
Abstract
Immune responses to parasitic pathogens are affected by the host physiological condition. High-density lipoprotein (HDL) and low-density lipoprotein (LDL) are transporters of lipids between the liver and peripheral tissues, and modulate pro-inflammatory immune responses. Pathogenic mycobacteria are parasitic intracellular bacteria that can survive within macrophages for a long period. Macrophage function is thus key for host defense against mycobacteria. These basic facts suggest possible effects of HDL and LDL on mycobacterial diseases, which have not been elucidated so far. In this study, we found that HDL and not LDL enhanced mycobacterial infections in human macrophages. Nevertheless, we observed that HDL remarkably suppressed production of tumor necrosis factor alpha (TNF-α) upon mycobacterial infections. TNF-α is a critical host-protective cytokine against mycobacterial diseases. We proved that toll-like receptor (TLR)-2 is responsible for TNF-α production by human macrophages infected with mycobacteria. Subsequent analysis showed that HDL downregulates TLR2 expression and suppresses its intracellular signaling pathways. This report demonstrates for the first time the substantial action of HDL in mycobacterial infections to human macrophages.
Similar content being viewed by others


Introduction
Low-density lipoprotein (LDL)-cholesterol (LDL-C) transfers cholesterol from the liver to peripheral tissues. However, a high level of LDL-C is a risk factor for cardiovascular diseases1,2 because it initiates atherosclerosis, leading to peripheral inflammations including the production of inflammatory cytokines and accumulation of macrophages and activated T cells. High-density lipoprotein (HDL), on the other hand, inversely transfers cholesterol from peripheral tissues to the liver. Indeed, high levels of serum HDL-cholesterol is correlated with a reduced risk of atherosclerosis and cardiovascular diseases3,4,5,6. When macrophages are filled with cholesterol, they become foam cells, which trigger massive inflammation during atherosclerosis. HDL removes cholesterol from macrophages through lipid transporter proteins, such as, ABCA17,8, ABCG18,9 and SR-B110. This is considered as a part of the mechanism of anti-inflammatory effects by HDL. However, accumulated evidence also suggests a direct role of HDL in the suppression of inflammation11,12,13,14,15. It is thus likely that LDL and HDL have impacts on human health status concomitant with inflammation, such as, in infectious diseases.
Mycobacterial infections are still a serious threat to human health, especially Mycobacterium tuberculosis complex, an etiologic agent of tuberculosis (TB), which is responsible for the highest mortality among all single pathogens. The World Health Organization (WHO) estimated that 10.4 million people newly developed TB and 1.7 million people died from this disease in 2015, as indicated in the most recent report (WHO; Global Tuberculosis Report, 2017).
M. tuberculosis is an intracellular pathogen that is well adapted to ensure its survival in macrophages. Therefore, the function of macrophages and the pro-inflammatory cytokines that activate them are critical for the host defense16. A key pro-inflammatory cytokine, tumor necrosis factor alpha (TNF-α), activates macrophages and is essential for granuloma formation. A granuloma is the hallmark of mycobacterial infections17. It is a roundish immunopathological structure made up of activated macrophages, which prevent the dissemination of mycobacteria. The significant role of TNF-α in granuloma formation and hence TB control in humans was proven with the administration of TNF-α-neutralizing therapy, which disrupted TB granuloma and increased TB reactivation18.
Thus, activated macrophages participate in the prevention of TB progression. However, M. tuberculosis can persist without complete sterilization, accounting for the huge TB reservoir19,20,21. During persistent infections, M. tuberculosis uses cholesterol as a carbon source22,23,24,25,26. Host cholesterol is also essential for phagocytosis of mycobacteria by macrophages27. However, the immunomodulatory effects of cholesterol transporters, LDL and HDL, on mycobacterial diseases remain to be elucidated. In this study, we assessed the action of LDL and HDL on mycobacteria-infected human macrophages.
Results
Mycobacteria-infected human macrophages produce a large amount of TNF-α, which is suppressed by HDL
We differentiated the THP1 human acute monocytic cell line to macrophages by using phorbol 12-myristate 13-acetate (PMA) (THP1 macrophages). Macrophages were then cultured with or without adding varying doses of HDL or LDL (5–50 µg/ml) for 24 hours. We confirmed no significant differences in the viability rates of cells by addition of 5 to 50 µg/ml HDL or LDL, based on the results of the trypan blue-exclusion assays or assessment of cytoplasmic lactate dehydrogenase (LDH) enzyme activity in the culture medium.
To assess the effects of human plasma-derived HDL and LDL on mycobacteria infection of human macrophages, THP1 macrophages were infected with M. tuberculosis complexes, such as Mycobacterium bovis BCG (BCG) or M. tuberculosis H37Rv. Twenty-four hours after infection, we assessed inflammatory responses of the macrophages by measuring the levels of cytokines, including granulocyte/macrophage colony-stimulating factor (GM-CSF), IFN-γ, interleukin (IL)-2, IL-4, IL-6, IL-8, IL-10 and TNF-α, secreted in the culture supernatant.
Upon infection with BCG, we found a robust production of TNF-α (14820.02 ± 165.82 pg/ml), whose level was remarkably reduced with HDL treatment (Supplementary Fig. S1d). We found insignificant levels of IL-6 (76.64 ± 15.797 pg/ml) and IL-10 (40.03 ± 5.404 pg/ml) production (Supplementary Fig. S1a and b) and moderate levels of IFN-γ production (287.266 ± 4.738 pg/ml) (Supplementary Fig. S1c), all of whose expression levels were suppressed with HDL treatment. However, LDL suppressed the production of TNF-α and IFN-γ, but its effect was lower than that of HDL treatment, and it did not suppress the production of IL-6 or IL-10 (Supplementary Fig. S1a and b). The levels of other tested cytokines, such as IL-4, GM-CSF, IL-2 or IL-8, were below the detection limits in all samples.
