically close, AMOVA revealed high between-species genetic differentiation (FST) 0.528 (Table 2). In C. gigantea, the highest partitioning of genetic variation was found among populations. Such partitioning is expected for species with mixed mating systems25 rather than for those with outcrossing systems such as is suggested to be the probable mating system in Calotropis. This could be as a result of the sampled populations of C. gigantea occurring within fragments due to natural barriers such as mountainous terrain. This restricts the movement of insect pollinators to far distances, resulting in pollination occurring only within clumps of close relatives. These results are comparable to those of Hippophae tibetana Schlect31, and Cycas simplicipinna32 populations, which were found to have high genetic differentiation as a result of barriers limiting gene flow. However, partitioning of genetic variance in C. procera found high within-population variation and lower FST than in C. gigantea. This is an indication of relatively unrestricted geneflow between most of the sampled populations, resulting in a low genetic differentiation because C. procera is continuously distributed in African regions. Mantel test analysis found a strong correlation between geographical and genetic distance in C. procera (r = 0.875, p = 0.020). Populations separated by greater distances were more genetically dissimilar than those populations that are geographically close - which led to stronger internal genetic differentiation. Therefore, it is likely that the genetic structure of this species is affected by geographical distance. This high isolation by distance implies that selection and use of genetically diverse genotypes are key factors in C. procera breeding program during domestication and development of varieties with a broad genetic base. However, genetic isolation by distance is not a static process and may change with time, resulting in changes in genetic composition of a given population. In C. gigantea, however, Mantel test showed no correlation between geographic distance and genetic distance pattern (r = 0.39, p = 0.21), which further supports our hypothesis that geographical isolation exists among studied populations of C. gigantea, which led to genetic high differentiation which is suggested to have resulted from population fragmentation, low gene flow and genetic drift, similar to that in white Jabon (Anthocephalus cadamba)33.
Conclusion
We developed the first EST–SSR markers, thus providing a strong impetus for genetic analysis in Calotropis spp. Although the EST-SSR markers were of moderate polymorphism they showed power in discriminating between two closely related Calotropis species. These markers are also linked to functional genes due to their location in the coding regions of the genome; thus, may be useful for functional analysis of traits of interest. We found moderate genetic diversity between the two species of Calotropis with no difference within them. Information on genetic diversity will ensure that the maximum genetic diversity can be captured during the domestication process. We found strong interspecies and intraspecies genetic structure in this genus, thus collection of germplasm efforts should focus on sampling the maximum number of populations in order to preserve a high level of genetic diversity. These markers of genetic differentiation will give insights into incorporating the most diverse populations into breeding programs. Overall this study will be a useful resource for further genetic research in Calotropis.
Materials and Methods
Sample Collection and DNA Extraction
We sampled 286 individuals of Calotropis from 10 natural populations from Africa and Asia, at a minimum distance of 50 meters. From each sampling point individuals’ GPS readings were taken which were then transformed into reference points and mapped (Table S4 and Fig. 1). Two young leaf samples were collected in replicates, then immediately put in silica gel to dry until DNA extraction. Total DNA was isolated following cetyltrimethyl ammonium bromide (CTAB) method34. The DNA integrity and quality was measured by Nanodrop 2000 spectrophotometer (Thermofischer scientific, wilmington, DE, USA) and also by running on 1.0% (w/v) agarose gel. The DNA for PCR amplification was then diluted to a final concentration of 50 ng/µL for each sample in TE buffer (10 mmol/L Tris-HCL, PH 8.0, 1 mmol/L EDTA).
SSR Marker Screening
We selected 170 primers pairs at random from those designed in our recent study16. The EST-SSR markers were screened for PCR amplification using eight individual samples selected at random, four samples from Asia (C. gigantea) and four from Africa (C. procera). Polymerase chain reactions (PCR) were performed in 12.5 µL reaction volumes containing 6.25 µL 2X easytaqPCR PAGE MasterMix (TransGen Biotech, Beijing, China), 0.5 µL of forward and reverse primers, 0.5 µL of genomic DNA (50 ng/µL) and ddH2O 4.75 µL. Amplification of the PCR products was carried out using a BIO-RAD T100TM Thermal cycler (Singapore) with the following cycle: initial denaturation at 95 °C for 5 min; 35 cycles of 30 s at 94 °C, 45 s at a range of annealing temperatures to attain the optimum (Table S1); 30 s of elongation at 72 °C, and a final extension for 10 min at 72 °C. The PCR products were then separated by electrophoresis at 180 V and 50 W on 8% non-denaturing polyacryramide gel and visualized by silver nitrate staining35. The polymorphic primers were then selected and utilized in genotyping 286 samples.
