
Abstract
Despite the broad distribution of M. ozzardi in Latin America and the Caribbean, there is still very little DNA sequence data available to study this neglected parasite’s epidemiology. Mitochondrial DNA (mtDNA) sequences, especially the cytochrome oxidase (CO1) gene’s barcoding region, have been targeted successfully for filarial diagnostics and for epidemiological, ecological and evolutionary studies. MtDNA-based studies can, however, be compromised by unrecognised mitochondrial pseudogenes, such as Numts. Here, we have used shot-gun Illumina-HiSeq sequencing to recover the first complete Mansonella genus mitogenome and to identify several mitochondrial-origin pseudogenes. Mitogenome phylogenetic analysis placed M. ozzardi in the Onchocercidae “ONC5” clade and suggested that Mansonella parasites are more closely related to Wuchereria and Brugia genera parasites than they are to Loa genus parasites. DNA sequence alignments, BLAST searches and conceptual translations have been used to compliment phylogenetic analysis showing that M. ozzardi from the Amazon and Caribbean regions are near-identical and that previously reported Peruvian M. ozzardi CO1 reference sequences are probably of pseudogene origin. In addition to adding a much-needed resource to the Mansonella genus’s molecular tool-kit and providing evidence that some M. ozzardi CO1 sequence deposits are pseudogenes, our results suggest that all Neotropical M. ozzardi parasites are closely related.
Similar content being viewed by others

Introduction
Mansonella ozzardi is one of three species of filarial parasite from its genus known to commonly use humans as a definitive host and to occur widely throughout Latin America and the Caribbean1,2,3. In the Brazilian Amazon, M. ozzardi is known to be transmitted by a wide range of insects including blackflies from the genus Simulium. In the Caribbean and elsewhere, however, the only known vectors are from the family Ceratopogonidae2,3. For this reason, it has been hypothesised that the M. ozzardi from the Caribbean region are distinct from the parasites of continental South America4,5,6.
Mitochondrial genes have been used extensively for diagnostics, systematics and population studies7,8,9,10,11. The haploid nature and fast rate of evolution of the CO1 gene has made it a particularly popular target for these studies and for these reasons also the target of an international DNA barcoding project that has been widely used for molecular taxonomy12,13. Mitochondrial DNA can, however, sometimes integrate into nuclear genomes and become pseudogenes, often referred to as Numts14,15,16. Unidentified mtDNA pseudogenes, like Numts, pose a threat to reliable molecular diagnostics, systematics and population analyses that include them17,18,19,20.
In this study, we report the complete mitochondrial genome of M. ozzardi parasites isolated from microfilariae obtained from an infected resident of Tefé, an Amazonas State town situated on the banks of the Rio Solimões, Brazil. In addition to providing a potentially useful new resource to the human filarial parasite community, this sequence has helped us to identify 14 novel mtDNA pseudogene sequences (MG913165- MG913178) and to incriminate previously published GenBank CO1 sequence deposits as potential pseudogenes (JF412329–JF412347). In the results we present here we also show that Caribbean and South American M. ozzardi are very closely related and most likely are not separate species as has been previously proposed4,5,6.
Methods
Mansonella ozzardi sample collection and identification
In previous work blood samples were taken from volunteers living in and around the Brazilian Amazon village of Tefé (3° 22′ 00″ S; 64° 42′ 00″ W), following a protocol (1504/10) approved by the Research Ethics Committee of the Fundação de Medicina Tropical Doutor Heitor Vieira Dourado, Manaus21. Mansonella ozzardi positive blood samples were identified as containing only M. ozzardi parasites by light microscopy (using methods described in Post et al.22) and by PCR amplification and sequencing of four taxonomic identifier sequences: a partial mitochondrial 12 S sequence, a partial CO1 gene sequence, a partial nuclear ribosomal 5 S sequence and a sequence spanning the complete ribosomal ITS-1 sequence. These PCR reactions followed previously published protocols and were all performed on microfilarial DNA extracts obtained from blood samples using QIAGEN blood and tissue kits8,21,23.
Shotgun Reversible Dye Terminator DNA sequencing of the M. ozzardi mitogenome
A single microfilarial DNA extract (containing a pool of microfilariae) obtained from a single blood collection taken from a single Tefé resident that tested positive for M. ozzardi (and no other filarial parasite) in both light-microscopy and PCR testing, was selected for M. ozzardi mitogenome characterisation. Shot-gun DNA sequencing of this DNA extract was performed on an Illumina HiSeq 2500 system (Oswaldo Cruz Foundation, high-throughput sequencing platform) using 2 × 100 nucleotide paired-end reads generated with Nextera Truseq libraries. Reads corresponding to human-host DNA were filtered out by mapping them against a human reference genome (accession number: GCA_000001405.19). The M. ozzardi mitogenome was assembled with the A5-miseq pipeline24. Mapping and short read post-processing was performed using Bowtie2 software25 and Samtools utilities26, respectively. The prediction of protein-coding genes, rRNAs and tRNAs was done using MITOS27 and Arwen software28 and validated manually by comparing homologous regions with a Loa loa mitochondria reference in Artemis29. A mitogenome map was generated using the BRIG software package30.