Similar to infection with BCG, we found a high level of TNF-α production upon M. tuberculosis infection (9057.91 ± 219.07 pg/ml, Fig. 1e). We also found moderate levels of IL-6 (498.01 ± 4.78 pg/ml), IL-10 (1203.29 ± 168 pg/ml) and IFN-γ (1046.18 ± 16.83 pg/ml) production, and an insignificant level of IL-4 production (79.16 ± 2.6 pg/ml) (Fig. 1a–d). Notably, treatment with HDL significantly reduced the production of all of these cytokines in a dose-dependent manner, except for IL-10. LDL treatment reduced the levels of IL-4, IL-6, IFN-γ and TNF-α production, but to a lesser extent than HDL treatment, and these changes did not show dose-responsiveness. The levels of other tested cytokines, such as GM-CSF, IL-2 and IL-8, were below the detection limits in all samples. These data suggest that among the tested cytokines, TNF-α is the most abundantly produced upon infection of THP1 macrophages with M. tuberculosis complex and HDL suppresses its production in a dose-dependent manner.

Effects of HDL and LDL on the expression of pro-inflammatory cytokines by M. tuberculosis-infected THP1macrophages. THP1-macrophages were cultured with or without adding HDL or LDL (5–50 µg/ml) for 24 hours. The treated macrophages were then infected with M. tuberculosis (multiplicity of infection = 10) for 24 hours (n = 4). The amounts of IL-4 (a), IL-6 (b), IL-10 (c), IFN-gamma (d), and TNF-α (e) from macrophages were measured using the Bio-Plex Multiplex System. ANOVA was used for test the differences in means.
We also assessed the effects of HDL on M. tuberculosis-infected human macrophages that were differentiated from CD14 positive monocytes derived from peripheral blood mononuclear cells (PBMC). As observed in THP1 macrophages, HDL but not LDL treatment of the M. tuberculosis-infected macrophages significantly suppressed TNF-α production by PBMC-derived human macrophages (Supplementary Fig. S2). This shows the common responses to HDL by both THP1 macrophages and PBMC-derived ones.
HDL but not LDL promotes entry of both BCG and M. tuberculosis into THP1 macrophages
HDL functions as a remover of cellular cholesterol in the periphery. In addition, previous reports showed that the removal of cholesterol suppressed BCG-infection of mouse macrophages27. This suggests the possibility that HDL inhibits phagocytosis of mycobacteria and in turn reduces infection-dependent TNF-α production. To investigate this possibility, we determined the infecting number of mycobacteria by counting colony forming units (CFUs) 24 hours after infection of THP1 macrophages. The data revealed that CFUs of BCG and M. tuberculosis in LDL-treated macrophages were similar to those in control macrophages. In contrast, HDL-treated macrophages harbored a significantly higher number of both BCG and M. tuberculosis (Supplementary Fig. S3). These data suggest that HDL enhances mycobacterial infections of macrophages contrary to prior expectations.
HDL inhibits the transcription of TNF-α and TNF receptors
Despite elevated mycobacterial entry into THP1 macrophage, the production of pro-inflammatory cytokines is inhibited by addition of HDL, suggesting some unknown systematic mechanism of HDL to this effect. We next analyzed whether the regulation occurs at the transcriptional level. In this experiment, the amount of TNF-α from BCG-infected THP1 macrophages in the control group, the group treated with 50 µg/mL HDL or LDL was 2067.2 pg/ml, 388.2 pg/ml, and 1161.3 pg/ml, respectively (Fig. 2a). The mRNA levels of TNF-α in BCG-infected THP1 macrophages pretreated with HDL were suppressed by 64% (Fig. 2b), suggesting the presence of transcriptional regulation.

Effect of HDL on the expression of TNF-α and its receptors in BCG-infected macrophages. (a) THP1 macrophages were cultured with or without adding HDL or LDL (50 µg/ml) for 24 hours. The treated macrophages were then infected with BCG (multiplicity of infection = 10) for 24 hours. The amounts of TNF-α in the cell culture supernatants were measured by ELISA. This assay was repeated three times. (b–d) THP1 macrophages were cultured with or without adding HDL or LDL (50 µg/ml) for 24 hours and were then infected with BCG (multiplicity of infection = 10) for 6 hours. TNF-α (b), TNFR-1 (c), and TNFR-2 (d) mRNA expression levels were quantified using real-time PCR (n = 3) and normalized to GAPDH expression. ANOVA was used to test for the differences of in means.
Prior work indicated that TNF-α transcription is regulated by autocrine systems through the activation of the NF-κB pathway by the bonding of TNF-α with TNF receptors (TNFR-1 and TNFR-2)28. We evaluated the effects of HDL on TNFR-1 and TNFR-2 mRNA expression levels. We found that these were both significantly suppressed by pretreatment with HDL (Fig. 2c and d), but to a lesser extent compared with LDL-treatment. This suggested the presence of a HDL-specific mechanism other than down-regulation of TNFRs.
HDL suppresses TLR2-dependent production of TNF-α by THP1 macrophages
A recent study showed that, in mouse bone marrow-derived macrophages, HDL induces transcription factor 3 (ATF3), which in turn suppresses the transcription of the genes encoding pro-inflammatory cytokines12. We, therefore, assessed the upregulation of ATF3 in BCG-infected THP1 macrophages. However, we could not find any significant increase in ATF3 with HDL-treatment compared with the untreated control and with LDL treatment (Supplementary Fig. S4).
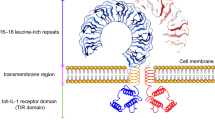
Stimulation of pattern recognition receptors, such as TLRs, is the predominant trigger of TNF-α transcription in mycobacteria-infected macrophages. TLR1/2, 2/6, 4 and 9 are involved in the recognition of mycobacterial components, and TLR2 is considered to be the most responsible receptor for induction of TNF-α29,30,31,32. We analyzed the effect of HDL on the mRNA levels of TLR2. In uninfected macrophages, HDL reduced the TLR2 mRNA level by around 80% compared with that in untreated or LDL-treated ones (Fig. 3a). Importantly, HDL treatment reduced the TLR2 mRNA level to around 40% in mycobacteria-infected macrophages compared with the untreated control (Fig. 3b). Flow cytometry data analysis showed that the level of surface TLR2 of BCG-infected THP1 macrophages was reduced from 55.8% to 22.4% by the treatment with HDL as shown in Fig. 3c.

