
Abstract
We performed comprehensive molecular analysis of five cases of metastasizing uveal malignant melanoma (UM) (fresh-frozen samples) with an NGS panel of 73 genes. A likely pathogenic germline TP53 mutation c.760A > G (p.I254V) was found in two tumor samples and matched nontumor tissue. In three cases, pathogenic BAP1 mutation was detected together with germline missense variants of uncertain significance in ATM. All cases carried recurrent activating GNAQ or GNA11 mutation. Moreover, we analyzed samples from another 16 patients with primary UM by direct Sanger sequencing focusing only on TP53 coding region. No other germline TP53 mutation was detected in these samples. Germline TP53 mutation, usually associated with Li-Fraumeni syndrome, is a rare event in UM. To the best of our knowledge, only one family with germline TP53 mutation has previously been described. In our study, we detected TP53 mutation in two patients without known family relationship. The identification of germline aberrations in TP53 or BAP1 is important to identify patients with Li-Fraumeni syndrome or BAP1 cancer syndrome, which is also crucial for proper genetic counseling.
Similar content being viewed by others


Introduction
Uveal melanoma (UM) is the most common primary intraocular tumor in adults and represents 3–5% of all melanomas1,2. The median age of diagnosis is approximately 62 years. The large majority of ocular melanoma originates from the choroid (95%), ciliary body (5%), and iris (5%)3. Over the past decades, the incidence rate has been stable in Central Europe with two cases per million inhabitants. However, this rate increases with latitude4,5. Up to half of all patients develop metastatic disease4,6. The most common metastatic site is liver (80%-90%), lung (24%), and bone (16%)2. Somatic and hereditary mutations of several genes have been described in patients with UM2,7,8,9,10,11. However, familial UM is rare and represents less than 1% of all UM, the majority of the families carry germline mutations in the BAP1 gene12.
In our study, we analyzed target region of 73 genes or gene parts by NGS approach in five patients with metastasizing uveal melanoma in order to describe the mutation spectrum and frequency of the variants. Moreover, we analyzed another 16 patients with UM by direct Sanger sequencing focusing only on germline alterations in the TP53 gene.
Results
Spectrum and frequency of genetic variants in mUMs
Nonsynonymous variants in the coding parts of genes and adjacent intronic sequences with variant allele fraction (VAF) ≥10% were evaluated. Genes with clinically relevant variants are listed in Table 1. All variants detected by NGS in five metastatic uveal melanomas (mUM) and nontumor tissue can be found in Supplementary Table S1. Recurrent activating somatic mutation in GNA11 or GNAQ (NM_002067.2: c.626A > T, p.Q209L or NM_002072.3: c.626A > C, p.Q209P, respectively), a typical molecular sign of uveal melanoma, have been detected in all five cases.
Patients with germline variants in TP53
In two of five patients with mUMs, identical germline TP53 mutation NM_001126112.2: c.760A > G, p.I254V was detected (representative visualization in IGV and confirmation by Sanger sequencing is in Supplementary Fig. S1). These two patients are unrelated according to family history, and the probability of kinship was furthermore excluded by a bioinformatics approach. One of the carriers was a male patient with primary diagnosis of uveal melanoma at the age of 54, his father had a brain tumor at 60 and his mother had breast cancer at 54. Both parents were not genetically tested and archive tissue for retrospective mutation analysis of TP53 is unavailable. The second carrier was a female patient diagnosed with uveal melanoma at the age of 39, a family history of cancer has not been demonstrated. Immunohistochemical (IHC) analysis performed on liver tissue slides showed wild-type expression of the p53 protein.
To screen germline TP53 mutations in a larger cohort, the TP53 mutation analysis by direct Sanger sequencing was performed in additional 16 nontumor tissue from unrelated patients with UM. No other patient with TP53 germline mutation was found.
Both mUM samples with germline TP53 mutation did not have a deletion of chromosome 3 and carried a duplication of 8q (Supplementary Fig. S2 and Supplementary Table S2). One of them also carried a somatic mutation in SF3B1 (NM_012433.2: c.1874G > T, p.R625L). Further, one of these patients carried a germline mutation in MSH2 (NM_000251.2: c.4G > A, p.A2T). This variant is probably damaging according to the in silico prediction programs, but IHC examination showed intact nuclear expression of all MMR proteins (MSH2, MSH6, MLH1, PMS2) and fragment analysis showed microsatellite stable phenotype.