EST-SSR Genotyping
Polymerase chain reaction (PCR) amplification of the 286 Calotropis samples using 20 developed loci was carried out in a 20 µL volume. The total volume contained 10X PCR buffer (2 µL), 25 mM MgCl2 (1.6 µL), 10 mM of dNTPs (0.4 µL), 5 unit/µL of Taq DNA polymerase (0.15 µL) TaKaRaTaqTM kit (TAKARA BIO INC., Dalian, China), 10 pmol each of forward and reverse primers (0.4 µL), 40–50 ng/µL of DNA template (1 µL) and 14 µL of sterile ddH2O. The forward primers of all the selected primers were fluorescent labeled with a 6-FAM, HEX and TAMRA (GENEray Biotech, Shanghai, China). PCR amplification conditions were as follows: 5 min at 95 °C, 35 cycles of 30 s at 94 °C, 45 s annealing temperatures 54 °C–62 °C (Table S1), 30 s at 72 °C and 10 min 72 °C final extension.
The success of amplification was determined by running the PCR products on 1% agarose in 1xTAE buffer. After successful selective amplification 1 µL of PCR product was mixed with 0.5 µL of size standard GeneScanTM 500 LIZ (Applied Biosystems) and 9 µL HI-DITM (Applied Biosystems), denatured and then separated on an ABI 3730xl Genetic Analyzer (Applied Biosystems, USA).
Data Analysis
The SSR allele sizes was estimated by Genemarker v4.0 (Softgenetics LLC, State College, PA, USA) for all populations, checked manually and entered in a spreadsheet. We used PowerMarker v3.2536 to calculate PIC, AR, number of alleles (NA), HO, HS. To determine average pair-wise between populations (FST) we used GenAlEx v. 6.537. Arlequin 3.1138 was used to determine FIS and FST per locus.
Analysis of molecular variance (AMOVA) to partition genetic variance was analysed in Arlequin 3.1138. Principal co-ordinate analysis (PCoA) to analyze genetic structure by covariance standardised approach of pairwise Nei’s genetic distances was conducted in GenAlEx version 6.537. The genetic relationships using neighbour-joining (NJ) was determined in PHYLIP3.6939 based on Nei’s genetic distance40. Nei’s genetic distance was calculated in MICROSATELLITE ANALYSER (MSA) v4.0541.The reliability of each node was tested using 1000 resamplings. FigTree v1.3.142 was used to visualize and edit the tree.
In order to determine the genetic groups among populations we used Bayesian clustering method in STRUCTURE V2.3.443. This analysis was run at 35 independent runs per K Value (K1–10) with a burn-in period of 100,000 iterations and 100,000 Markov chain Monte Carlo (MCMC). Structure Harvester44 was used to visualize the best K value based on delta K (ΔK)45 and maximum log likelihood L (K)46. We used Mantel tests18 to determine the pattern of isolation by distance at 1000 permutations using GenAlEx version 6.537. The comparison of the ten geographic populations distance matrix was calculated according to latitude and longitude from GPS coordinates using Vincenty’s formula, http://www.movabletype.co.uk/scripts/latlong.html.
References
Maji, S., Mehrotra, R. & Mehrotra, S. Extraction of high quality cellulose from the stem of Calotropis procera. South Asian. Journal of Experimental Biology 3, 113–118 (2013).
Babu, G. D., Babu, K. S. & Kishore, P. N. Tensile and Wear Behavior of Calotropis gigentea Fruit Fiber Reinforced Polyester Composites. Procedia Engineering 97, 531–535 (2014).
Ahmed, K. M., Rana, A. & Dixit, V. Calotropis Species (Ascelpediaceace)-A Comprehensive Review. Pharmacognosy Magazine 1, 48 (2005).
Orwa, C., Mutua, A., Kindt, R., Jamnadass, R. & Simons, A. Agroforestree database: a tree species reference and selection guide version 4.0. World Agroforestry Centre ICRAF, Nairobi, KE (2009).
Sobrinho, M. S., Tabatinga, G. M., Machado, I. C. & Lopes, A. V. Reproductive phenological pattern of Calotropis procera (Apocynaceae), an invasive species in Brazil: annual in native areas; continuous in invaded areas of caatinga. Acta Botanica Brasilica 27, 456–459 (2013).