HDL suppresses TLR2 expression of THP1 macrophages. (a) Macrophages were cultured with or without adding HDL or LDL (50 µg/ml) for 6 hours. The TLR2 mRNA expression levels were then quantified using real-time PCR (n = 3) and normalized to GAPDH expression. (b) Macrophages were cultured with or without adding HDL or LDL (50 µg/ml) for 24 hours and then infected with BCG (multiplicity of infection = 10) for 6 hours. The TLR2 mRNA expression level was quantified by real-time PCR (n = 3). ANOVA was used to test for the differences of in means. These assays were repeated twice. (c) THP1macrophages were cultured with (merged with pink) or without (filled with purple) addition of HDL (50 µg/ml) for 6 hours. Surface TLR2 was detected with PE-labeled anti-TLR2 antibody and analyzed by FACScan. The border of M1 and M2 was set at the peak of untreated THP1 cells stained by TLR2 antibody. Calculated % of cells in the M2 area of control cells and HDL-treated cells were 55.8% and 22.4%, respectively. (d) THP1 macrophages were cultured with or without addition of HDL (50 µg/ml) for 24 hours and were then stimulated with the TLR2 ligand Pam3CSK4 (50 ng/ml) for 24 hours. The amounts of TNF-α in the cell culture supernatants (n = 2) were measured by ELISA. ANOVA was used to test for the differences of in means. This assay was repeated twice.
We next assessed whether TLR2-dependent TNF-α production was impaired by HDL. We found that, indeed, HDL reduced TNF-α production by macrophages stimulated with 50 ng/ml of PAM3CSK4, a ligand for the TLR2/TLR1 homodimer (Fig. 3d).
To confirm that TLR2-mediated signaling pathways were impaired by HDL, we analyzed the activation (phosphorylation) of NF-κB (p65) and p38, ERK and JNK mitogen-activated protein kinases (MAPKs) by immunoblot of the cell lysates derived from HDL-treated BCG-infected macrophages. The data indicated 29%, 38% and 45% reduced phosphorylation of P65, P38 and JNK, respectively. This showed that HDL indeed suppressed activation of both major cellular signaling pathways for the transcription of the TNF-α gene30,33,34,35 (Fig. 4 and Supplementary Fig. S5). By contrast, HDL did not influence the phosphorylation of ERK.

HDL impairs activation of TLR2-mediated intracellular signalings. THP1 macrophages were cultured with or without (control) adding HDL (50 µg/ml) for 24 hours. The macrophages were then infected with BCG (multiplicity of infection = 10) for 15 minutes. Immunoblot of p65 phosphorylation (phospho-p65), total p65, p38 phosphorylation (phospho-p38), total p38, ERK phosphorylation (phospho-ERK), total ERK, JNK phosphorylation (phospho-JNK) and total JNK (relative to total β-actin) were detected. Each whole cell lysate was fractionated by SDS-PAGE and transferred on a membrane.Immunoblot experiments were then performed to validate the level of each target protein. The western blot bands of phospho-p65 (65 kDa) total p65 (65 kDa), phospho-p38 (43 kDa), total p38 (43 kDa), phospho-ERK (44 and 42 kDa), total ERK (44 and 42 kDa), phospho-JNK (46 and 54 kDa) and total JNK(46 and 54 kDa) and total beta-actin (43 kDa)are displayed in the figure. Wholemembranes were also displayed in Supplemental Fig. S5. The immunoblots are representative of three independent experiments.
TLR2 is responsible for TNF-α production by THP1 macrophages upon infection with mycobacteria
TLR2 is a receptor responsible for TNF-α production upon mycobacterial infection of mouse macrophages36,37. While its similar role in human macrophages has been implied38, its exact responsibility has not been elucidated36,37,38. To establish its exact role, we generated TLR-2 knock-out (KO) THP1 cells using the CRISPR-Cas9 system (Supplementary Fig. S6). As expected, the TLR-2 KO THP1 macrophage could not produce TNF-α when stimulated with PAM3CSK4. In this experimental setting, we observed that TNF-α production was impeded because of TLR2-deficiency upon BCG (Fig. 5a) and M. tuberculosis (Fig. 5b) infection. This demonstrated the critical role of TLR2 in TNF-α production by mycobacteria-infected human macrophages.

TLR2 is critical for TNF-α production from THP1 macrophages upon mycobacterial infection. (a) THP1 macrophages and its TLR2-KO cells were stimulated with the TLR2 ligand, Pam3CSK4 (50 ng/ml) or infected with BCG (multiplicity of infection = 10) for 24 hours, and the amount of TNF-α in the cell culture supernatant (n = 3) was measured by ELISA. (b) THP1 macrophages and TLR2-KO cells were infected M. tuberculosis (multiplicity of infection = 10) for 24 hours, and the amount of TNF-α in the cell culture supernatant (n = 3) was measured by ELISA. ANOVA was used to test for the differences of in means.
Discussion
This study demonstrates for the first time that HDL dose-dependently suppresses the production of pro-inflammatory cytokines, such as, IL-6, TNF-α and IFN-γ by THP1 macrophages infected with M. tuberculosis (Fig. 1). The same was true when macrophages differentiated from CD14+ monocytes derived from human blood were used (Supplementary Fig. S2). LDL treatment also reduced the production of cytokines but its effect was lower than that of HDL and it did not show dose responsiveness (Fig. 1), suggesting its minor function or non-functional artifacts. TNF-α was the most abundantly produced cytokine among those tested. It plays a key role in the immune defense during both active and latent TB39,40,41,42,43,44. IL-6 and IFN-γ also contribute to protection against mycobacterial diseases40,45,46,47,48. Inversely, however, massive production of these pro-inflammatory cytokines is harmful to the body. Our data thus suggest that HDL has the potential to modulate immune responses against mycobacterial infections.
