
Abstract
Oxidative phosphorylation (OXPHOS) generates ROS as a byproduct of mitochondrial complex I activity. ROS-detoxifying enzymes are made available through the activation of their antioxidant response elements (ARE) in their gene promoters. NRF2 binds to AREs and induces this anti-oxidant response. We show that cells from multiple origins performing OXPHOS induced NRF2 expression and its transcriptional activity. The NRF2 promoter contains MEF2 binding sites and the MAPK ERK5 induced MEF2-dependent NRF2 expression. Blocking OXPHOS in a mouse model decreased Erk5 and Nrf2 expression. Furthermore, fibroblasts derived from patients with mitochondrial disorders also showed low expression of ERK5 and NRF2 mRNAs. Notably, in cells lacking functional mitochondrial complex I activity OXPHOS did not induce ERK5 expression and failed to generate this anti-oxidant response. Complex I activity induces ERK5 expression through fumarate accumulation. Eukaryotic cells have evolved a genetic program to prevent oxidative stress directly linked to OXPHOS and not requiring ROS.
Similar content being viewed by others
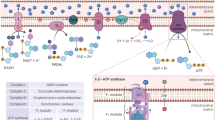


Introduction
Energy consumption in organisms should be finely regulated to spare resources. The vast majority of eukaryotic cells perform oxidative phosphorylation (OXPHOS), which uses the energy generated by mitochondrial oxidation to produce adenosine triphosphate (ATP). This metabolic pathway is highly efficient in releasing energy but it produces reactive oxygen species (ROS) as a byproduct. ROS are involved in normal cell signaling and homeostasis. However, under stress conditions levels may rapidly increase resulting in cell damage, a process known as oxidative stress. Hence, cells using mitochondria as first energy source must regulate ROS levels. Logically, ROS and mitochondria are functionally linked in several ways. First, ROS in the short-term regulate mitochondrial morphology and function via non-transcriptional pathways1. Second, ROS lead to Kelch-like ECH-associated protein 1 (KEAP-1) degradation, thereby activating nuclear factor (erythroid-derived 2)-like 2 (NFE2L2 or NRF2)2,3, which regulates expression of mitochondrial genes4. In addition, NRF2 controls ROS production by mitochondria5 and mitochondrial function6,7. NRF2 arguably mediates the strongest anti-oxidant cellular response by binding to anti-oxidant response elements (ARE) in gene promoters and, consequently, regulates oxidative stress2,3. On the other hand, mitochondrial activity induced by acute exercise promotes Ref1/Nrf2 signaling and increases mitochondrial antioxidant activity and capacity in myocardial and skeletal muscle8,9. Remarkably, restraining OXPHOS in vivo in the liver strongly decreases Nrf2 levels10. Moreover, tumor cells forced to perform OXPHOS generate a NRF2-mediated anti-ROS response11. However, how mitochondria transcriptionally signal the genetic program to block the ROS they produce remains unknown.
NRF2 activation depends on its dissociation from the repressor protein KEAP1 and its subsequent translocation into the nucleus2. In hematopoietic cells, the MAPK extracellular signal-regulated kinase-5 (ERK5), through the transcription factor MEF2, induces expression of miR-23 that inhibits KEAP-1 mRNA leading to NRF2 activation11. Several types of oxidative stress activate ERK512, notably in leukemic cells11,13,14. In fact, ERK5 is considered a redox MAPK15. In endothelial cells, steady laminar blood flow (s-flow) activates ERK5 that induces up-regulation of NRF2-dependent gene expression, although the mechanism is not fully elucidated16,17.
Growing evidence indicates that there are alternative pathways leading to de novo production of NRF23. In this context, KEAP-1 inhibition only partially accounts for OXPHOS-induced antioxidant response11. Chip-seq experiments performed by the ENCODE consortium have shown that the NRF2 promoter contains MEF2 binding sites18. Moreover, predicted networks of transcription factor interactions in skeletal muscle unveil direct regulation of NRF2 by MEF2A19 and MEF2D binds and activates the Nrf2 promoter20. Hence, ERK5 could transcriptionally induce NRF2 expression through MEF2, a transcription factor that mediates some of the metabolic effects of ERK511,13,14,21,22,23,24. In fact, ERK5 regulates the choice of catabolic substrates in hematopoietic cells11,13,14,21,22,23, suggesting that is a good candidate to mediate the link between OXPHOS and the antioxidant response. We hypothesize that mitochondrial activity triggers the ERK5 pathway that, through MEF2, induces NRF2 expression and NRF2-mediated antioxidant response. We validate this by showing that mitochondrial complex I activity and fumarate accumulation induce the transcriptional expression of ERK5. ERK5 through MEF2 induces NRF2 de novo expression. Therefore, mitochondrial activity is directly linked to the most important antioxidant response in the absence of de novo increase in ROS levels. This implies that eukaryotic cells have evolved a genetic program to prevent oxidative stress directly linked to OXPHOS and not requiring ROS.