Patients with somatic mutations in BAP1 and germline variants in ATM
A germline mutation of ATM and a somatic mutation in BAP1 were found in three patients. None of these patients have a mutation of TP53. One patient carried a mutation in ATM (NM_000051.3:c.5975A > C, p.K1992T) and had a somatic frameshift mutation in BAP1 (NM_004656.2:c.79delG, p.V27Cfs*45, VAF 29.21%). Other patient had a somatic mutation in the BAP1 first coding amino acid that leads to the change of methionine to lysine (c.2T > A, p.?, VAF 60.78%). CNV analyses suggested a partial deletion of chromosome 3, and the patient carried a germline missense mutation in ATM (c.5558A > T, p.D1853V) and a germline tandem duplication of 56 bp in exon 48 of the ATM gene (c.7010_7065dup56, p.?) (Supplementary Fig. S3). Exon 48 of the ATM gene is a part of the regulatory FAT domain that inhibits ATM kinase activity until the occurrence of DNA damage13. This patient developed uveal melanoma relatively late at age 66, and breast cancer was diagnosed at 70.
The third patient without TP53 mutation carried germline mutation in the ATM gene (c.1522C > T, p.L508F), and somatic mutation in BAP1 (c.505_506insC, p.H169Pfs*13, VAF 77.59%) and CNV analyses suggested a partial deletion of chromosome 3.
Discussion
The Cancer Genome Atlas (TCGA) has recently published comprehensive data of 80 uveal melanomas and suggested four prognostic subtypes11. The most prevalent somatic mutations were GNAQ (guanine nucleotide-binding protein G(q) subunit α) (50%), GNA11 (guanine nucleotide-binding protein subunit α-11) (45%), BAP1 (BRCA1 associated protein-1 (ubiquitin carboxy-terminal hydrolase) (32.5%), SF3B1 (splicing factor 3b, subunit 1) (22.5%), and EIF1AX (Eukaryotic Translation Initiation Factor 1 A, X-Linked) (12.5%). Mutant GNAQ was shown to activate the MAPK pathway and it may also have important effects on other pathways such as the phosphatidylinositol-calcium second messenger system2. Hot-spot mutations affecting amino acid Glutamine at codon 209 in GNAQ or its paralog GNA11 are mutually exclusive, they have also been detected in benign uveal nevi and are not sufficient for full malignant transformation to melanoma2,10. Total loss of function of BAP1, that codes the protein involved in DNA damage control, correlates with increased metastatic potential. Partial or complete monosomy of chromosome 3, where BAP1 (3p21.31-p21.2) is located, is a relatively common event in metastasizing uveal melanoma. Other common chromosomal changes include gain or loss of 1p, 6q, 8q, 8p and less frequently 9p, and 16q. There is a myriad of combinations of such cytogenetic changes. Monosomy of chromosome 3, gain of 8q, epithelioid–mixed cell type and/or larger tumor diameter were strongly associated with a poor prognosis and potential to metastasize2,14,15.
In our study, comprehensive molecular analysis of five cases of mUMs was performed. We detected germline TP53 mutation in 2/5 patients and germline ATM mutation together with somatic BAP1 alteration in another 3/5 patients. Somatic GNAQ/GNA11 recurrent mutation was detected in all 5 cases. Further, somatic or germline variants were detected in BARD1, CDH1, MET, MMR genes, PARD3, SF3B1, SNAI3 (Table 1). Based on the unexpected finding of germline TP53 mutation, we analyzed another 16 patients with UM focusing only on germline TP53 mutations. However, no other TP53 mutation was found.
The TP53 gene is highly polymorphic in coding and noncoding regions and some of these polymorphisms have been shown to increase cancer susceptibility16. The frequency of de novo TP53 germline mutation has been estimated up to 30%, which is very high compared with the frequency of mutations in other tumor suppressor genes17,18. Germline mutations in TP53 are linked to Li-Fraumeni syndrome (LFS). Germline missense mutations are the most common TP53 variants, occurring in approximately 70% of cases and mainly altering residues within the DNA-binding domain19. Patients with LFS are predisposed to a wide variety of cancer types, with early onset, and with the potential for multiple primary cancer sites, including breast cancer, brain tumor, soft tissues cancer, adrenocortical carcinoma and other types16. Association of cutaneous malignant melanoma with LFS is relatively rare20,21,22. Moreover, a case of mucosal melanoma has been associated with LFS23.