HDL-dependent suppression of pro-inflammatory cytokine production was not owing to inhibition of mycobacterial infection of macrophages. Rather, HDL treatment enhanced these infections (Supplementary Fig. S3). In the presence of sera, mycobacteria enters macrophages mainly through the complement receptor49. The contribution of other receptors, such as mannose receptor and the scavenger receptor B1 (SR-B1) have been reported as well29,50. We investigated the expression of these receptors but could not find any upregulation in HDL-treated THP1 macrophages. It is thus considered that other uncharacterized receptors may contribute to this process. The other possibility relates to the dynamic quantitative change of the macrophage membrane fluidity as a result of a change in cholesterol levels that should affect phagocytosis51.
Recently ATF3 was proposed to be the master transcriptional regulator in HDL-dependent suppression of pro-inflammatory cytokines in mice bone marrow-derived macrophages12. However, we could not find any upregulation of ATF3 in THP1 macrophages treated with HDL (Supplementary Fig. S4). This discrepancy may be due to the difference in responses between mouse and human macrophages.
We investigated other possible molecular mechanisms to explain the HDL-dependent suppression of TNF-α production. TLR2 is a host-protective molecule against mycobacterial infections in both mouse and human52,53,54,55,56 and is considered a predominant receptor for TNF-α production. We thus next focused on TLR238. We found that HDL suppressed TLR2 expression (Fig. 3), cellular response to the TLR2-specific ligand (Fig. 3d) and TLR2-related cellular signaling pathways (Fig. 4). We then proved that TLR2 was a key receptor for TNF-α production upon mycobacterial infection by creating a TLR2-KO THP1 cell (Fig. 5). Taken together these data indicate the significant role of TLR2 in TNF-α production upon mycobacterial infection and the involvement of down-regulation of TLR2 by HDL in the suppression of TNF-α production by mycobacteria-infected THP1 macrophages.
Previously, Tang et al. showed that apolipoprotein A-1, a component of HDL, interacts with ABCA1 and activates STAT3, which in turn suppresses the production of pro-inflammatory cytokines57. ABCA1 transcription was remained in HDL-treated THP1 macrophages, although it was significantly reduced upon LDL treatment (Fig. S7). This implies the possibility of interaction between down-regulation of TLR2 and activation of the JAK/STAT3 signaling pathway, which should be clarified in future studies.
There are few epidemiological cohort studies that estimate the relationship between HDL levels and tuberculosis. Two papers showed that decrease of HDL-cholesterol levels was correlated with severity of the disease in active TB patients58. Another study showed that the level of HDL-c, but not LDL-c or total cholesterol was significantly lower in 115 TB patients compared to 70 pneumonia patients and 30 healthy control59. In contrast, hypercholesterolemia impairs protective immunity to tuberculosis in mice model60. A recent adolescent cohort study with 6,363 enrolled participants identified the upregulation of apolipoprotein A-1 as a risk factor of TB progression from latent infection61. It is also known that M. tuberculosis uses a host cholesterol as a major carbon source during infection22,23,24,25,26, implying that higher cellular cholesterol levels may activate TB disease progression. Taken together, these suggest the important but controversial role of HDL in the determination of TB disease outcome. Further studies are in demand to clarify the effect of HDL and LDL in clinical situations of mycobacterial diseases including initial infection, asymptomatic states, and active diseases.
HDL is a highly condensed lipoprotein fraction in plasma. The most abundant apolipoprotein is A-I and the second one is apo A-II, but it is composed of over different 80 kinds of proteins and their variants. To identify which components and sub-fractions of HDL are responsible for their activity are next important issues to be clarified.
The downregulation of TLR2 by HDL interferes with the general recognition of pathogen-associated molecular patterns and is not limited to mycobacterial diseases. It is thus likely that HDL modulates host protective responses against other pathogens. While HDL is related to lower risk of vascular disorders contributing to human longevity, this study suggests the importance of taking into account the risk of immune suppression posed by HDL.
Methods
Isolation of HDL and LDL from healthy donors
HDL and LDL were isolated by a method previously developed62. Briefly, freshly drawn blood samples were collected (1 mM EDTA as anti-coagulant) and centrifuged at 2,000 × g for 15 min. Chylomicrons were then removed from plasma by additional centrifugation at 100,000 × g for 10 min. For each sample, four volumes of plasma were mixed with one volume of OptiPrep™ (Axis-Shield, Dundee, Scotland), and 2.8 ml of the resulting mixture was transferred to an OptiPrep™ tube. Solution B (0.85 g of NaCl dissolved in 50 ml water) was layered on top of this mixture, followed by a layer of 10 ml of 100-mM Hepes stock solution. After sealing the tubes, the samples were centrifuged at approximately 350,000 × g for 3 hours at 16 °C. The HDL and LDL fractions were collected by using disposable syringes with 23-gauge needles.
Cells
THP1 cells, a human acute monocytic cell line that was purchased from the Human Science Foundation (Tokyo, Japan), were cultured in RPMI-1640 supplemented with 10% HI-FBS (Thermo Fisher Scientific, Waltham, MA, USA). Differentiation was achieved by re-suspending the cells at 8 × 105 cells/ml in Dulbecco’s modified Eagle’s medium (Wako) supplemented with 10% heat-inactivated fetal bovine serum and penicillin-streptomycin with the addition of 100 nM phorbol 12-myristate 13-acetate (Sigma) for 48 hours. Non-adherent cells were removed by aspiration of the supernatant followed by replacement with fresh medium.