Mitogenome sequence assembly verification
A provisional mitogenome nucleotide sequence assembly was aligned to a set of 21 previously published nematode mitogenome sequences using CLUSTAL X31 (See supplementary information, Fig. 1). This identified an anomalous region in our provisional mitogenome assembly which was PCR amplified using primers designed from one conserved part of the NADH dehydrogenase subunit 2 gene (NADHDS2: TGAATGTGGTTTTGTTGTTCCT) and one conserved part of the NADH dehydrogenase subunit 5 gene (NADHDS5: GGGCTGCTATAGCCTTTGGT). Amplification was performed with a PCR reaction mix containing: 0.5 µl of each primer at a concentration of 100 mM, 5 µl of M. ozzardi DNA extract and 16.5 µl of PCR master mix in a 50 µl reaction mix, topped-up with 27.5 µl ultrapure sterilised water. The chemical composition of the PCR master mix was: 10 µl of 5 × Promega GoTaq PCR buffer; 4 µl of MgCl2 at a concentration of 25 mM and 1 µl of a DNTP mix, containing each DNTP at a concentration of 10 mM, and 1.5 µl of promega GoTaq at a concentration of 5Uµl-1. PCR amplification used the following cycling conditions: 95 °C for 2 minutes followed by 5 cycles of: 95 °C for 1 minute then 58–68 °C for 1 minute then 72 °C for 1 minute and 30 seconds; followed by 40 cycles of: 95 °C for 1 minute then 58 °C for 1 minute then 72 °C for 1 minute and 30 seconds. The protocol was completed with a final extension step of 72 °C for 10 minutes. Amplified PCR products were Sanger sequenced in both directions using the forward and reverse NADHDS2 NADHDS5 primers used to amplify them as well as a set of internal primers: TGTGGTTTTGTTGTTCCTAGTTT and AAAACCCAATCACAGACAATGAA. Prior to sequencing, the amplicons were cleaned-up using a Promega wizard PCR kit and protocol. Sequencing was done with an Applied Biosystems BigDye Terminator kit and ABI PRISM® 3100 Genetic Analyzer.

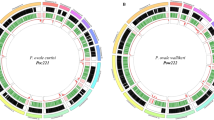
The genomic architecture of the M. ozzardi mitogenome shows a graphical representation of the M. ozzardi genome. The mitogenome’s annotated with gene content, gene order and orientation. Inside the circular genome proposed mitogenome Numt sequences (described in detail in Table 1) are mapped to the genome.
Phylogenetic analysis
DNA and conceptually translated amino acid sequences obtained for this study were aligned with sequences downloaded from GenBank using the program Clustal X 2.031. Sequence alignment editing was done in GenDoc32. Phylogenetic analysis was performed using software from the PHYLIP package (version 3.67)33. Because analysis of a similar set of filarial parasite mitogenomes found evidence that variation in the third codon base position of mitochondrial protein coding regions had reached saturation (see Yilmaz et al.34), our whole mitogenome tree construction was done using an alignment of conceptually translated amino acid sequence concatemers and the Maximum likelihood tree-building program “proml” of Felsenstein33. The mitogenome alignment used for this tree was prepared with 22 mitogenome sequences and all 12 mitochondrial protein codon gene sequences (see supplementary information, Fig. 2). In order to make our phylogenetic analysis easily comparable with previous analysis of Yilmaz et al.34 we used mitogenome sequences from all of the species that they used in their analysis and thus have included mitogenome sequences from two nematode species that are not from the filarial worm taxa Onchocercidae. Yilmaz et al.34 used Ascris lumbricoides and Dracunculus medinensis mitogenome sequences as outgroups in the construction of their phylogenetic tree and so we have also included these non-Onchocercidae nematode-origin sequences in our analysis. Maximum likelihood trees from partial mitochondrial CO1 and 12 S sequences were prepared using nucleotide-level alignments and the PHYLIP program “dnaml”, as was the nuclear 5 S ribosomal tree33. The robustness of all four tree topologies was tested with 1,000 bootstrap pseudo replicate alignments that were generated with the PHYLIP program “seqboot”33.
Results
The complete mitogenome of M. ozzardi has an architecture typical of a filarial parasite mitogenome
Our initial assembly of the M. ozzardi mitogenome used standard illumina Hiseq sequence reads and sequence assembly software. The resultant provisional M. ozzardi mitogenome was of 13,681 nucleotides in length and was seen to share remarkable genomic architectural similarities with previously reported filarial parasite mitogenomes. The M. ozzardi mitogenome encodes exactly the same set of genes, arranged in the same order and orientation, as all hitherto published Onchocercidae mitogenomes (see Fig. 1). This astonishing level of synteny, allowed for us to easily align the raw nucleotide sequence of our provisional M. ozzardi mitogenome to the 21 previously determined filarial parasite mitogenomes. This suggested that our provisional M. ozzardi genome contains a 52 nucleotide sequence insert that is not present in any of the other previously published filarial parasite mitogenomes. The apparent insert was observed to occur between the two genes that encode the NADHDS2 and NADHDS5 proteins (see supplementary figure 3). As the Illumina Hiseq sequence run had also detected the occurrence of pseudogenes sequences covering this region (see Fig. 1 and Table 1), which we recognised could potentially corrupt our mitogenome assembly, we decided to Sanger sequence across this region to determine whether this 52 nucleotide region represented an artefact of mitogenome assembly. PCR primers designed from conserved regions of the NADH dehydrogenase subunits 2 and 5 were used to amplify and sequence this region and determined that this 52 nucleotide sequence was indeed an artefact of sequence assembly. Figure 1 shows the mitogenome organisation of M. ozzardi, which has the same architecture as all hitherto reported filarial parasite genomes34. The complete and verified mitogenome sequence of M. ozzardi is 13,629 base pairs and has been deposited at GenBank and assigned the accession number KX822021.