Somatic mutations in the TP53 gene are one of the most frequent alterations in human cancers. Somatic TP53 mutations were detected in 12–19% cases of cutaneous malignant melanoma24,25,26 and also described in melanocytic tumor originating in the central nervous system27. In uveal melanoma, the occurrence of a somatic mutation is rare28,29,30,31,32,33. However, most researchers use immunostaining with an antibody against p53 protein to detect aberrant protein expression in UM instead of mutation analysis by sequencing of the TP53 gene. According to the dataset from IARC TP53 database, detected missense mutation show positive IHC results in 88% of cases. Not only TP53 mutations but also a disturbed p53 pathway can result in abnormal p53 expression34.
On the contrary, germline TP53 mutations in UM is, according to the literature, exceedingly rare and has been described only once in a British family with four generations burdened with uveal melanoma35. Here, we describe two other unrelated Czech patients with uveal melanoma that are carriers of germline TP53 p.I254V mutation. However, pathogenicity of the TP53 variant p.I254V is not fully elucidated. Foretova et al. described a Czech family with LFS (family without evidence of uveal melanoma) and causal TP53 mutation p.I254V36. The result of functional analysis of p53 transactivation ability in yeast (FASAY)37 has confirmed that p.I254V is a fully inactivating mutation. Codon 254 is buried in DNA binding domain and located in conserved beta-sheet structure according to the 3D model of p53 (Supplementary Fig. S4)38. Any change in this conserved domain could potentially lead to change of conformation, even though amino acids Isoleucine and Valine has similar physicochemical properties. On the contrary, in silico analyses suggested deleterious effect of this variant due to its impact on protein structure and function. The ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, accessed November 2017) contains data from two submitters and indicates uncertain significance of this germline TP53 variant. Halvorsen et al. detected germline variant p.I254V in TP53 in 3 out of 394 patients with lung cancer and designated this variant as polymorphism. Nevertheless, we need to take into consideration that this opinion is not based on strong arguments39. On the other hand, in the same codon 254 another two missense variants p.I254T and p.I254L were described, either somatic or germline in LFS family40, that were considered pathogenic throughout the databases and the literature. Somatic mutation p.I254V has been described according to the IARC TP53 database (http://p53.iarc.fr; assessed December 2017) in seven different tumors so far (Table 2).
Inherited pathogenic ATM mutation is the cause of autosomal recessive disease ataxia-telangiectasia which predisposes the individuals for increased cancer risk41,42. Germline or somatic mutations in ATM have not been described in uveal melanoma so far. In our study, we detected germline ATM variants in three patients with metastatic uveal melanoma. Nevertheless, all variants were of unknown significance or likely benign. Three of them have been described previously and their population mutant allele frequency (MAF) is low: p.K1992T, MAF(gnomAD) = 0.0003; p.D1853V, MAF(gnomAD) = 0.004915, and p.L508F, which is only described in database FLOSSIES MAF = 0.0001. In all three of our cases, ATM mutations cooccurred with pathogenic somatic variants in the BAP1 gene and two tumors have partial loss of chromosome 3, which are also relatively common event in metastasizing uveal melanoma, both associated with worse prognosis. Not only somatic but also germline mutation of BAP1 gene may occur in UM and the majority of familial UM with germline BAP1 mutation is associated with BAP1-tumor predisposition syndrome, which also increases the risk of the atypical Spitz tumors, malignant mesothelioma, and cutaneous melanoma43.