The human blood PBMC was obtained by using Optiprep (Axis-Shield, Dundee, Scotland) in accordance with the manufacturer’s instructions. CD14+ monocytes were then isolated from PBMC by the AutoMacs separation system using anti-CD14 magnetic microbeads (Miltenyi Biotec, Germany). The CD14+ human monocytes were differentiated to macrophages by commercially available macrophage differentiation kit (R&D systems, Minnesota).
Reagents
Salmonella-derived LPS and Pam3CSK4 were purchased from Sigma and R&D Systems (Minneapolis, USA), respectively.
Cell viability
We assessed the cell viability by using two methods: 1) trypan blue-exclusion assay; and 2) Cytotoxicity Detection Kitplus, which measures the LDH release from dead cells. Differentiated macrophages were cultured with or without adding HDL or LDL (5–50 µg/ml) for 24 hours. In trypan blue-exclusion assays, cells were stained with trypan blue solution, loaded into a hemocytometer and immediately examined under a microscope. When using the Cytotoxicity Detection Kitsplus (LDH), the cytoplasmic LDH enzyme activity in the culture medium was measured according to the manufacturer’s instructions.
Measurement of mycobacterial infection by CFU assays
In vitro infection experiments using BCG and M. tuberculosis were conducted in BSL2 and BSL 3 level facilities in the Department of Bacteriology, Niigata University School of Medicine after approval by the Institutional Biosafety Committee of Niigata University. Differentiated macrophages were infected with M. tuberculosis strain H37Rv or BCG at a multiplicity of infection of 10 for 3 hours, washed, and lysed with PBS containing 0.5% Triton X-100. CFU assays were performed by harvesting serially diluted suspensions on Middlebrook 7H11 agar plates supplemented with oleic, albumin, dextrose, and catalase (OADC) at 37 °C for 3 weeks to quantify the number of mycobacteria that had infected the macrophages.
Quantitative measurement of TNF-α, TNFR-1, TNFR-2, TLR2, and ATF3 mRNA expression levels in THP1 macrophages
Differentiated macrophages were harvested, and total RNA was extracted using TRIzol® (Thermo Fisher Scientific) according to the manufacturer’s instructions. The resulting RNA was then treated with RNase-free DNase (Ambion, California, USA) to remove any potential DNA contamination. After being reverse-transcribed to cDNA using the Super Script First Strand Synthesis System for reverse-transcription PCR (Invitrogen, Massachusetts, USA), triplicate cDNA aliquots were amplified with sequence-specific primers and SYBR green (Applied Biosystems by Life Technologies, California, USA) using 7500 Fast Real-Time PCR Systems (Applied Biosystems by Life Technologies). All reactions were repeated independently at least three times to ensure the reproducibility of the results. All primers were purchased from Sigma (Supplementary Table S1).
Measurement of secreted TNF-α level by enzyme-linked immunoabsorbent assay (ELISA)
TNF-α levels in the cell-culture supernatant were measured using human TNF-α ELISA kits (R&D Systems) according to the manufacturer’s instructions.
Measurement of secreted IL-4, IL-6, IL-10, IFN-γ and TNF-α level by the Bio-Plex Multiplex System
IL-4, IL-6, IL-10, IFN-γ and TNF-α levels in cell-culture supernatants were measured using the Bio-Plex Multiplex System (Bio-Rad, California, USA) according to the manufacturer’s instructions.
Flow cytometry
Following treatment with HDL (50 µg/ml), cells were washed with ice-cold PBS and then fixed with 80% methanol for 5 min and washed again with ice-cold PBS. Cells were then incubated with unlabeled anti-Fc receptor antibody (eBioscience, San Diego, USA) in PBS in the dark at 4 °C for 20 min to block the Fc receptor. The cells were subsequently incubated with PE-labeled anti-human CD282 (TLR2) (clone: TL2.1) (eBioscience) in PBS at room temperature in the dark for 20 min. Cells were then washed twice with ice-cold PBS, resuspended in ice-cold PBS, and analyzed on a flow cytometer (FACSCalibur HG, Becton Dickinson). An isotype-matched antibody, mouse IhG2aκ Iso Control PE (clone: eBM2a) (eBioscience), of irrelevant specificity was used as a control. The control cells were analyzed using FACScan (BD Bioscience, Franklin Lakes, USA).
Gene editing of TLR2 by CRISPR-Cas9 system
To generate TLR2-KO THP1 cells, the CRISPR-Cas9 system was applied. Niigata University Recombinant DNA Advisory Committee approved CRISPR/Cas9 gene knock out of TLR2 gene in THP1 cells in the BSL2 laboratory of Department of Bacteriology, Niigata University School of Medicine. The sequence of guide RNA (gRNA) synthesis, which targets the human TLR2 gene, was GACTGTACCCTTAATGGAGT(TGG). It was inserted into pSpCas9 (BB)-2A-Puro (PX459) V2.0 (Addgene, Cambridge, MA, USA) as previously described63. After insertion of the gRNA sequence, the pX459 vector was transfected into THP1 cells by electroporation with Gene Pulser Xcell (Bio-Rad, Hercules, CA, USA). For electroporation, THP1 cells were collected by centrifugation from culture and were suspended by the serum-free RPMI-1640 medium at 1 × 107 cells/ml. In a 0.4-cm cuvette, 10 μg of plasmid vector and 0.4 ml of cell suspension were mixed and incubated for 10 min, and then pulsed once with 260 V and 1050 μF. After electroporation, cells were grown in RPMI-1640 medium supplemented with 10% of FBS. Forty-eight hours after electroporation, cells were selected by 1 μg of puromycin (Invivogen, San Diego, CA, USA) for 48 hours. To obtain a monoclonal cell population, cloning by limiting dilution was performed. Gene editing of TLR2 was confirmed by the PAGE-based genotyping protocol64 and by determining the DNA sequence of a targeted region of the TLR2 exon.