Priya, T. A., Manimekalai, V. & Ravichandran, P. Intra Specific Genetic Diversity Studies on Calotropis gigantea (L) R. Br.-Using RAPD Markers (2015).
El-Bakry, A. A., Hammad, I. A. & Rafat, F. A. Polymorphism in Calotropis procera: preliminary genetic variation in plants from different phytogeographical regions of Egypt. Rendiconti Lincei 25, 471–477 (2014).
Mahmood, T., Aslam, R. & Rehmann, N. S. Molecular markers assisted genetic characterization of different salt tolerant plant species. J Anim Plant Sci 23, 1441–1447 (2013).
Pandeya, S., Chandra, A. & Pathak, P. Genetic diversity in some perennial plant species with-in short distances. Journal of Environmental Biology 28, 83–86 (2007).
Hassan, A. M., El-Shawaf, I. I. S., Bekhit, M. M. M., El-Saied, F. M. & Masoud, I. M. Genetic variation within Ushaar (Calotropis procera (ait) F.) genotypes using SDS-PAGE for protein and isozyme analysis. The fourth Comf. of sustain. Agric. Develop., Fac. of Agric., Fayoum Univ., 20–22 Oct., 2008, 103–114 (2008).
Agossou, Y. D., Angelo, R., Sprycha, Y., Porembski, S. & Horn, R. AFLP assessment of the genetic diversity of Calotropis procera (Apocynaceae) in the West Africa region (Benin). Genetic Resources and Crop Evolution 62, 863–878 (2015).
Qian, W., Ge, S. & Hong, D.-Y. Genetic variation within and among populations of a wild rice Oryza granulata from China detected by RAPD and ISSR markers. TAG Theoretical and Applied Genetics 102, 440–449 (2001).
Sun, G.-L., Diaz, O., Salomon, B. & Von Bothmer, R. Genetic diversity and structure in a natural Elymus caninus population from Denmark based on microsatellite and isozyme analyses. Plant Systematics and Evolution 227, 235–244 (2001).
Powell, W., Machray, G. C. & Provan, J. Polymorphism revealed by simple sequence repeats. Trends in plant science 1, 215–222 (1996).
Ellis, J. & Burke, J. as a resource for population genetic analysis. Heredity. 99, 125–132 (2007).
Lind, J. F. & Gailing, O. Genetic structure of Quercus rubra L. and Quercus ellipsoidalis EJ Hill populations at gene-based EST-SSR and nuclear SSR markers. Tree genetics & genomes 9, 707–722 (2013).
Logan, S. A., Phuekvilai, P. & Wolff, K. Ancient woodlands in the limelight: delineation and genetic structure of ancient woodland species Tilia cordata and Tilia platyphyllos (Tiliaceae) in the UK. Tree Genetics & Genomes 11, 1–12 (2015).
Muriira, N. G., Xu, W., Muchugi, A., Xu, J. & Liu, A. De novo sequencing and assembly analysis of transcriptome in the Sodom apple (Calotropis gigantea). BMC genomics 16, 723 (2015).
Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer research 27, 209–220 (1967).
Ellis, J. & Burke, J. EST-SSRs as a resource for population genetic analyses. Heredity 99, 125–132 (2007).
Varshney, R. K., Graner, A. & Sorrells, M. E. Genic microsatellite markers in plants: features and applications. TRENDS in Biotechnology 23, 48–55 (2005).
Jia, X., Deng, Y., Sun, X., Liang, L. & Su, J. De novo assembly of the transcriptome of Neottopteris nidus. Molecular Breeding 36, 1–12 (2016).
Zhang, H., Wei, L., Miao, H., Zhang, T. & Wang, C. Development and validation of genic-SSR markers in sesame by RNA-seq. BMC genomics 13, 316 (2012).
Botstein, D., White, R. L., Skolnick, M. & Davis, R. W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American journal of human genetics 32, 314 (1980).
Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Molecular ecology 13, 1143–1155 (2004).
Ekué, M. R., Gailing, O., Vornam, B. & Finkeldey, R. Assessment of the domestication state of ackee (Blighia sapida KD Koenig) in Benin based on AFLP and microsatellite markers. Conservation Genetics 12, 475–489 (2011).