Results
OXPHOS-induced de novo expression of NRF2
We have previously described that leukemic cells performing OXPHOS generated an anti-oxidant response independently of ROS11. This response was partially mediated by an ERK5-induced increase in miR-23 that impairs expression of KEAP-111. In parallel experiments, we found that NRF2 mRNA was also increased in three hematopoietic cell lines and in primary cells obtained from a B-cell lymphoma (BCL) patient growing in OXPHOS medium (Fig. 1A). This glucose-free culture medium has final concentrations of 4 mM glutamine and 10 mM galactose. Glutamine is used to drive mitochondria to utilize OXPHOS and galactose allows cells to synthesize nucleic acids through the pentose phosphate pathway13,14,25,26. We called it ‘OXPHOS medium’, because it forced leukemic cells to use OXPHOS as primary ATP producer13,24,27. The PDK1 inhibitor dichloroacetate (DCA), which stimulates OXPHOS in all tested leukemic cells11,13,14,22,27,28, also increased NRF2 mRNA (Fig. 1A). Both ways to stimulate OXPHOS also induced NRF2 protein (Fig. 1B). The effect of DCA on NRF2 mRNA and protein is reproduced in two hepatic cell lines (Supplemental Fig. 1A) and in a group of primary leukemic cells from 4 patients (Supplemental Fig. 1B). Of relevance, we observed that in primary human hepatocytes DCA also increased ERK5 and NRF2 mRNA as well as that of the NRF2 targets HO-1 and NQO-1 (Fig. 1C). In summary OXPHOS induced expression of NRF2 in multiple cell contexts.

Cells performing OXPHOS upregulated NRF2 expression. (A) Different hematopoietic cells were incubated in OXPHOS medium or treated with 5 mM DCA for 2 weeks. NRF2 mRNA was quantified by qPCR and values normalized to β-actin mRNA. Results were represented as the % of mRNA compared to cells growing only in glucose medium. Bars show average ± SD of 3 independent experiments performed in triplicate. (B) NRF2 protein expression was analyzed in several cell lines by western blotting (upper panel) or by flow cytometry in OCI-AML3 cells. (C) Hepatocytes from 4 donors were treated with the indicated concentration of DCA for 24 h and ERK5, NRF2, NQO-1 and HO-1 mRNA were analyzed. Bars show average ± SD of the four donors performed in duplicate.
OXPHOS induced NRF2 translocation to the nucleus
NRF2 must translocate to the nucleus to activate its target genes and generate the antioxidant response. HuH7 hepatic cells treated with DCA showed NRF2 accumulation in the nucleus (Fig. 2A). These results were reproduced in non-adherent Jurkat cells by western blotting (Fig. 2B) and in the hepatic cell line HepG2C3A (Supplemental Fig. 2A). We observed a total increase in NRF2 that was more predominant in the nuclear fraction. OXPHOS medium also induced NRF2 translocation to the nucleus in Jurkat cells (Supplemental Fig. 2B).

OXPHOS induced NRF2 translocation into the nucleus. (A) Huh7 cells were treated with 10 mM DCA for 48 h and nuclear translocation was revealed by immunofluorescence. (B) Jurkat cells were treated with 10 mM DCA for 48 h and NRF2 nuclear translocation was revealed by subcellular fractionation and western blotting.
OXPHOS induced de novo expression of NRF2 in vivo
To test if enhanced OXPHOS could exert a similar effect on NRF2 expression in vivo, we engrafted AML primary cells in non-obese diabetic/severe combined immunodeficient (NOD/SCID)-interleukin-2 receptor γ null (NSG) mice, as previously described27. Mice with established tumors (day 80 post-graft) were treated with DCA. The treatment was not toxic and did not show any notable effect on mouse survival27. Human tumor AML cells gather in mouse spleen and bone marrow, hence we isolated mRNA from these organs. We used human-specific primers to analyze the expression of the selected mRNAs and found an increase in NRF2 mRNA (Fig. 3A). This increase paralleled that of ERK5 and NQO-1 under similar conditions11.

Cells performing OXPHOS induce NRF2 expression in vivo. (A) NSG mice were engrafted with primary human AML cells. At day 80 post-graft, they were treated with DCA (n = 4) or leave untreated (n = 4). At day 140, mRNA from bone marrow or spleen was isolated and the expression of different human mRNA was quantified by qPCR. (B) B6 wt mice (n = 4/5 per group) were treated with a dose of DCA (50 mg/kg) everyday intraperitoneally and mouse Erk5, Nrf2 and Nqo-1 mRNA was analyzed in spleen and liver at different times. The data represent means ± SD; statistics were performed using student t-test (A) or One-way ANOVA with post-hoc Tukey test (B); *p < 0.05, **p < 0.01, ***p < 0.001. Different times posttreatment were compared to non-treated mice (control) if not specified in the graph.
DCA also induced mouse Erk5, Nrf2 and Nqo1 mRNA in liver and spleen in a separate experiment in which C57BL/6 wild type mice were treated for different periods of time, 1 to 3 days, with DCA (Fig. 3B). The effect was first observed in spleen and later in liver tissue. Nrf2 was likely active because we observed an increase in its target gene Nqo-1 (Fig. 3B). Hence DCA induced NRF2 expression in multiple cell populations in vitro and in vivo.