Conclusion
In conclusion, the results of our study have shown that genetic changes occurring in UM can be very heterogeneous. In three tumors and/or metastases we detected pathogenic somatic mutations in BAP1, which is not an unexpected finding, but these mutations occur in all three patients together with the germline missense variant in ATM. Despite the fact that detected ATM variants are of uncertain significance, this finding deserves further research because ATM mutations have never been reported in UM to date. Moreover, we have found germline TP53 mutation in two patients. Germline TP53 aberration has been described in UM in only one family so far, but the true incidence of TP53 mutations in UM is difficult to estimate due to the sparse studies focusing on this topic. However, the identification of germline TP53 or BAP1 mutations is important to be able to identify patients with Li-Fraumeni syndrome or BAP1 cancer syndrome and is the first step for proper genetic counseling and management of the patients and family members. We are well aware of the limitations of our study, which are mainly due to the small sample set. Nevertheless, we believe that the results of our study broaden the knowledge of molecular changes occurring in such a rare tumor as UM.
Material and Methods
Patients and samples
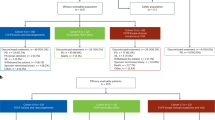
Archive files of the Bank of Biological Material of the First Faculty of Medicine, Charles University, Prague, were searched for uveal malignant melanomas or their metastasis. Five fresh frozen liver metastasis (stored in liquid nitrogen at −180 °C) of uveal melanoma (mUM) from 5 patients were found. For all 5 mUM cases, formalin-fixed paraffin-embedded tissue (FFPE) blocks of liver metastasis were also available for subsequent IHC analysis. Corresponding fresh-frozen nontumor tissue was available in 4/5 patients and genomic DNA from blood was available in 1/5 patient. Characteristics of the patients and tumors are summarized in Table 1. For 2/5 patients, FFPE blocks of matching primary uveal tumors were available in the archive files of the Institute of Pathology, First Faculty of Medicine, Charles University and General University Hospital in Prague. For extended analysis of TP53 mutation status in UM, the same archive files (between 2007 and 2017) were searched and another 16 patients (mean age 50, range 20–79 years) with available FFPE tissue were found. Review of the hematoxylin and eosin stained slides was performed in all cases and areas of nontumor or tumor tissues for macrodissection were marked (with estimation of tumor cell percentage in the selected area; ranges between 60–99%).
Ethics Statement
In compliance with the Helsinki Declaration, the study has been approved by The Ethics Committee of General University Hospital in Prague and an informed consent document was signed by participants. The methods were carried out according to the approved ethical guidelines.
DNA isolation
DNA from fresh-frozen tissue or FFPE blocks was isolated using standard procedures implementing QIAamp DNA Tissue kit (Qiagen) or cobas® DNA Sample Preparation kit (Roche; Germany), respectively.
NGS
Whole project and all auxiliary files are designed for genome build GRCh37 (hg19) coordinates. Samples for sequence capture NGS (massive parallel sequencing) were prepared using the KAPA HyperPlus kit. Target sequences were enriched using commercial hybridization probes (Nimblegen, Roche) designed to human DNA regions of our interest (Supplementary Table S3; 219 kbp). Library was pair-end sequenced by MiSeq instrument (Illumina). Processing of raw sequencing data was performed to analyze spectrum of genetic variants, such as single nucleotide variants and short insertions or deletions, and copy number variations (CNVs) using NextGENe software (Softgenetics, State College, PA) according to standardized biostatistical methods for NGS data (detailed setting in Supplementary Methods). Nonsynonymous variants in exons and adjacent intronic regions with minimal average coverage 100x and frequency ≥10% were evaluated and manually controlled using IGV viewer (Broad Institute). The data of average coverage of each region can be found as Supplementary Table S4.
Sanger sequencing
Detected variants with a frequency higher than 10% were confirmed by direct Sanger sequencing using BigDye v3.1 and ABI3500 analyzer (ThermoFisher). Selected variants were also checked in primary tumors from respective cases, available for 2/5 cases. Nontumor tissue or genomic DNA from blood was used to determine germline or somatic state of detected variants.
Further, direct sequencing of the TP53 coding- and adjacent intronic sequences (NM_001126112.2: exons 2–11) was performed in 16 primary uveal melanomas. Primers used for amplification and direct Sanger sequencing are available in Supplementary Table S5.
Microsatellite instability
Analysis of microsatellite instability (MSI) was performed with the set of five quasimonomorphic mononucleotide microsatellite markers BAT-26, BAT-25, NR-21, NR-22, NR-24. Fragmentation analysis was performed on ABI 3500 (ThermoFisher). MSI-high or MSI-low phenotype were defined as the presence of two or more- or one instable loci, respectively. MSI stable tumors (MSS) show no instability.