Ouborg, N., Vergeer, P. & Mix, C. The rough edges of the conservation genetics paradigm for plants. Journal of Ecology 94, 1233–1248 (2006).
Leonardi, S. et al. Effect of habitat fragmentation on the genetic diversity and structure of peripheral populations of beech in Central Italy. Journal of Heredity 103, 408–417 (2012).
Schaal, B., Hayworth, D., Olsen, K. M., Rauscher, J. & Smith, W. Phylogeographic studies in plants: problems and prospects. Molecular Ecology 7, 465–474 (1998).
Richards, C. M., Antolin, M. F., Reilley, A., Poole, J. & Walters, C. Capturing genetic diversity of wild populations for ex situ conservation: Texas wild rice (Zizania texana) as a model. Genetic resources and crop evolution 54, 837–848 (2007).
Qiong, L. et al. Testing the effect of the Himalayan mountains as a physical barrier to gene flow in Hippophae tibetana Schlect.(Elaeagnaceae). Plos One 12, e0172948 (2017).
Feng, X., Wang, Y. & Gong, X. Genetic diversity, genetic structure and demographic history of Cycas simplicipinna (Cycadaceae) assessed by DNA sequences and SSR markers. BMC Plant Biology 14, 187, https://doi.org/10.1186/1471-2229-14-187 (2014).
Sudrajat, D. J. Genetic variation of fruit, seed, and seedling characteristics among 11 populations of white jabon in Indonesia. Forest Science and Technology 12, 9–15 (2016).
Doyle, J. J. & Doyle, J. Isolation of plant DNA from fresh tissue. Focus 12 12, 13–15 (1990).
Bassam, B. J. & Gresshoff, P. M. Silver staining DNA in polyacrylamide gels. Nature protocols 2, 2649–2654 (2007).
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Peakall, R. & Smouse, P. K. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics (2012).
Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary bioinformatics 1 (2005).
Felsenstein, J. “PHYLIP, version 3.6 [computer progam]. Seattle: Department of Genome Sciences, University of Washington, Seattle” (2004).
Nei, M., Tajima, F. & Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. Journal of molecular evolution 19, 153–170 (1983).
Dieringer, D. & Schlötterer, C. Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes 3, 167–169 (2003).
Rambaut, A. “FigTree ver. 1.3. 1. Edinburgh: Institute of Evolutionary Biology, University of Edinburgh” (2008).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Earl, D. A. Structure Harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conservation genetics resources 4, 359–361 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software Structure: a simulation study. Molecular ecology 14, 2611–2620 (2005).
Rosenberg, N. A. et al. Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics 159, 699–713 (2001).
Acknowledgements
We thank Catherine Dembele and Sailesh Ranjiktar for their help in sample collection, and Moses Wambulwa for advice on data analysis. The experiments were performed at the Laboratory of Genetic Engineering and Molecular Breeding, in Kunming Institute of Botany, Chinese Academy of Sciences. This work was jointly funded by National Natural Science Foundation of China (31661143002) and the local science and technology development program (sponsored by Chinese central government).
Author information
Authors and Affiliations
Contributions
J.X. and A.M. conceived the project, A.L. designed the experiments; J.X., A.M., N.G. collected the samples; N.G. performed the experiments, analysed the data and interpreted the results; A.Y. analysed the data; N.G. and A.L. wrote the manuscript. All the authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muriira, N.G., Muchugi, A., Yu, A. et al. Genetic Diversity Analysis Reveals Genetic Differentiation and Strong Population Structure in Calotropis Plants. Sci Rep 8, 7832 (2018). https://doi.org/10.1038/s41598-018-26275-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26275-x
This article is cited by
-
Studies on genetic diversity, gene flow and landscape genetic in Avicennia marina: Spatial PCA, Random Forest, and phylogeography approaches
BMC Plant Biology (2023)
-
Microsatellites reveal divergence in population genetic diversity, and structure of osyris lanceolata (santalaceae) in Uganda and Kenya
BMC Ecology and Evolution (2023)
-
Spatial PCA and Random Forest Analyses of Spatial Patterns in Genetic Diversity of Calotropis procera
Iranian Journal of Science (2023)
-
Climate change will disproportionally affect the most genetically diverse lineages of a widespread African tree species
Scientific Reports (2022)
-
Transcriptome sequencing and microsatellite marker discovery in Ailanthus altissima (Mill.) Swingle (Simaroubaceae)
Molecular Biology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