Increase in ROS levels is not essential for NRF2 expression
The cellular oxidative state can regulate NRF2 expression2. Therefore, we investigated whether NRF2 expression is regulated by ROS in our setting. DCA induces ROS production in some hematopoietic cell lines, e.g. OCI-AML3, but not all, e.g. Jurkat11,27,29. In contrast, both cell lines increased NRF2 expression suggesting that ROS production was not essential for this induction (Fig. 4). Next, we incubated both cell lines with the antioxidant N-acetyl-cysteine (NAC), which failed to consistently reduce DCA-induced ERK5, NRF2 or NQO-1 mRNA (Fig. 4), although efficiently blocked DCA-induced ROS increase11,29. We observed similar results in primary leukemic cells from a BCL patient (BCL-P2). DCA does not increase ROS in the hepatic cell line HepG2C3A, but it did in Huh729. However, DCA significantly increased ERK5, NRF2 or NQO-1 mRNA in both cell lines and in the presence of NAC (Supplemental Fig. 3). These results excluded a major role of ROS in NRF2 expression after DCA treatment. Normally ROS activate NRF2. Unexpectedly in AML cells, there is no relationship between high ROS levels and high nuclear NRF230. Furthermore, the use of NAC, which successfully sequesters endogenous ROS in AML, has no effect on nuclear NRF2 levels30. Taken together, this excludes ROS as causing nuclear accumulation of NRF2 in resting human AML cells30.

Increase in ROS levels is not required for NRF2 expression. OCI-AML and HuH7 cell lines and primary leukemic cells from a BCL patient were treated with 2 mM NAC 1 h before adding DCA (10 mM) for 24 h. mRNA was analyzed as described in Fig. 1. Experiments were done in triplicate and data represent means ± SD; statistics were performed using One-way ANOVA with post-hoc Tukey test; *p < 0.05, **p < 0.01, ***p < 0.001. Treatments were compared to non-treated cells (control) if not specified in the graph.
ERK5/MEF2 controls NRF2 expression
Next, we investigated the mechanism responsible for mitochondria activity-induced NRF2 expression. Reducing expression of ERK5 with a small hairpin RNA (shERK5) diminished NRF2 mRNA expression in hematopoietic cells under resting conditions (Fig. 5A). NRF2 protein levels were also reduced after shERK5 transfection (Fig. 5B). We could not treat shERK5-expressing cells with DCA because they die due to lack of appropriate mitochondrial functions and antioxidant response11,13,14,21,22,23. Conversely, overexpression of ERK5 increased NRF2 mRNA (Fig. 5A). Reducing expression of ERK5 in primary human hepatocytes (Fig. 5C) and hepatic cell lines, HuH7 and HepG2C3A (Supplemental Fig. 1A,B), with small interference RNA for ERK5 (siERK5) also impaired expression of NRF2 and its target genes NQO-1 and HO-1.

ERK5 controls NRF2 expression. (A) 107 Jurkat-TAg cells were transfected with 5 µg of the empty pSUPER Neo vector or with this vector containing a small hairpin RNA for ERK5 (shERK5) or with a pcDNA vector expressing ERK5. Forty-eight hours later mRNA expression was analyzed by qPCR and represented as the % of mRNA compared to cells transfected with the control vector. (B) Cell transfected with control (Neo) or shERK5 were analyzed for protein expression by western blotting at 24 and 48 h post-transfection. Graphic bars show the NRF2/actin ratio of the depicted experiment. (C) Primary human hepatocytes were double transfected with control siRNA or with siRNA against ERK5 (siERK5). 96 h later mRNA was collected and mRNA expression was analyzed by qPCR. (D) 107 Jurkat-TAg cells were co-transfected with 5 μg of the following vectors ERK5 wild type, a constitutively active MEK5 mutant (MEK5D, M5), MEF2C and MEF2C with dominant negative function (MEF2DN) together with 2 μg of a luciferase reporter plasmid driven by the NRF2 promoter along with 1 μg of β-galactosidase expression vector. Cells were incubated in regular glucose media (gray bars) or containing 10 mM DCA (black bars) 24 h after transfection and analyzed 2 days later for luciferase and β-galactosidase activities. The graphic represents the relative luciferase units (RLU). (E) OCI-AML3 cells were transfected with siRNA for MEF2A and C and 24 h later treated with 10 mM DCA for 36 h. NRF2 mRNA and NRF2 protein were analyzed as in in (A) and (B) respectively. Experiments were done in triplicate. The data represent means ± SD; statistics were performed using student t-test (C) or One-way ANOVA with post-hoc Tukey test (A,D and E); *p < 0.05, **p < 0.01, ***p < 0.001. Treatments were compared to empty vector transfected cells (control) if not specified in the graph.