Mansonella ozzardi belongs to the Onchocercidae “ONC5” clade and maybe more closely related to the lymphatic filariasis causing parasites than previously thought
Figure 2 shows a phylogenetic tree constructed from the protein coding region of 21 parasitic nematode worms. This tree was constructed using a similar approach to Yilmaz et al.34 and is very similar to theirs. Our analysis, like the analysis of Yilmaz et al.34, supports the seven-gene multi locus sequence typing (MLST) analysis of Lefoulon et al.35 and therefore strongly supports the monophyly of Onchocercidae and the existence of at least two of the phylogenetic Onchocercidae clades (ONC4 and ONC5) within it. Consistent also with the MLST analysis of Lefoulon et al.35, our mitogenome analysis has placed M. ozzardi in the “ONC5” clade. As in both the MLST analysis of Lefoulon and the mitogenome analysis of Yilmaz et al.34, in our analysis all four lymphatic filariasis causing parasites (W. bancrofti, B malayi, B. timori, B. pahangi) have clustered together in a highly boot-strap supported monophyletic group. In the mitogenome analysis of Yilmaz et al.34 the relationship between this grouping and the other ONC5 clade filarial parasites was not resolved. In our analysis the L. loa parasite forms an out-group to a weekly bootstrap-supported monophyletic group containing two highly bootstrap-supported sister clades. One of these sister clades contains the four parasites that cause lymphatic filariasis and the other contains M. ozzardi and the chicken filarial parasite Chandlerella quiscali. Our analysis therefore suggests that M. ozzardi is more closely related to the four parasite species that cause lymphatic filariasis than it is to the L. Loa parasite (which causes Loiasis). It should, however, be noted that while the clustering supporting this observation has bootstrap support (52.2%), such weak support is not always considered significant and is certainly far from the >90% bootstrap support that would typically be considered as robust support.

A phylogenetic tree constructed from filarial parasite mitogenome sequences. A representative maximum likelihood tree constructed from a 3378 amino acid residue alignment prepared by concatenating all 12 mitochondrial protein sequences from the M. ozzardi mitogenome and aligning to their orthologous protein sequences from 21 other whole mitogenome protein sequences from allied parasitic nematodes, (this alignment is available in the supplementary information, Fig. 3). Bootstrap-supported nodes referred to in the text are indicating with percentage values that were determined from 1000 pseudoreplicates. Branch labels provide source species names and GenBank accession numbers from which whole mitogenome sequence can be retrieved.
The identification of mitochondrial pseudogenes from a M. ozzardi DNA shot-gun sequence contig library
Using the provisionally assembled M. ozzardi mitogenome as a query in a BLASTn search of a M. ozzardi DNA shot-gun sequence contig library we identified 24 contigs that we have classified as pseudogenes. Fourteen of these contigs range between ~200 and 800 nucleotides in length and not only share significant nucleotide identity with our M. ozzardi mitogenome sequence, but also display the characteristic hallmarks of Numt pseudogenes (i.e they encode frame-shift mutations or stop codons or contain indels— see Table 1). We have deposited these sequences as putative mtDNA pseudogenes M. ozzardi sequences at GenBank, where can be retrieved using their assigned accession numbers: MG913165-MG913178. Table 1 provides an inventory of the mtDNA pseudogenes sequences and their accession numbers and also provides details on what basis each sequence was categorised as a pseudogene sequence. In Fig. 1 these contigs are displayed mapped to the circular mitogenome of M. ozzardi. As can be seen in Fig. 1 (and also Table 1) the pseudogene sequences identified in this study cover a broad range of M. ozzardi mitogenome. Notably, half of the mtDNA pseudogene contigs listed in Table 1 share identify with the CO1 sequence cytochrome oxidase I (CO1) gene, a gene which is often used as a barcode for species identifications (see below). Notably too, two putative pseudogene sequences with sequence identity spanning the region between NADHDS2 to NADHDS5 (the region which contained a putative 52 nucleotide insertion in our provisional mitogenome assembly) were also identified. We did not identify mtDNA pseudogenes sharing identity with the 12 S mitochondrial gene.
The characterisation of CO1 pseudogenes from GenBank sequence deposits: Peruvian CO1 sequences are genetically distinct from Haitian and Brazilian CO1 sequences
Figure 3 show a filarial parasite phylogenetic tree constructed with a partial mitochondrial DNA sequence from the cytochrome oxidase gene (CO1). This tree has been constructed with an alignment made using a partial M. ozzardi CO1 sequence obtained from its mitogenome (KX822021) and various reference sequences retrieved from GenBank sequence deposits. As can be seen in Fig. 3, the M. ozzardi CO1 sequence obtained for this study (KX822021) clusters in a 100% bootstrap supported monophyletic group with a M. ozzardi CO1 reference sequence (KP760340) obtained from Haitian M. ozzardi. This Brazilian-Haitian group forms a sister clade to a 100% bootstrap supported cluster of 21 CO1 sequences obtained from Peruvian M. ozzardi samples and collectively these two clusters form a bootstrap-supported monophyletic group that is a sister clade to a M. perstans specific cluster. The topology of the tree thus suggest that the M. ozzardi CO1 sequences from Haiti and Brazil share common ancestry before either does with the Peruvian CO1 sequences and also that all the M. ozzardi CO1 sequence share common ancestry with one another before any do with M. perstans CO1 sequences.