Immunoblotting of p65, p38, ERK1/2 and JNK
More than 5 × 106 differentiated THP1 macrophages were lysed with 1 Xlysis buffer containing 1% NP-40, 20 mM Tris-HCl pH 7.5, 2 mM EDTA and 150 mM NaCl, supplemented with a protease inhibitor cocktail (Nacalai Tesque) and 100 mM NaF, on ice for 10 min. The samples were then centrifuged at 16,000 g for 5 min at 4 °C, and the supernatant was analyzed by immunoblotting of whole cell lysates. A total of 5 µg of each lysate, of which the protein concentration was determined by Advanced Protein Assay (Cytoskeleton, Inc.), was run on 12.5% SDS-PAGE gel. After SDS-PAGE, fractionated proteins were transferred onto PVDF membranes (Millipore) and blocked in Tris-buffered saline (pH 7.5) containing 1% (wt/vol) skim milk and 0.05% Tween 20 before overnight incubation with each specific primary antibody. The following antibodies were purchased from Cell Signaling TECHNOLOGY and used in this study: anti-phospho-NF-κB p65 (Ser536) (93H1) Rabbit mAb (1:1000 dilution), anti-NF-κB p65 (D14E12) XP Rabbit mAb (1:1000 dilution), anti-phospho-p38 MAP Kinase (Thr180/Tyr182) antibody (1:1000 dilution), anti-p38 MAPK antibody (1:1000 dilution), anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP Rabbit mAb (1:2000 dilution), anti-p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb (1:1000 dilution), anti-phospho-SAPK/JNK (Thr183/Tyr185) (81E11) Rabbit mAb (1:1000 dilution), anti-SAPK/JNK antibody (1:1000 dilution) and anti-β-Actin (C4): sc-47778 antibody (1:2500 dilution). The membranes were then washed and incubated with appropriate secondary antibodies, and the antibody reactions were detected with Immobilon™ western (Millipore).
Data analysis
ANOVA was used to analyze in vitro assay data. All statistical analyses were carried out using SPSS (ver. 21, IBM, NY, USA), and p-values less than 0.05 were considered significant65,66,67,68,69.
References
Kannel, W. B., Dawber, T. R., Kagan, A., Revotskie, N. & Stokes, J. 3rd. Factors of risk in the development of coronary heart disease–six year follow-up experience. The Framingham Study. Annals of internal medicine 55, 33–50 (1961).
Wilson, P. W. et al. Prediction of coronary heart disease using risk factor categories. Circulation 97, 1837–1847 (1998).
Gordon, T., Castelli, W. P., Hjortland, M. C., Kannel, W. B. & Dawber, T. R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. The American journal of medicine 62, 707–714 (1977).
Gordon, D. J. & Rifkind, B. M. High-density lipoprotein–the clinical implications of recent studies. The New England journal of medicine 321, 1311–1316, https://doi.org/10.1056/nejm198911093211907 (1989).
Assmann, G., Schulte, H., von Eckardstein, A. & Huang, Y. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis 124(Suppl), S11–20 (1996).
Tanne, D., Yaari, S. & Goldbourt, U. High-density lipoprotein cholesterol and risk of ischemic stroke mortality. A 21-year follow-up of 8586 men from the Israeli Ischemic Heart Disease Study. Stroke 28, 83–87 (1997).
Oram, J. F., Lawn, R. M., Garvin, M. R. & Wade, D. P. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. The Journal of biological chemistry 275, 34508–34511, https://doi.org/10.1074/jbc.M006738200 (2000).
Wang, X. et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. Journal of Clinical Investigation 117, 2216–2224, https://doi.org/10.1172/Jc32057 (2007).
Wang, N., Lan, D., Chen, W., Matsuura, F. & Tall, A. R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proceedings of the National Academy of Sciences of the United States of America 101, 9774–9779, https://doi.org/10.1073/pnas.0403506101 (2004).
Zhang, Y. et al. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. The Journal of clinical investigation 115, 2870–2874, https://doi.org/10.1172/jci25327 (2005).
Cockerill, G. W. et al. Elevation of plasma high-density lipoprotein concentration reduces interleukin-1-induced expression of E-selectin in an in vivo model of acute inflammation. Circulation 103, 108–112 (2001).
De Nardo, D. et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nature Immunology 15, 152–160, https://doi.org/10.1038/Ni.2784 (2014).
Libby, P., Ridker, P. M. & Hansson, G. K. Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325, https://doi.org/10.1038/nature10146 (2011).
Murphy, A. J., Westerterp, M., Yvan-Charvet, L. & Tall, A. R. Anti-atherogenic mechanisms of high density lipoprotein: effects on myeloid cells. Biochimica et biophysica acta 1821, 513–521, https://doi.org/10.1016/j.bbalip.2011.08.003 (2012).
Norata, G. D., Pirillo, A., Ammirati, E. & Catapano, A. L. Emerging role of high density lipoproteins as a player in the immune system. Atherosclerosis 220, 11–21, https://doi.org/10.1016/j.atherosclerosis.2011.06.045 (2012).
Flynn, J. L. & Chan, J. Immunology of tuberculosis. Annual review of immunology 19, 93–129, https://doi.org/10.1146/annurev.immunol.19.1.93 (2001).
Flynn, J. L. et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572 (1995).
Keane, J. et al. Tuberculosis associated with infliximab, a tumor necrosis factor (alpha)-neutralizing agent. New England Journal of Medicine 345, 1098–1104, https://doi.org/10.1056/Nejmoa011110 (2001).
Manabe, Y. C. & Bishai, W. R. Latent Mycobacterium tuberculosis-persistence, patience, and winning by waiting. Nature medicine 6, 1327–1329, https://doi.org/10.1038/82139 (2000).
Getahun, H. et al. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. The European respiratory journal 46, 1563–1576, https://doi.org/10.1183/13993003.01245-2015 (2015).
Getahun, H., Matteelli, A., Chaisson, R. E. & Raviglione, M. Latent Mycobacterium tuberculosis Infection. New England Journal of Medicine 372, 2127–2135, https://doi.org/10.1056/Nejmra1405427 (2015).