To further study the role of the ERK5/MEF2 pathway in NRF2 expression, we overexpressed several proteins of this pathway. Strong activation of the ERK5 pathway by co-overexpression of a constitutively active mutant of MEK5 (MEK5D), the upstream kinase of ERK5, and ERK5 induced a greater increase in NRF2 mRNA (Supplemental Fig. 4C). In those experiments, only 30–60% of the cells are effectively transfected. To overcome this issue, we use a luciferase reporter plasmid driven by a DNA fragment of 1.5 kb of the human NRF2 promoter30. In this context, cells expressing the reporter plasmid also contain the overexpressed proteins. ERK5 significantly activated the reporter and MEK5D increased this effect (Fig. 5D). Expression of a dominant negative form of MEF2C (MEF2C-DN) decreased the effect of ERK5 and MEK5D. This DN construct also diminished basal or DCA-stimulated reporter expression. In contrast, MEF2C overexpression increased both basal and DCA-induced activity (Fig. 5D). DCA, which induced strong activation, did not show a synergistic, but rather an additive, effect with the activating proteins. These results suggested that ERK5 controls NRF2 expression through MEF2. To test this, we transfected a small interference RNA for MEF2 (siMEF2) in the hepatic cell line HepG2C3A (Supplemental Fig. 4D) and the AML cell line OCI-AML3 (Fig. 5E). This efficiently decreased MEF2 mRNA and protein29 and also decreased both basal and DCA-induced NRF2 mRNA (Fig. 5E).
Mitochondrial complex I activity signals ERK5 expression
The previous experiments had shown that OXPHOS generates a signal that induces ERK5 expression, which contributes to the NRF2-mediated antioxidant response. We confirmed this in vivo by using a transgenic Tet-Off mouse that express a mutant active form of the ATPase Inhibitory Factor 1 (IF1) in hepatocytes to restrain OXPHOS in the liver10. Interestingly Santacatterina et al. describe in the Fig. 8C of their MS that liver Nrf2 levels are lower in mice expressing IF1. We confirmed it by analyzing expression of Nrf2 mRNA (Fig. 6A). This correlated with lower expression of Erk5 mRNA as compared to wild type mice (Fig. 6A). This shows that OXPHOS also induces Erk5 mRNA expression in vivo.

Inhibition of mitochondrial complex I and II signals ERK5 expression. (A) Erk5 and Nrf2 mRNA expression in the liver of wild-type and T/H (Tet-Off-H49K (h-IF1) mice. mRNA from 3 mice of each genotype was quantified by qPCR and represented as the % of mRNA compared to wild-type mice. (B) ERK5 and NRF2 mRNA expression in fibroblasts derived from a group of 8 healthy donors or 8 patients suffering from mitochondrial defects (Supplemental Table 1). (C) Different hematopoietic cell lines were incubated for 24 h with 5 mM metformin. mRNA expression was quantified by qPCR and represented as the % of mRNA compared non-treated cells. ERK5 and NRF2 protein expression was analyzed in these cell lines by western blotting (lower panel). (D) Jurkat and OCI-AML3 cells were treated with 10 mM DCA and 300 µM TTFA for 24 h. NRF2 mRNA expression was quantified by qPCR and represented as the % of mRNA compared to control cells. (E) Different cell lines described in Supplemental Table 2 were treated with 20 mM DCA during 24 hours. ERK5 mRNA was quantified by qPCR and represented as the % of mRNA compared to non-mutant control cells. Experiments were done in triplicate and data represent means ± SD; statistics were performed using student t-test (A–C) or One-way ANOVA with post-hoc Tukey test (D and E); *p < 0.05, **p < 0.01, ***p < 0.001; §p < 0.05, §§p < 0.01 compare to the respective control cell lines. Treatments were compared to non-treated cells (control) if not specified in the graph.
We confirmed the essential role of mitochondrial function on ERK5 expression by using fibroblasts derived from patients with strong mitochondrial disorders (Supplemental Table 1). ERK5 and NRF2 mRNA were significantly reduced in these patients (Fig. 6B).
We next focused on the molecular mechanisms underlying our observations. When we inhibited the mitochondrial complex I with metformin, we observed a decrease on ERK5, NRF2 and NQO-1 mRNA and protein expression (Fig. 6C). Both metformin and DCA induce AMPK activation27, however, they blocked and induced ERK5 expression, respectively. This suggested that AMPK and its associated metabolic changes were not involved in ERK5 expression. We confirmed this by reducing the expression of the catalytic subunit of AMPK, AMPKα, with 2 different siRNA that effectively blocked several AMPK-mediated metabolic changes27. This did not affect expression of ERK5 or NRF2 mRNA (Supplemental Fig. 5). Therefore, AMPK activation was not responsible for generating the antioxidant response in cells performing OXPHOS.
In Fig. 6C we showed that complex I inhibition decreased ERK5 mRNA. The electron transport chain complex III removes electrons from ubiquinol (QH2) and sequentially transfer them to cytochrome c. The reduction of ubiquinone (Q) to QH2 could either be due to mitochondrial complex I, which removed electrons from NADH, or mitochondrial complex II, which removed them from succinate and transferred through FAD. Then, we investigated the effect of the complex II inhibitor thenoyltrifluoroacetone (TTFA). This drug strongly induced ERK5 mRNA expression (Fig. 6D). DCA did not increase TTFA effects suggesting that both shared the same target.