A phylogenetic tree constructed from filarial parasite CO1 sequences. A representative maximum likelihood tree constructed from an alignment of partial mitochondrial and Numt CO1sequence spanning 601 nucleotide positions (available in the supplementary information, Fig. 4). Bootstrap-supported nodes, referred to in the text, are indicated with percentage values that were determined from 1000 pseudoreplicates. Branch labels provide source species names and GenBank accession numbers, which can be used to retrieve source sequences. National flags are used to indicate the geographical origin of where M. ozzardi CO1 sequences derive.
The genetic distance between the Peruvian and the Brazilian and Haitian CO1 sequence clusters is, however, comparable to the genetic distance between species and even genera. Across the 601 nucleotide positions used in the alignment to construct the tree shown in Fig. 2, the M. ozzardi from Haiti and Brazil share > 99% identity, but neither the Haitian or the Brazilian sequences share >84% identity with their closest CO1 relatives from Peru. The genetic distance between the Brazilian-Haitian and the Peruvian M. ozzardi CO1 cluster is thus greater than the distance between the Wuchereria bancrofti CO1 sequence and its closest relative (AP017686) from the Brugia genus, which share >92% identity with one another. In Supplementary Fig. 3, part of this genetic distance between the M. ozzardi CO1 clusters can been seen to be attributable to what we contend here are three indels occurring in the CO1 sequences of Peruvian M. ozzardi CO1 sequences. None of these proposed indels occur in the alignment of any of the other filarial parasite CO1 sequences. The first of these is a nine nucleotide insertion, which does not produce a frame shift in the corresponding amino acid sequence. The second is a single nucleotide deletion and the third is a 16 nucleotide deletion. Using conceptual translation of each of the variant M. ozzardi CO1 sequences (ie the Brazilian, Haitian and Peruvian CO1 sequences) and aligning them to a reference M. perstans CO1 amino acid sequence (ANE06625) the single nucleotide deletion can be seen to correspond with a marked fall-off in amino-acid identity. Conceptual translation of the 354 nucleotides that immediately precede this gap, results in a 114 amino acid sequence that share at least 91% identity with the M. perstans CO1 amino acid (ANE06625) reference sequence (See Fig. 4). Although conceptual translation of the 230 nucleotides in the Peruvian CO1 sequences immediately following this deletion does not result in the introduction of a stop codon the resultant amino acid sequence shares almost no identity with the M. perstans CO1 reference sequence. In contrast to this, conceptual translation of the corresponding 246 nucleotides of the Brazilian and the Haitian CO1 M. ozzardi sequences results in amino acid sequences that share at least 96% identity with the M. perstans CO1 sequences. The CO1 sequences of Peruvian M. ozzardi can thus be regarded as containing a cryptic frame-shift mutation and displaying many of the hallmarks of non-coding mtDNA pseudogenes Numt sequences, which have integrated into the genome of M. ozzardi after its divergence from M. perstans.

Conceptual translations of M. ozzardi CO1 sequence deposits Shows an amino acid level alignment of conceptually translated M. ozzardi CO1 sequences. Variant M. ozzardi CO1 sequences from Peru are labelled with their source nucleotide sequence accession numbers and are shown aligned to a conceptually translated M. perstans CO1 reference sequence and the M. ozzardi CO1 sequence generated for this study. The source nucleotide sequences used to create the amino acid level alignment are shown below it. Three indels discussed in the text of the paper are highlight, as is a break in identity caused by a frame-shift mutation. Yellow highlighting has been used to make the grouping of Peruvian CO1 sequences more obvious.
The characterisation of CO1 pseudogenes from GenBank sequence deposits: 5 S and 12 S reference sequences suggest Peruvian M. ozzardi are almost identical to Brazilian, Bolivian and Haitian parasites
Although the Peruvian M. ozzardi CO1 sequences show many of the hallmarks of mtDNA pseudogene Numt origin, their discrete grouping in Fig. 3 could also be explained by them deriving from an a-typical strain of M. ozzardi, a novel species or M. ozzardi supporting multiple mitochondrial haplotypes. A-typical forms of M. ozzardi from the Peruvian villages in which Marcos et al.8 collected their samples have been previously reported36 and thus we felt it important to try to eliminate the possibility that the observed differences in CO1 barcodes was on account of them deriving from distinct strains or species. We did this by obtaining 12 S (mitochondrial DNA sequences) and 5 S nuclear ribosomal DNA sequences from our Brazilian M. ozzardi samples and using them in phylogenetic analysis with the 12 S and 5 S sequences that Marcos et al.8 obtained from the same Peruvian M. ozzardi samples which they recovered what we are contending here maybe CO1 Numts. Figure 5 shows a phylogenetic tree constructed using the mitochondrial 12 S sequences from a similar set of filarial parasite species samples as was used to prepare the CO1 tree. In contrast to the tree constructed with the CO1 gene, the 12 S mitochondrial sequence tree does not separate the Peruvian, Haitian and Brazilian M. ozzardi sequences into discrete clusters and displays very short branch lengths (genetic distances) between the M. ozzardi 12 S sequences. Across the 389 nucleotide positions used to construct the tree in Fig. 4, none of the M. ozzardi derived sequences (from Haiti, Brazil and Peru) share less than 97% identity (see supplementary information Fig. 4). Across the same 389 nucleotide sequence region Wuchereria bancrofti shares 92–95% identity with Brugia genus 12 S sequences. Consistent with the conclusions of Marcos et al.8, thus our mitochondrial 12 S mitochondrial DNA phylogenetic analysis suggest that none of the Peruvian M. ozzardi from their analysis are from an a-typical form of M. ozzardi and that M. ozzardi from across the Amazon region and the Caribbean are all very closely related. The nuclear ribosomal 5 S gene tree shown in Fig. 6 also strongly supports these conclusions. All of the M. ozzardi 5 S sequences (from Brazil, Peru and Bolivia) used to construct the tree in Fig. 5 cluster together tightly in a single bootstrap-supported group, in which no two sequences share less than 96% identity (see supplementary information Fig. 5).