Pandey, A. K. & Sassetti, C. M. Mycobacterial persistence requires the utilization of host cholesterol. Proceedings of the National Academy of Sciences of the United States of America 105, 4376–4380, https://doi.org/10.1073/pnas.0711159105 (2008).
Brzostek, A., Pawelczyk, J., Rumijowska-Galewicz, A., Dziadek, B. & Dziadek, J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. Journal of bacteriology 191, 6584–6591, https://doi.org/10.1128/jb.00488-09 (2009).
Griffin, J. E. et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS pathogens 7, e1002251, https://doi.org/10.1371/journal.ppat.1002251 (2011).
Marques, M. A. et al. The Essential Role of Cholesterol Metabolism in the Intracellular Survival of Mycobacterium leprae Is Not Coupled to Central Carbon Metabolism and Energy Production. Journal of bacteriology 197, 3698–3707, https://doi.org/10.1128/jb.00625-15 (2015).
Soto-Ramirez, M. D. et al. Cholesterol plays a larger role during Mycobacterium tuberculosis in vitro dormancy and reactivation than previously suspected. Tuberculosis (Edinburgh, Scotland) 103, 1–9, https://doi.org/10.1016/j.tube.2016.12.004 (2017).
Gatfield, J. & Pieters, J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science (New York, N.Y.) 288, 1647–1650 (2000).
Pekalski, J. et al. Spontaneous NF-kappaB activation by autocrine TNFalpha signaling: a computational analysis. PloS one 8, e78887, https://doi.org/10.1371/journal.pone.0078887 (2013).
van Crevel, R., Ottenhoff, T. H. & van der Meer, J. W. Innate immunity to Mycobacterium tuberculosis. Clinical microbiology reviews 15, 294–309, https://doi.org/10.1128/Cmr.15.2.294-309.2002 (2002).
Jo, E. K., Yang, C. S., Choi, C. H. & Harding, C. V. Intracellular signalling cascades regulating innate immune responses to Mycobacteria: branching out from Toll-like receptors. Cell Microbiol 9, 1087–1098, https://doi.org/10.1128/Cmr.15.2.294-309.2002 (2007).
Carmona, J. et al. Mycobacterium tuberculosis Strains Are Differentially Recognized by TLRs with an Impact on the Immune Response. PloS one 8, e67277, https://doi.org/10.1371/journal.pone.0067277 (2013).
Stamm, C. E., Collins, A. C. & Shiloh, M. U. Sensing of Mycobacterium tuberculosis and consequences to both host and bacillus. Immunological reviews 264, 204–219, https://doi.org/10.1111/imr.12263 (2015).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nat Rev Immunol 4, 499–511 (2004).
Kleinnijenhuis, J., Oosting, M., Joosten, L. A., Netea, M. G. & Van Crevel, R. Innate immune recognition of Mycobacterium tuberculosis. Clinical & developmental immunology 2011, 405310, https://doi.org/10.1155/2011/405310 (2011).
Rajaram, M. V. et al. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. Journal of immunology (Baltimore, Md.: 1950) 185, 929–942, https://doi.org/10.4049/jimmunol.1000866 (2010).
Harding, C. V. & Boom, W. H. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol 8, 296–307, https://doi.org/10.1038/nrmicro2321 (2010).
Saraav, I., Singh, S. & Sharma, S. Outcome of Mycobacterium tuberculosis and Toll-like receptor interaction: immune response or immune evasion? Immunol Cell Biol 92, 741–746, https://doi.org/10.1038/icb.2014.52 (2014).
Thoma-Uszynski, S. et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science (New York, N.Y.) 291, 1544–1547, https://doi.org/10.1126/science.291.5508.1544 (2001).
Bean, A. G. D. et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. Journal of Immunology 162, 3504–3511 (1999).
Flynn, J. L. et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. The Journal of experimental medicine 178, 2249–2254 (1993).
Kaneko, S. et al. Health and Demographic Surveillance System in the Western and coastal areas of Kenya: an infrastructure for epidemiologic studies in Africa. J Epidemiol 22, 276–285 (2012).
Mohan, V. P. et al. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: Possible role for limiting pathology. Infection and immunity 69, 1847–1855, https://doi.org/10.1128/Iai.69.3.1847-1855.2001 (2001).
Saunders, B. M. & Britton, W. J. Life and death in the granuloma: immunopathology of tuberculosis. Immunology and Cell Biology 85, 103–111, https://doi.org/10.1038/Sj.Icb.7100027 (2007).
Stenger, S. Immunological control of tuberculosis: Role of tumour necrosis factor and more. Annals of the Rheumatic Diseases 64, 24–28, https://doi.org/10.1136/Ard.2005.042531 (2005).
Cooper, A. M. et al. Disseminated tuberculosis in interferon gamma gene-disrupted mice. The Journal of experimental medicine 178, 2243–2247 (1993).
Flesch, I. E. & Kaufmann, S. H. Role of cytokines in tuberculosis. Immunobiology 189, 316–339, https://doi.org/10.1016/s0171-2985(11)80364-5 (1993).
Jouanguy, E. et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. The New England journal of medicine 335, 1956–1961, https://doi.org/10.1056/nejm199612263352604 (1996).
Ladel, C. H. et al. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infection and immunity 65, 4843–4849 (1997).
Ernst, J. D. Macrophage receptors for Mycobacterium tuberculosis. Infection and immunity 66, 1277–1281 (1998).
Aderem, A. & Underhill, D. M. Mechanisms of phagocytosis in macrophages. Annual review of immunology 17, 593–623, https://doi.org/10.1146/annurev.immunol.17.1.593 (1999).
Goluszko, P. & Nowicki, B. Membrane cholesterol: a crucial molecule affecting interactions of microbial pathogens with mammalian cells. Infection and immunity 73, 7791–7796, https://doi.org/10.1128/iai.73.12.7791-7796.2005 (2005).