TTFA was slightly toxic (Supplemental Fig. 6A) and, like metformin, could have off-target effects. Therefore, we used an array of cell lines with impaired activity of the different mitochondrial complexes (Supplemental table 2). ρ0 cells that lack mitochondrial DNA and thus a functional ETC, did not induce ERK5 expression after DCA treatment (Fig. 6E, right lower panel), in agreement with our previous results showing that mitochondrial activity induced ERK5 expression (Fig. 6A,B13. We next used 3 different cell lines in 2 different mitochondrial backgrounds with defects in mitochondrial complex I and observed that DCA treatment did not induce ERK5 expression (Fig. 6E, left panels). In agreement with Fig. 6D, mutation in mitochondrial complex II did not inhibit DCA-induced ERK5 expression (Fig. 6E, top right panel). Cells with mutations in complex III and V, but not in complex IV, increased ERK5 expression after DCA treatment (Fig. 6E, right panels). However, complex V mutant show lower basal ERK5 mRNA levels in agreement with in vivo experiments (Fig. 6A). Mutation in the mitochondrial tRNA Ile in the L929 cell line (mB77), which produces more ROS31, did not increase ERK5 mRNA (Fig. 6E, upper right panel). This supported our results in Fig. 4 showing that de novo ROS production was not involved in ERK5 expression.
Mitochondria adapt the organization of the different complexes and supercomplexes to optimize the use of the available substrates, mainly regulating the proportion of respiratory complex III superassembled with complex I for electron transport. This is needed to avoid competition between FADH2- and NADH-derived electrons31. DCA, by inhibiting PDK1, activates PDH and the formation of acetyl-CoA from pyruvate. This generates 3 NADH per 1 FADH2 (through succinate) molecules in the TCA cycle. Other substrates however, generate a different proportion of NADH/FADH2 electrons and therefore a different demand of CI/CII dependent oxidation. Therefore, while both complexes are always delivering electrons to the ETC simultaneously, the requirement of complex I seems relatively favored by DCA. Complex II, or succinate dehydrogenase (SDH), is also part of the Krebs cycle and catalyzes the conversion of succinate to fumarate. Hence, if complex II is outcompeted by complex I activity, succinate accumulation and fumarate reduction may be induced. Both phenomena are well known in intracellular signaling. Fumarate and succinate, in their acid form, acidify culture media. Hence, we used monomethylsuccinate (MMS) and dimethylfumarate (DMF) to investigate the impact of fumarate and succinate accumulation on ERK5 expression. MMS decreased ERK5 levels (Fig. 7A). In contrast, DMF increased them (Fig. 7A). Next, we used metformin to inhibit complex I, forcing the use of complex II, and added MMS to increase complex II activity. When used together, they decreased even further ERK5 expression (Fig. 7B). This suggested that complex II activity reduced ERK5 expression, probably by inhibiting complex I activity. In summary, whereas succinate probably does not play any role per se on ERK5 expression, fumarate induces its expression. This suggested that DCA, by accumulating fumarate, induces ERK5 mRNA.

Fumarate/succinate regulate ERK5 expression. OCI-AML3 were treated with different drug combinations and the expression of ERK5 mRNA was analyzed by qPCR. (A) OCI-AML3 cells were treated with 10 mM DCA, 5 mM MMS and/or 300 µM DMF for 24 h. (B) OCI-AML3 cells were treated with 5 mM metformin and/or 5 mM MMS for 24 h. (C) OCI-AML3 cells were treated with 5 mM DCA and/or 100 µM Etomoxir for 48 h. The data represent means ± SD; *p < 0.05, **p < 0.01, ***p < 0.005 ANOVA with post-hoc Tukey test. Treatments were compared to non-treated cells (control) if not specified in the graph.
Complex I receive electrons from NADH and deliver them to CoQ. Complex II employs FADH as co-factor to deliver electrons from succinate to CoQ.FADH2. Therefore, the ratio of NADH/FADH2 electrons changes with different substrates and the requirement of complex I for NADH oxidation vary according to the NADH/FADH2 ratio. Oxidative metabolism of one molecule of glucose generates ten NADH and two FADH2, a NADH:FADH2 electron ratio of 5. Fatty acids (FA), e.g. palmitate, generate a ratio of 232. Etomoxir inhibits FA transport into the mitochondria and blocks fatty acid oxidation (FAO), resulting in an increase of the ratio NADH/FADH2. Interestingly, etomoxir decreased basal and DCA-induced increase ERK5 mRNA (Fig. 7C). Therefore, we found no correlation between ERK5 expression and the expected changes in NADH/FADH2 ratio. Etomoxir and DCA were not toxic to OCI-AML3 cells, although they decreased cell proliferation (Supplemental Fig. 6B). However when combined they induced cell death suggesting that DCA treatment requires FAO for cell survival as suggested by our previous results29. In summary complex I activity, through accumulating fumarate, induces ERK5 expression leading to NRF2-mediated antioxidant response.