A phylogenetic tree constructed from filarial parasite 12 S sequences. A representative maximum likelihood tree constructed from an alignment of partial mitochondrial 12 S sequences spanning 380 nucleotide positions (available in the supplementary information, Fig. 4). Bootstrap-supported nodes, referred to in the text, are indicated with percentage values that were determined from 1000 pseudoreplicates. Branch labels provide source species names and GenBank accession numbers, which can be used to retrieve source sequences. National flags are used to indicate the geographical origin of where M. ozzardi 12 S sequences derive.

A phylogenetic tree constructed from filarial parasite 5 S sequences. A representative maximum likelihood tree constructed from an alignment of partial ribosomal 5 S sequences spanning 380 nucleotide positions (available in the supplementary information, Fig. 5). Bootstrap-supported nodes, referred to in the text, are indicated with percentage values that were determined from 1000 pseudoreplicates. Branch labels provide source species names and GenBank accession numbers, which can be used to retrieve source sequences. National flags are used to indicate the geographical origin of where M. ozzardi 5 S sequences derive.
Discussion
The complete mitogenome of M. ozzardi is a potentially valuable new resource for the filarial parasite research
Thus far almost all the molecular tools that have been developed to assist with filarial parasite research have been based on the PCR amplification and sequencing of a few select mitochondrial and ribosomal DNA taxonomic marker sequences2,3. In the field of mansonellosis research tools of this type have been used to: confirm the existence of M. perstans in Latin America11; invalidate the legitimacy of reports of a-typical forms of M. ozzardi parasites8,30,37 and to reveal the existence of a previously unrecognised subspecies of M. perstans, e.g. M. perstans “deux”38. Existing tools have not, however, yet provided much help in understanding of the phylogeography or population genetics of filarial parasites because the sequences that have been historically targeted have uncovered so little within species diversity2,3. With many intergenic and poorly conserved sequence regions, mitogenome sequences have the potential to be a resource from which to develop new molecular tools to study the phylogeography and population genetics of filarial parasites34,39. After determining and then aligning the whole mitogenome sequence of D. repens to all previously published Onchocercidae mitogenomes, Yilmaz et al.27 identified hypervariable regions and then used the mitogenome sequence of D. repens to design PCR primers to amplify these regions for a population genetics study. The M. ozzardi mitogenome reported here could, thus, be used to develop similar tools to the study the phylogeography and population genetics of M. ozzardi in much the same way. Furthermore, our finding here that M. ozzardi shares near perfect gene content, gene order and gene orientation with all previously published Onchocercidae mitogenomes suggests that generic pan-Mansonella genus or even pan-Onchocercidae family population genetics tools with the ability to amplify variable regions from a range of species could be designed from filarial parasite mitogenome alignments. Our discovery of multiple M. ozzardi pseudogene sequences (explained in more detail below), however, also shows that any such tools must be designed with extreme care.
Whole mitogenome phylogenetic analysis raises questions about the placement of the Mansonella genus within ONC5
Overall, our phylogenetic analysis agrees well with the previous MLST analysis of Lefoulon et al.35, which found strong support for the monophyly of Onchocercidae and reported the existence of five well supported clades within it (ONC1 to ONC5). Our analysis, like the analysis of Yilmaz et al.34, provides strong support for at least the ONC4 and ONC5 clades that Lefoulon et al.35 reported and provides no indication that the other three clades that Lefoulon et al.35 reported are not equally valid. There are, however, a few minor within clade incongruences between our phylogeny and that of Lefoulon et al.35. Most notably, from a public health perspective, our mitogenome phylogeny (see Fig. 2) provides weak bootstrap support for the existence of monophyletic group that includes M. ozzardi and the filarial parasites that cause lymphatic filariasis (i.e those from the Wuchereria and Brugia genera)35, but which excludes L. loa. Our analysis thus suggests that M. ozzardi is more closely related to the lymphatic filariasis causing parasites than L. loa is, which contrasts with the MLST analysis of Lefoulon et al.35 that suggested they are equally distant (by very robustly clustering the L. loa parasite and Mansonella parasites together in a monophyletic group that excluded the lymphatic filariasis causing parasites). As our analysis was inferred entirely from mitochondrial DNA sequences, whereas the analysis of Lefoulon et al.35 was based on a mixture of mitochondrial and nuclear DNA sequences, their analysis can be considered less susceptible to the corrupting effects of the parasite´s maternally inherited endosymbiont, Wolbachia. It should, however, be noted that there are other important differences between our analysis and the analysis of Lefoulon et al.35 which could also be contributing to incongruences. Lefoulon et al.35 used nucleotide sequence alignments, which when used to infer phylogenies of distant taxa, can be more susceptible to effects of homoplasy than amino acid alignments. Yilmaz et al.34 who used some of the same gene sequences that Lefoulon et al.35 used, found significant support for saturation of nucleotide variation at the third codon position of the protein coding mitochondrial genes they analysed and for this reason chose (like us) to construct their trees with amino acid sequence alignments34. As L. loa parasites can compromise the reliability of the ICT cards, which are a popular immunodiagnostic tool that has been used extensively for the design and implementation of Lymphatic filariasis control and elimination programmes, the relationship these parasites have with Mansonella parasites (which are among the most common causes of human parasitaemias) is arguably thus a relationship that merits further investigation40,41,42.