Immunohistochemical analysis
Immunohistochemical analysis (IHC) was performed using the avidin-biotin complex method with an antibody against p53 (BP 53–12, dilution 1:200, Zytomed Systems, Berlin, Germany). Additional IHC in the case mUM_2 (carrier of the mutation in MSH2 c.4G > A, p.A2T) was performed with antibodies against mismatch repair (MMR) proteins including MSH2 (clone FE 11, dilution 1:50, Zytomed Systems), MSH6 (clone 44, dilution 1:50, Zytomed Systems), MLH1 (clone G168-15, dilution 1:200, Spring Bioscience, Pleasanton, CA), PMS2 (EPR3947, ready-to-use, Zytomed Systems). Antigen retrieval was performed in 0.01 M citrate buffer (pH 6.0) for 40 minutes in a water bath at 98 °C for p53; in Dako Target Retrieval Solution (pH 9.0) overnight at 98 °C for MSH2, MSH6, MLH1 and for 50 minutes at 98 °C for PMS2.
Biostatistical analysis
NextGENe® Software was used for the analysis of the sequencing data, and CNV analysis. The Pindel tool was used to detect break points of large deletions and medium sized insertions44. The R package SNPRelate was performed to exclude unknown family relationships45.
Copy number variation
Used custom NGS panel was not primarily designed for analysis of copy number variation (CNV), so only regions with sufficient coverage by target genes were analyzed. Each region of our custom panel and their chromosomal location are listed in Supplementary Table S6. CNV analysis was performed focused on chromosome 3, 6, 8, 16, according to the current knowledge about cytogenetic changes in uveal melanoma. CNV tool Dispersion and Hidden Markov Model (HMM; part of NextGENe Software) was used to evaluate CNV variations between tumor tissue and corresponding nontumor tissue (mUM_1, mUM_2, mUM_3, mUM_4) and in case mUM_5 for comparison were used normalized coverage of the pool of other 4 nontumor samples.
In silico prediction tools
In order to assess the impact of detected missense variants, we employed several widely used in silico prediction programs or databases, which are imported in NextGENe Software.
Clinical significance and ensemble prediction scores from the ClinVar database, COSMIC database, and dbNSFP database (MetaSVM, MetaLR, RS_DBSNP141, SIFT, Polyphen2_HDIV, Polyphen2_HVAR, LRT, MutationTaster, MutationAssessor, FATHMM, PROVEAN GERP++, phyloP46, SiPhy) were imported to NextGENe mutation report (Supplementary Table S1).
Mutant allele frequencies for ATM aberrations were searched in databases The Genome Aggregation Database (gnomAD) and Fabulous Ladies Over Seventy database (FLOSSIES).
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Files).
References
Jovanovic, P. et al. Ocular melanoma: an overview of the current status. Int J Clin Exp Pathol 6, 1230–44 (2013).
Harbour, J.W. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell & Melanoma Research 25 (2012).
Pandiani, C., Beranger, G. E., Leclerc, J., Ballotti, R. & Bertolotto, C. Focus on cutaneous and uveal melanoma specificities. Genes Dev 31, 724–743 (2017).
Krantz, B. A., Dave, N., Komatsubara, K. M., Marr, B. P. & Carvajal, R. D. Uveal melanoma: epidemiology, etiology, and treatment of primary disease. Clin Ophthalmol 11, 279–289 (2017).
Singh, A. D., Turell, M. E. & Topham, A. K. Uveal Melanoma: Trends in Incidence, Treatment, and Survival. Ophthalmology 118, 1881–1885 (2011).
Goh, A. Y. & Layton, C. J. Evolving systemic targeted therapy strategies in uveal melanoma and implications for ophthalmic management: a review. Clin Exp Ophthalmol 44, 509–19 (2016).
Coupland, S. E., Lake, S. L., Zeschnigk, M. & Damato, B. E. Molecular pathology of uveal melanoma. Eye 27, 230–242 (2013).
Dogrusoz, M. & Jager, M.J. Genetic prognostication in uveal melanoma. Acta Ophthalmol (2017).
Harbour, J. W. et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–3 (2010).
Onken, M. D. et al. Oncogenic Mutations in GNAQ Occur Early in Uveal Melanoma. Investigative Ophthalmology & Visual Science 49, 5230–5234 (2008).