Discussion
ROS generation is inherent to the activity of the electron transport chain, with Complex I being considered one of the main sites at which premature electron leakage to oxygen occurs and give rise to superoxide anion33. We show here that complex I activity initiates an antioxidant response mediated by ERK5-induced NRF2 expression. It is interesting to note that the main generator of ROS is at the same time responsible of triggering the mechanism to eliminate them. Of relevance, ROS de novo production is not required for this response. The cell “anticipates” ROS formation and activates the pathway to avoid their uncontrolled increase. Once produced, ROS quickly originate biochemical reactions that generate damage to cell structures. Hence, it is on the cell’s own benefit to create the antioxidant response when ROS production is going to occur. However, new data are challenging the “only” deleterious view of mitochondrial ROS. For example ROS increase with aging, but increasing mitochondrial ROS production specifically through the respiratory complex I reverse electron transport (RET) extends Drosophila lifespan34.
We show that mitochondrial complex I activity is required for ERK5-induced NRF2 expression. We have examined several possibilities that could account for our observation. DCA inhibits glycolysis and increases FAO as suggested by the high toxicity of the combined etomoxir plus DCA treatment ((Supplemental Fig. 6) and29). Then, this switch would change NADH:FADH2 electron ratio. When electron flux from FAD overwhelms the oxidation capacity of CoQFAD, CI is degraded, releasing CIII from CI-containing complexes to receive FAD-derived electrons35. The increased electron flux through FAD could saturate the oxidation capacity of the dedicated coenzyme Q (CoQ) pool and result in the generation of ROS36. However, in our experiments ROS do not mediate OXPHOS-induced ERK5 expression (Figs 4 and 6). Moreover, etomoxir decreases basal ERK5 expression but it does not block DCA-induced increase. Finally, if FAO is inducing ERK5 expression, complex II inhibition should decrease it, but we found an increase with TTFA and basically no effect by genetic approaches. This suggests that another mechanism is responsible for triggering ERK5 expression.
In non-transformed cells inhibition of OXPHOS by IF1 induces AMPK activation10, which could lead to ERK5 activation37. DCA also induces AMPK27. However, our pharmacological (metformin, Fig. 6C) and genetic (siRNA, Supplemental Fig. 5) approaches suggest that AMPK is not involved on ERK5 expression during OXPHOS.
Strong mitochondrial complex I activity could decrease electron transport through complex II and the subsequent accumulation of succinate or reduced fumarate be responsible for ERK5 expression. Our results using TTFA or genetically-modified cells support this conclusion (Fig. 6). However, MMS fails to induce ERK5 even if complex I was blocked by metformin (Fig. 7). In contrast, we found that DMF induced ERK5 (Fig. 7). Interestingly, DMF induces Nrf2 expression through a PD98059-sensitive pathway38. Although this MAPKK inhibitor was initially described as a specific MEK1 inhibitor, it also inhibits the ERK5 upstream kinase MEK539. Therefore, fumarate indeed mediates ERK5 expression. Accumulated fumarate can covalently modify cysteine residues of proteins, in an uncatalyzed process termed succination, modifying cellular signaling40. Succination occurs on KEAP141 and results in constitutive NRF2 activation and increased expression of its target genes41. Therefore, fumarate induces the NRF2-mediated antioxidant response by directly affecting KEAP-141 and by inducing de novo NRF2 expression (our results).
An alternative is that succinate promotes CII activity and induces RET thereby decreasing mitochondrial membrane potential. In reverse, fumarate blocks CII thereby increasing complex I activity and this could trigger ERK5 expression.
Although it is well-established that ROS induce NRF2 activation2, recent data support that alternative pathways independent of ROS are also operative. For example, OXPHOS decreases KEAP-1 expression independently of ROS11 and NRF2 expression in AML depends on NF-κB but not on ROS30. Interestingly, ERK5 activates NF-κB in leukemic cells42. Hence, ERK5 could handle the NRF2-mediated antioxidant response by at least 3 mechanisms independently of de novo ROS generation: i) direct transcription through MEF2 (the results presented here); ii) direct transcription through NF-κB30,42; iii) upregulation of miR-23 and downregulation of KEAP1 mRNA11. This emphasizes the central role of ERK5 in the antioxidant response11,12,13,14,15,16,17.
Transcriptome analysis shows that the ERK5 pathway regulates in normoxia several genes involved in metabolic remodeling, including some controlled by hypoxia inducible factor-1α (HIF-1α under hypoxia13,43. Also, like HIF-1α, ERK5 is degraded by a process depending on the tumor suppressor von Hippel-Lindau (VHL), through a prolyl hydroxylation-dependent mechanism44. Hence, mitochondrial complex I activity through fumarate accumulation could also protect ERK5 from VHL-induced degradation. This is based on the fact that succinate and fumarate (and succinate) outcompete α-ketoglutarate, an essential co-factor of prolyl hydroxylase domain enzymes45.
How ERK5 induces NRF2 mRNA expression is not totally elucidated. ERK5 directly phosphorylates MEF2A, C and D at different serines and threonines46,47. It activates MEF2A and D by direct interaction because ERK5 serves as a MEF2 coactivator through its signal-dependent direct association with the MEF2 MADS domain; although, at least, MEF2A-dependent transcription requires ERK5 kinase activity48,49.
Finally, forcing cells to produce energy through OXPHOS also affects cell viability and proliferation independently of ROS. This is rather related to energy depletion. In this sense OXPHOS requires mitochondrial function and DCA induces cell death in ρ0 cells, while in other cells it just inhibits growth11,23,50. In summary forcing OXPHOS in vitro is cytostatic in “normal” tumor cells and cytotoxic in cells with major mitochondrial dysfunctions.