The identification of cryptic CO1 pseudogenes among GenBank sequence deposits
Using ribosomal DNA ITS-1 sequence it has been shown that M. ozzardi from Argentina, Roraima state (in the extreme north of Brazil), Acre and various parts of the Brazilian state of Amazonas are also very closely related23,37,43. Using the ribosomal 5 S gene here we have shown that M. ozzardi parasites from Peru, Bolivia and the Brazilian state of Amazonas are all very closely related too (see Fig. 4). In Fig. 2 our analysis of the 12 S mitochondrial gene shows that M. ozzardi from the Caribbean are also very closely related to M. ozzardi parasites from the Amazon region suggesting that M. ozzardi from the Caribbean are not a distinct species as it was once proposed they might be4,5. Our analysis of the Brazilian and the Haitian CO1, which suggested they are almost identical, adds further weight to this notion and thus our data can be taken collectively to strongly support the notion that there is just one species of M. ozzardi in the New World.
Given that both the mitochondrial 12 S gene and the nuclear 5 S genes of the Peruvian M. ozzardi parasites so strongly suggest they are the same parasite species, the distinct clustering of the Peruvian CO1 sequences in Fig. 2 must be viewed as anomalous. Reports of a single species, with very little nuclear or morphological variation, having multiple distinct CO1 barcodes have been made44,45. These reports are usually attributed to the effects of maternally inherited endosymbionts (like Wolbachia) which cause indirect selective sweeps on mitochondrial diversity45. Such phenomena, however, are expected to affect the whole mitogenome sequence and thus would not be expected to create M. ozzardi populations with distinct CO1 sequences, but near identical 12 S sequences and thus cannot easily explain our observed results45. More than 4 million morphologically identified species have had their CO1 barcodes deposited in public databases, but we are unaware of a single report of a species that has two or more distinct CO1 haplotypes (i.e CO1 sequences that vary by >15%) and which also has < 1% diversity across its mitochondrial 12 S gene sequence46. We believe that this and the fact that Peruvian M. ozzardi CO1 sequence deposits showed all the hall marks of mtDNA pseudogene (including possible frame-shift mutations) and half of the pseudogene contigs over 200 nucleotides (see table one) that we identified in our shot-gun sequence library screen were of CO1-origin, means that the most parsimonious explanation of the phylogenetic grouping of Peruvian CO1 sequences shown in Fig. 3 is that they are of non-coding mtDNA pseudogene origin. As, however, these Peruvian CO1 sequences appear to encode open reading frame sequences, it is not presently possible for us to entirely exclude the possibility that these Peruvian CO1 sequences encode functional proteins and thus that they represent a distinct variant CO1 allele rather than a cryptic CO1 pseudogene.
Most CO1 pseudogene sequences, which occur quite commonly across a range of species17,18,19,20 are of Numt origin. Because Numts that do not encode stop codons can be easily miss-identified as coding sequences19, we believe our results highlight the need for the careful screening of mitochondrial sequence for cryptic pseudogenes prior to their depositing in public databases. We believe that they also highlight the risk that mtDNA pseudogenes, like Numts, pose to the popular single-gene mtDNA diagnostics system of CO1 barcoding13. In our study half of the mtDNA pseudogenes we identified from our contig library were of CO1 sequence origin. By contrast we did not: detect any mitochondrial 12 S Numt sequences in our contig library; encounter difficulties Sanger sequencing this gene or indeed encounter sequence variants in GenBank. This suggests that the 12 S gene is a more reliable target for M. ozzardi diagnostics and systematics than the CO1 gene is. In Lamberton et al.46 they amplified three mitochondrial DNA targets (ND5 12 S and 16 S) to detect the presence of O. volvulus parasites in their insect vectors. Because of difficulties in sequencing the ND5 gene, which they did not encounter with the 12 S and 16 S genes, they stopped targeting the ND5 gene sequence. Such a scenario could be explained by the occurrence of Numts in the O. volvulus and it may thus be that there is something special about filarial parasite mtDNA 12 S genes that makes them less susceptible to forming the kind of Numts that affect their utility as population genetics tools. Given the potential value of mitogenomes and mtDNA-based tools to the field of filarial parasite research, we believe further investigations into filarial Numt diversity and prevalence are warranted. We believe also that it is worth noting that Numt sequences themselves can be used for the design of population genetics studies47,48. From our work presented here, for example, it appears that the Peruvian CO1 sequences of Marcos et al.8, may not just be cryptic Numts but might also be Numts which occur only in a geographically restricted range of parasites and thus could be a potentially useful resource for future phylogeographic and population genetics studies of M. ozzardi.