Bafica, A. et al. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. The Journal of experimental medicine 202, 1715–1724, https://doi.org/10.1084/jem.20051782 (2005).
Drennan, M. B. et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol 164, 49–57 (2004).
Heldwein, K. A. et al. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J Leukoc Biol 74, 277–286 (2003).
Ogus, A. C. et al. The Arg753GLn polymorphism of the human toll-like receptor 2 gene in tuberculosis disease. The European respiratory journal 23, 219–223 (2004).
Schroder, N. W. et al. Heterozygous Arg753Gln polymorphism of human TLR-2 impairs immune activation by Borrelia burgdorferi and protects from late stage Lyme disease. Journal of immunology (Baltimore, Md.: 1950) 175, 2534–2540 (2005).
Tang, C., Liu, Y., Kessler, P. S., Vaughan, A. M. & Oram, J. F. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. The Journal of biological chemistry 284, 32336–32343, https://doi.org/10.1074/jbc.M109.047472 (2009).
Deniz, O. et al. Serum total cholesterol, HDL-C and LDL-C concentrations significantly correlate with the radiological extent of disease and the degree of smear positivity in patients with pulmonary tuberculosis. Clin Biochem 40, 162–166, https://doi.org/10.1016/j.clinbiochem.2006.10.015 (2007).
Sahin, F. & Yildiz, P. Distinctive biochemical changes in pulmonary tuberculosis and pneumonia. Arch Med Sci 9, 656–661, https://doi.org/10.5114/aoms.2013.34403 (2013).
Martens, G. W. et al. Hypercholesterolemia impairs immunity to tuberculosis. Infection and immunity 76, 3464–3472, https://doi.org/10.1128/iai.00037-08 (2008).
Zak, D. E. et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet 387, 2312–2322, https://doi.org/10.1016/s0140-6736(15)01316-1 (2016).
Graham, J. M. et al. A novel method for the rapid separation of plasma lipoproteins using self-generating gradients of iodixanol. Atherosclerosis 124, 125–135 (1996).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, https://doi.org/10.1038/nprot.2013.143 (2013).
Zhu, X. et al. An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci Rep 4, 6420, https://doi.org/10.1038/srep06420 (2014).
Ma, M. J. et al. Toll-like receptors, tumor necrosis factor-alpha, and interleukin-10 gene polymorphisms in risk of pulmonary tuberculosis and disease severity. Hum Immunol 71, 1005–1010, https://doi.org/10.1016/j.humimm.2010.07.009 (2010).
Jafari, M. et al. TheNRAMP1, VDR, TNF-alpha, ICAM1, TLR2 and TLR4 gene polymorphisms in Iranian patients with pulmonary tuberculosis: A case-control study. Infect Genet Evol 39, 92–98, https://doi.org/10.1016/j.meegid.2016.01.013 (2016).
Thuong, N. T. et al. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun 8, 422–428, https://doi.org/10.1038/sj.gene.6364405 (2007).
Velez, D. R. et al. Variants in toll-like receptors 2 and 9 influence susceptibility to pulmonary tuberculosis in Caucasians, African-Americans, and West Africans. Human genetics 127, 65–73, https://doi.org/10.1007/s00439-009-0741-7 (2010).
Nagi, S. et al. Risk factors and spatial distribution of Schistosoma mansoni infection among primary school children in Mbita District, Western Kenya. PLoS neglected tropical diseases 8, e2991, https://doi.org/10.1371/journal.pntd.0002991 (2014).
Acknowledgements
We are grateful to Ms. Yuko Kobayashi, Sara Matsumoto, and Shaban Amina Kaboso for their assistance, English editing, and heartfelt encouragement. This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Japanese Ministry of Health, Labour and Welfare (Research on Emerging and Re-emerging Infectious Diseases, Health Sciences Research Grants), the Japan Health Science Foundation, the United States-Japan Cooperative Medical Science Program against Tuberculosis and Leprosy, and the Collaborative Research Foundation of Otsuka. This paper is published with the permission of the Director of the Kenya Medical Research Institute.
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.O., S.M.N., S.H., and S.M. Formal analysis: M.I., M.N., Y.O., S.N., E.C., M.O.O., T.Y., K.O., T.O., F.M., and Y.K. Funding acquisition: S.H. and S.M. Investigation: M.I., M.N., Y.O., S.N., E.C., M.O.O., T.Y., K.O., T.O., F.M., S.H., and S.M. Methodology: K.O., T.O., and M.M. Project administration: S.K., Y.I., S.M.N., and S.H. Supervision: S.K., Y.I., S.M.N., and S.H. Validation: M.I., M.N., Y.O., S.M.N., S.H., and S.M. Writing – original draft: M.I., Y.O., T.Y., S.H., and S.M. Writing – review and editing: M.I., Y.O., T.Y., S.H., S.K., Y.I., and S.M.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Inoue, M., Niki, M., Ozeki, Y. et al. High-density lipoprotein suppresses tumor necrosis factor alpha production by mycobacteria-infected human macrophages. Sci Rep 8, 6736 (2018). https://doi.org/10.1038/s41598-018-24233-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-24233-1
This article is cited by
-
Associations between type 1 diabetes and pulmonary tuberculosis: a bidirectional mendelian randomization study
Diabetology & Metabolic Syndrome (2024)
-
Association between biomarkers of inflammation and dyslipidemia in drug resistant tuberculosis in Uganda
Lipids in Health and Disease (2024)
-
An Appraisal of the Preventive Effect of Statins on the Development of Graves’ Ophthalmopathy: A Hospital-Based Cohort Study
Ophthalmology and Therapy (2024)
-
Global-scale GWAS associates a subset of SNPs with animal-adapted variants in M. tuberculosis complex
BMC Medical Genomics (2023)
-
Monitoring IgG against Mycobacterium tuberculosis proteins in an Asian elephant cured of tuberculosis that developed from long-term latency
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