Experimental Procedures
Ethical statement
Experimental procedures were conducted according to the European guidelines for animal welfare (2010/63/EU). Protocols were approved by the Animal Care and Use Committee “Languedoc-Roussillon” (approval number: CEEA-LR-12163). The use of human specimens for scientific purposes was approved by the French National Ethics Committee. All methods were carried out in accordance with the approved guidelines and regulations of this committee. Written informed consent was obtained from each patient prior to surgery.
Reagents and antibodies
DCA was from Santa Cruz Technologies. Galactose and glutamine were from GIBCO. RIPA buffer to prepare protein extracts was from Euromedex. The complete protease inhibitor cocktail (Complete EDTA-free) and the phosphatase inhibitor cocktail (PhosSTOP) were from Roche. H2O2, DMF and MMS were from Sigma. ERK5 and NRF2 antibodies were from Cell Signaling Technology and Santa Cruz respectively. The antibody against β-Actin and HRP-labeled secondary antibodies were from Sigma.
In vivo mouse experiments
In vivo experiments were carried out using 6 to 8 weeks/old male NSG mice. Mice were bred and housed in pathogen-free conditions in the animal facility of the European Institute of Oncology–Italian Foundation for Cancer Research (FIRC), Institute of Molecular Oncology (Milan, Italy). For engraftment of human cells, 1 million AML cells were injected intravenously (i.v.) through the lateral tail vein in non-irradiated mice. NSG mice with established human AML tumors (day 80 post-graft) were treated with DCA (50 mg/kg, 1 dose/day by gavage, starting at day 1 for 16 consecutive days). Human tumor AML cells gather in mouse spleen and bone marrow, hence we isolated mRNA from these organs. We used human-specific primers to visualize expression of human mRNA. In a different experiment B6 wild type mice were treated with a daily single dose of DCA (50 mg/kg/day) intraperitoneally and mouse mRNA was analyzed in spleen and liver after different times.
hIF1 Transgenic mice
The samples from transgenic mice containing the mutant H49K version of hIF1 have been described10. mRNA was analyzed in liver of these mice.
Cell lines and culture conditions
The leukemic human cell lines T Jurkat Tag, NB4 and OCI-AML3 were grown in RPMI 1640–Glutamax (GIBCO) supplemented with 5% (Jurkat) or 10% (OCI and NB4) FBS. Primary cells from a lymphoma B cell patient (BCL-P2) were grown in the same medium with 10% FBS. In certain experiments cells were grown in RPMI 1640 without glucose (GIBCO 11879) with the addition of 2 mM glutamine and 10 mM galactose (OXPHOS medium). The Jurkat TAg cells carry the SV40 large T Ag to facilitate cell transfection. HepG2C3A and HuH7 cells were grown in MEM and DMEM respectively supplemented with 10% FBS, sodium pyruvate, glutamine, penicillin and streptomycin. The HCT116 human colon cancer cells were cultured in low glucose (5 mM) DMEM medium supplemented with 10% FBS. Cellular confluence during experiments was between 80–85%.
Primary Leukemic Cells
Data and samples from patients with different hematological cancers were collected at the Oncology and Clinical Hematology Department of the CHU Montpellier, France, after patient’s informed consent. Patients were enrolled in two independent clinical programs approved by the “Comités de Protection des Personnes Sud Méditerranée I (ref 1324)” and ID-RCB: 2011-A00924-37. All samples from cancer patients were collected at diagnosis.
Human liver samples and preparation of primary human hepatocytes (PHHs) cultures
PHHs were isolated as described previously51 from donor organs unsuitable for transplantation or from liver resections performed in adult patients for medical reasons unrelated to our research program. Liver samples were obtained from the Biological Resource Center of Montpellier University Hospital (CRB-CHUM; http://www.chu-montpellier.fr; Biobank ID: BB-0033-00031) and this study benefitted from the expertise of Dr Jeanne Ramos (hepatogastroenterology sample collection) and Prof Sylvain Lehmann (CRB-CHUM manager). The procedure was approved by the French Ethics Committee and written or oral consent was obtained from the patients or their families.
Human hepatocytes isolation and culture were performed as described previously51. Briefly, after liver perfusion, hepatocytes were counted and cell viability was assessed by trypan blue exclusion test. A suspension of 1 × 106 cells/mL per well was added in 12-well plates pre-coated with type I collagen (Beckton Dickinson) and cells were allowed to attach for 12 h. Then, the supernatant containing dead cells and debris was removed and replaced with 1 mL of serum-free long-term culture medium (Lanford medium, LNF). The number of confluent attached cells was estimated at ~1.5 × 105 cells/cm2.
Plasmids
The luciferase reported plasmid driven by a DNA fragment of 1.5 kb of the human NRF2 promoter was a kind gift from Stuart Rushworth30. The expression vectors for ERK5, the pSUPER expression vector for GFP alone or GFP plus shERK5 and the pSiren-retroQ-puro (BD Biosciences) retroviral vectors for shERK5 and control have been previously described52. Control, MEF2A and C and ERK5 siRNA were ON-TARGETplus SMARTpools (mixture of 4 siRNA) from Dharmacon.