References
Bain, O. et al. Review of the genus Mansonella Faust, 1929 sensu lato (Nematoda: Onchocercidae), with descriptions of a new subgenus and a new subspecies. Zootaxa 3918, 151–93 (2015).
Medeiros, J. F., Crainey, J. L., Pessoa, F. A., & Luz, S. L. Mansonelliasis. In Arthropod borne diseases (ed. Marcondes, C. B.) 405–426 (Springer International Publishing, 2017).
Ta-Tang, T. H., Crainey, J. L., Post, R. J., Luz, S. B. L. & Rubio, J. M. Mansonellosis: current perspectives. Research and Reports in Tropical Medicine 9, 9–24 (2018).
Nelson, G. S. & Pester, F. R. N. The identification of infective filarial larvae in Simuliidae. Bulletin of the World Health Organization 27, 473–481 (1962).
Shelley, A. J. & Shelley, A. Further evidence for the transmission of Mansonella ozzardi by Simulium amazonicum in Brazil. Annals of Tropical Medicine and Parasitology 70, 213–7 (1976).
Lima, N. F., Veggiani Aybar, C. A., Dantur Juri, M. J. & Ferreira, M. U. Mansonella ozzardi: a neglected New World filarial nematode. Pathogens and Global Health 110, 97–107 (2016).
Ferri, E. et al. Integrated taxonomy: traditional approach and DNA barcoding for the identification of filarioid worms and related parasites (Nematoda). Frontiers in Zoology 6, 1–12 (2009).
Marcos, L. A. et al. Genetic characterization of atypical Mansonella (Mansonella) ozzardi microfilariae in human blood samples from northeastern Peru. The American Journal of Tropical Medicine and Hygiene 87, 491–4 (2012).
Small, S. T. et al. Population genetics of the filarial worm Wuchereria bancrofti in a post-treatment region of Papua New Guinea: Insights into diversity and life history. PLOS Neglected Tropical Diseases 7, e2308 (2013).
Small, S. T., Tisch, D. J. & Zimmerman, P. A. Molecular epidemiology, phylogeny and evolution of the filarial nematode Wuchereria bancrofti. Infect Genet Evol. 28, 33–43 (2014).
Tavares da Silva, L. B. et al. Molecular Verification of New World Mansonella perstans Parasitemias. Emerging Infectious Diseases 23, 545–547 (2017).
Prosser, S. W. J., Velarde-Aguilar, M. G., León-Règagnon, V. & Hebert, P. D. N. Advancing nematode barcoding: A primer cocktail for the cytochrome c oxidase subunit I gene from vertebrate parasitic nematodes. Molecular Ecology Resources 13, 1108–1115 (2013).
Ratnasingham, S. & Hebert, P. D. N. SBARCODING BOLD: The Barcode of Life Data System (www.barcodinglife.org) Molecular Ecology Notes, 7, 355–364 (2007).
Lopez, J. V., Yuhki, N., Masuda, R., Modi, W. & O’Brien, S. J. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. Journal of Molecular Evolution 39, 174–90 (1994).
Richly, E. & Leister, D. Numts in sequenced eukaryotic genomes. Molecular Biology and Evolution 21, 1081–4 (2004).
Chaves, B. R. et al. Barcoding Neotropical birds: assessing the impact of nonmonophyly in a highly diverse group. Molecular Ecology Resources 15, 921–931 (2015).
Zhang, D. X. & Hewitt, G. M. Nuclear integrations: challenges for mitochondrial DNA markers. Trends in Ecology & Evolution 11, 247–51 (1996).
Bensasson, D., Zhang, D., Hartl, D. L. & Hewitt, G. M. Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends in Ecology & Evolution 16, 314–321 (2001).
Bertheau, C., Schuler, H., Krumböck, S., Arthofer, W. & Stauffer, C. Hit or miss in phylogeographic analyses: the case of the cryptic Numts. Molecular Ecology Resources 11, 1056–9 (2011).
Haran, J., Koutroumpa, F., Magnoux, E., Roques, A. & Roux, R. Ghost mtDNA haplotypes generated by fortuitous Numts can deeply disturb infra-specific genetic diversity and phylogeographic pattern. Journal of Zoological Systematics and Evolutionary Research 53, 109–115 (2015).
Medeiros, J. F. et al. A field trial of a PCR-based Mansonella ozzardi diagnosis assay detects high-levels of submicroscopic M. ozzardi infections in both venous blood samples and FTA card dried blood spots. Parasites & Vectors 8, 1–8 (2015).
Post, R. J. et al. The morphological discrimination of microfilariae of Onchocerca volvulus from Mansonella ozzardi. Parasitology 127(1), 21–27 (2003).
Ta-Tang, T. H. et al. Nested PCR to detect and distinguish the sympatric filarial species Onchocerca volvulus, Mansonella ozzardi and Mansonella perstans in the Amazon Region. Memórias do Instituto Oswaldo Cruz 105, 823–8 (2010).
Coil, D., Jospin, G. & Darling, A. E. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31(4), 587–9 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009).
Bernt, M. et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution. 69(2), 313–9 (2013).
Laslett, D. & Canbäck, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24(2), 172–5 (2008).
Carver, T., Harris, S. R., Berriman, M., Parkhill, J. & McQuillan, J. A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28(4), 464–9 (2012).
Alikhan, N. F., Petty, N. K., Ben-Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12(402), 1–10 (2011).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 25, 876–4882 (1997).