Robertson, A. G. et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 32, 204–220 e15 (2017).
Rai, K. et al. Germline BAP1 alterations in familial uveal melanoma. Genes Chromosomes Cancer 56, 168–174 (2017).
Bakkenist, C. J. & Kastan, M. B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 (2003).
Shields, C.L. et al. Personalized Prognosis of Uveal Melanoma Based on Cytogenetic Profile in 1059 Patients over an 8-Year Period: The 2017 Harry S. Gradle Lecture. Ophthalmology (2017).
Kilic, E. et al. Clinical and cytogenetic analyses in uveal melanoma. Invest Ophthalmol Vis Sci 47, 3703–7 (2006).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harbor Perspectives in Biology 2 (2010).
Gonzalez, K. D. et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 46, 689–93 (2009).
Leroy, B. et al. Recommended Guidelines for Validation, Quality Control, and Reporting of TP53 Variants in Clinical Practice. Cancer Res 77, 1250–1260 (2017).
Kratz, C. P. et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res 23, e38–e45 (2017).
Curiel-Lewandrowski, C., Speetzen, L. S., Cranmer, L., Warneke, J. A. & Loescher, L. J. Multiple primary cutaneous melanomas in Li-Fraumeni syndrome. Arch Dermatol 147, 248–50 (2011).
Kollipara, R. et al. Spitzoid melanoma in a child with Li-Fraumeni syndrome. Pediatr Dev Pathol 17, 64–9 (2014).
Potzsch, C., Voigtlander, T. & Lubbert, M. p53 Germline mutation in a patient with Li-Fraumeni Syndrome and three metachronous malignancies. J Cancer Res Clin Oncol 128, 456–60 (2002).
Klein, J. D. & Kupferman, M. E. Li-Fraumeni syndrome presenting as mucosal melanoma: Case report and treatment considerations. Head Neck 39, E20–E22 (2017).
Berger, M. F. et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485, 502–6 (2012).
Hodis, E. et al. A landscape of driver mutations in melanoma. Cell 150, 251–63 (2012).
Hugo, W. et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44 (2016).
van de Nes, J. et al. Targeted next generation sequencing reveals unique mutation profile of primary melanocytic tumors of the central nervous system. J Neurooncol 127, 435–44 (2016).
Brantley, M. A. & Harbour, J.W. Deregulation of the Rb and p53 pathways in uveal melanoma. American Journal of Pathology 157, 1795–1801 (2000).
Chana, J. S. et al. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. British Journal of Ophthalmology 83, 110–114 (1999).
Kishore, K., Ghazvini, S., Char, D. H., Kroll, S. & Selle, J. p53 gene and cell cycling in uveal melanoma. American Journal of Ophthalmology 121, 561–567 (1996).
Tokosova, E., Hermanova, M., Uhmannova, R., Smardova, J. & Hlinomazova, Z. [Immunohistochemical detection of the gene p53 and p21 expression in cells of the malignant melanoma of the uvea]. Cesk Slov Oftalmol 64, 153–6 (2008).
Liu, H. & Zhou, M. Evaluation of p53 gene expression and prognosis characteristics in uveal melanoma cases. Onco Targets Ther 10, 3429–3434 (2017).
Tobal, K. et al. Increased expression and mutation of p53 in choroidal melanoma. Br J Cancer 66, 900–4 (1992).
Kong, Y. et al. Analysis of mTOR Gene Aberrations in Melanoma Patients and Evaluation of Their Sensitivity to PI3K-AKT-mTOR Pathway Inhibitors. Clin Cancer Res 22, 1018–27 (2016).
Jay, M. & McCartney, A. C. Familial malignant melanoma of the uvea andp53: a Victorian detective story. Surv Ophthalmol 37, 457–62 (1993).
Foretova, L. et al. Genetic testing and prevention of hereditary cancer at the MMCI–over 10 years of experience. Klin Onkol 23, 388–400 (2010).
Smardova, J., Pavlova, S. & Koukalova, H. Determination of optimal conditions for analysis of p53 status in leukemic cells using functional analysis of separated alleles in yeast. Pathol Oncol Res 8, 245–51 (2002).