Nicholas, K. B., Nicholas, H. B. & Deerfield, D. W. GeneDoc Analysis and Visualization of Genetic Variation. Embnet news 4 (1997).
Felsenstein, J. PHYLIP (Phylogeny Inference Package). Version 3.6. Distributed by the Author. Department of Genetics, University of Washington, Seattle, USA (2002).
Yilmaz, E. et al. The Mitochondrial Genomes of the Zoonotic Canine Filarial Parasites Dirofilaria (Nochtiella) repens and Candidatus Dirofilaria (Nochtiella). Honkongensis Provide Evidence for Presence of Cryptic Species. PLOS Neglected Tropical Diseases 10, 0005028 (2016).
Lefoulon, E. et al. Shaking the Tree: Multi-locus Sequence Typing Usurps Current Onchocercid (Filarial Nematode) Phylogeny. PLOS Neglected Tropical Diseases 9, e0004233 (2015).
Arrospide, N. et al. Atypical microfilaria in coinfection with Mansonella ozzardi and Plasmodium vivax in Peruvian amazon. Revista Peruana de Medicina Experimental y Salud Pública 26, 408–416 (2009).
Ta-Tang, T. H. et al. Atypical Mansonella ozzardi Microfilariae from an Endemic Area of Brazilian Amazonia. The American Journal of Tropical Medicine and Hygiene 95, 629–32 (2016).
Mourembou, G. et al. Mansonella, including a Potential New Species, as Common Parasites in Children in Gabon. PLOS Neglected Tropical Diseases 9, e0004155 (2015).
Crainey, J. L. et al. The mitogenome of Onchocerca volvulus from the Brazilian Amazonia focus. Memórias do Instituto Oswaldo Cruz 111, 79–81 (2016).
Weil, G. J. & Ramzy, R. M. Diagnostic tools for filariasis elimination programs. Trends in Parasitology 23(2), 78–82 (2007).
Wanji, S. et al. Further evidence of the cross-reactivity of the Binax NOW® Filariasis ICT cards to non-Wuchereria bancrofti filariae: experimental studies with Loa loa and Onchocerca ochengi. Parasites & Vectors 9, 1–10 (2016).
Pion, S. & Chesnais, C. Loasis. In Arthropod borne diseases (ed. Marcondes, C. B.) 427–444 (Springer International Publishing, 2017).
Morales-Hojas, R. et al. Characterisation of nuclear ribosomal DNA sequences from Onchocerca volvulus and Mansonella ozzardi (Nematoda: Filarioidea) and development of a PCR-based method for their detection in skin biopsies. International Journal for Parasitology 31, 169–77 (2001).
Ballard, J. W., Chernoff, B. & James, A. C. Divergence of mitochondrial dna is not corroborated by nuclear dna, morphology, or behavior in. Drosophila simulans. Evolution 56(3), 527–45 (2002).
Hurst, G. D. & Jiggins, F. M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc Biol Sci. 272(1572), 1525–1534 (2005).
Pentinsaari, M., Salmela, H., Mutanen, M., & Roslin T. Molecular evolution of a widely-adopted taxonomic marker (COI) across the animal tree of life. Scientific Reports, 6 (35275), 1–12 (2016).
Lamberton, P. H. et al. Onchocerciasis transmission in Ghana: Persistence under different control strategies and the role of the simuliid vectors. PLOS Neglected Tropical Diseases 9, e0003688 (2015).
Miraldo, A., Hewitt, G. M., Dear, P. H., Paulo, O. S. & Emerson, B. C. Numts help to reconstruct the demographic history of the ocellated lizard (Lacerta lepida) in a secondary contact zone. Molecular Ecology 21, 1005–18 (2012).
Acknowledgements
The authors would like to gratefully acknowledge financial support from Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM), which provided financial support for this work with the grant award: 062.02005/2014. We would also like to gratefully acknowledge financial support from the Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM) and Programa Pesquisa Sistema Único de Saúde (PPSUS) which provided additional financial support with grant award: 062.00647/2014.
Author information
Authors and Affiliations
Contributions
S.L.B., A.C.P.V. designed the study; S.L.B. and F.A.C.P. secured funding. F.A.C.P. and J.F.M. collected and identified the samples. T.T.R.S., Y.V.S., M.A.B. performed all the laboratory work. J.L.C. and M.A.M. performed the D.N.A. sequence analysis. J.L.C. and A.C.P.V. drafted and edited the manuscript. All author saw and approved the final draft of the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crainey, J.L., Marín, M.A., Silva, T.R.R.d. et al. Mansonella ozzardi mitogenome and pseudogene characterisation provides new perspectives on filarial parasite systematics and CO-1 barcoding. Sci Rep 8, 6158 (2018). https://doi.org/10.1038/s41598-018-24382-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24382-3
This article is cited by
-
Development and validation of a long-read metabarcoding platform for the detection of filarial worm pathogens of animals and humans
BMC Microbiology (2024)
-
Genetic variability, cryptic species and phylogenetic relationship of six cyathostomin species based on mitochondrial and nuclear sequences
Scientific Reports (2021)
-
Detection of DNA of filariae closely related to Mansonella perstans in faecal samples from wild non-human primates from Cameroon and Gabon
Parasites & Vectors (2020)
-
First report of an Onchocercidae worm infecting Psychodopygus carrerai carrerai sandfly, a putative vector of Leishmania braziliensis in the Amazon
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