Cho, Y. J., Gorina, S., Jeffrey, P. D. & Pavletich, N. P. Crystal-Structure of a P53 Tumor-Suppressor DNA Complex - Understanding Tumorigenic Mutations. Science 265, 346–355 (1994).
Halvorsen, A. R. et al. TP53 Mutation Spectrum in Smokers and Never Smoking Lung Cancer Patients. Front Genet 7, 85 (2016).
Bougeard, G. et al. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 45, 535–8 (2008).
Choi, M., Kipps, T. & Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol Cancer Ther 15, 1781–91 (2016).
Soukupova, J., Dundr, P., Kleibl, Z. & Pohlreich, P. Contribution of mutations in ATM to breast cancer development in the Czech population. Oncol Rep 19, 1505–10 (2008).
Rai, K., Pilarski, R., Cebulla, C. M. & Abdel-Rahman, M. H. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet 89, 285–94 (2016).
Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–71 (2009).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–8 (2012).
Wiesner, T. et al. Toward an improved definition of the tumor spectrum associated with BAP1 germline mutations. J Clin Oncol 30, e337–40 (2012).
Muzeau, F. et al. [Profile of p53 mutations and abnormal expression of P53 protein in 2 forms of esophageal cancer]. Gastroenterol Clin Biol 20, 430–7 (1996).
Ibrahim, S. O. et al. Mutations of the p53 gene in oral squamous-cell carcinomas from Sudanese dippers of nitrosamine-rich toombak and non-snuff-dippers from the Sudan and Scandinavia. Int J Cancer 81, 527–34 (1999).
Iwamoto, K. S. et al. p53 mutations in tumor and non-tumor tissues of thorotrast recipients: a model for cellular selection during radiation carcinogenesis in the liver. Carcinogenesis 20, 1283–91 (1999).
Foulkes, W. D. et al. Primary node negative breast cancer in BRCA1 mutation carriers has a poor outcome. Annals of Oncology 11, 307–313 (2000).
Kerbauy, F. R. et al. Detection and possible prognostic relevance of p53 gene mutations in diffuse large B-cell lymphoma. An analysis of 51 cases and review of the literature. Leukemia & Lymphoma 45, 2071–2078 (2004).
Kurniawan, A. N. et al. Gene mutation analysis of sinonasal lymphomas in Indonesia. Oncology Reports 15, 1257–1263 (2006).
Samuels, Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004).
Acknowledgements
This work was supported by Ministry of Health, Czech Republic (Conceptual development of research organization 64165, General University Hospital in Prague, and by project AZV 16-30954A), by Charles University (Project Progres Q28/LF1 and SVV 260367), by Bank of Biological Material project BBMRI_CZ_LM2015089, and by OPPK (Research Laboratory of Tumor Diseases, CZ.2.16/3.1.00/24509). We thank dr. Cynthia Veenstra and M.S. Liam Ward for editing of English writing.
Author information
Authors and Affiliations
Contributions
Study concept and design: P.D., N.H., I.T. Acquisition of data: N.H., J.H., I.T., K.N., P.D. Analysis of genomic data: N.H., J.H. Preparation and analysis of samples, and interpretation of data: N.H., J.H., I.T. Contribution of the patient material with clinical data: P.D., K.N., J.U., K.J., J.G. Drafting of the manuscript: N.H., I.T., P.D. Proofread of the manuscript: P.D., I.T. All authors discussed the results, commented on the manuscript, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hajkova, N., Hojny, J., Nemejcova, K. et al. Germline mutation in the TP53 gene in uveal melanoma. Sci Rep 8, 7618 (2018). https://doi.org/10.1038/s41598-018-26040-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26040-0
This article is cited by
-
Targeting ferroptosis in melanoma: cancer therapeutics
Cell Communication and Signaling (2023)
-
Current understanding of epigenetics role in melanoma treatment and resistance
Cancer Cell International (2022)
-
Ovarian mesonephric-like adenocarcinoma arising in serous borderline tumor: a case report with complex morphological and molecular analysis
Diagnostic Pathology (2020)
-
Tumour predisposition and cancer syndromes as models to study gene–environment interactions
Nature Reviews Cancer (2020)
-
A comprehensive evaluation of pathogenic mutations in primary cutaneous melanomas, including the identification of novel loss-of-function variants
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