Transient transfection
Jurkat cells in logarithmic growth phase were transfected with the indicated amounts of plasmid by electroporation42,53. In each experiment, cells were transfected with the same total amount of DNA by supplementing with empty vector. Cells were incubated for 10 min at RT with the DNA mix and electroporated using the Gene Pulser Xcell™ Electroporation system (Bio-Rad) at 260 mV, 960 mF in 400 µl of RPMI 1640. Expression of the different proteins was confirmed by western blot. The transfection efficiency in Jurkat TAg cells is between 60 and 80%. OC-AML-3 cells were transfected using Amaxa TM D-Nucleofector TM Lonza Kit according to manufactured protocol. In HuH7 and HCT116 cells, transfection of 30–50 nM siRNAs was carried out using Lipofectamine RNAiMAX (Invitrogen) in Opti-MEM (Invitrogen), according to the manufacturer’s instructions. Adherent primary hepatocytes were transfected twice at day first and third post-seeding with 20 nM siRNA. Cells were harvested 48 to 96 h post-transfection.
Reporter assay
In all experiments, Jurkat cells were transfected with β-galactosidase reporter plasmid42,53. The transfected cells were harvested after 2 days and centrifuged at 1000 g for 5 min. The cell pellet was suspended in 1 ml cold PBS and transferred to 1.5 ml Eppendorf tube for washing. Cells were lysed with 100 μl luciferase lysis buffer (Promega) and incubated at room temperature for 10 min. The lysates were centrifuged and luciferase assays (40 µl) performed according to the manufacturer’s instructions (Promega, Charbonnières, France) using a Berthold luminometer. For β-Galactosidase assays, 40 µl of lysates were added to 200 µl of β-Galactosidase assay buffer (50 mM phosphate buffer pH 7.4; ONPG 200 µg; 1 mM MgCl2; 50 mM β-Mercaptoethanol) and the absorbance measured at 405 nm. The results were expressed as luciferase units normalized to the corresponding β-galactosidase activity. The expression level of the transfected proteins was routinely control by immunoblot analysis.
Subcellular fractionation
For preparation of nuclear extracts, Jurkat cells were grown in indicated medium. Ten million cells were taken and washed twice in cold PBS54. Nuclear and cytoplasmic proteins were extracted using according to manufactured instruction of Bio Basic Inc®. Extracted soluble proteins were analyzed by immunoblotting.
Counting and determination of cell viability
Cell number, viability and cell death was analyzed with the Muse Cell Analyzer (Millipore) by incubating cells with Muse Count & Viability and Annexin V and Dead Cell kits respectively, following manufacturer’s instructions27.
Immunofluorescent assay
Control or treated cells were washed with cold buffer and fixed with paraformaldehyde (3.2% in PBS) for 20 minutes. Cells were washed 3 times with PBS and stored at 4 °C until labelling. Cells were permeabilized with Triton (0.1% in TBS) for 5 minutes and washed with TBS-T (TBS + Tween 0.05%). Cells were labelled with primary antibody (for one hour at room temperature (dilution in TBS + 2% SVF) and washed with TBS-T. Cells were labeled with secondary antibody and Hoechst or DAPI (1/1000 dilution in TBS + 2% SVF) for 30 minutes. Cells were washed with TBS-T and finally washed with H2O before montage. Immunofluorescent labeling was examined under a fluorescent microscope (Leica Microsystem, Rueil-Malmaison, France) and images were analyzed using Metamorph software (Universal Imaging Corporation, Downington, PA).
RT-PCR
Total RNA was extracted using NucleoSpin RNA isolation columns (Macherey-Nagel), reverse transcription was carried out using iScript™ cDNA Synthesis Kit (Biorad). Quantitative PCR was performed with KAPA SYBR Green qPCR SuperMix (Cliniscience) and a CFX Connect™ Real-Time qPCR machine (Biorad) with ERK5, NRF2, NQO1, HO-1 and actin primers. Supplemental Table 3 shows all primers used in this study. All samples were normalized to β-actin mRNA levels. Results are expressed relative to control values arbitrarily set at 10027.
Immunoblotting
Protein analysis by immunoblotting was performed essentially as previously described27. Briefly, samples were collected, washed out with PBS and lysed with RIPA buffer. Protein concentration was determined by BCA assay (Pierce) before electrophoresis in 4–15% TGX gels (BioRad) and equal amount of protein was loaded in each well. Protein transfer was performed in TransTurbo system (BioRad) in PVDF membranes. After blocking for 1 h with 5% non-fat milk, membranes were incubated overnight at 4 °C in agitation with primary antibodies, washed three times with PBS-Tween 0,1% and incubated with the appropriate HRP-labeled secondary antibody for 1 h. Membranes were washed out three times with PBS-Tween 0,1% and developed with Substrat HRP Immobilon Western (Millipore). Band quantification was performed using the “ImageLab” software from BioRad and represented as the ratio between the protein of interest and a control protein i.e. actin. The value of 1 is arbitrarily given to control cells. One blot representative of several experiments is shown.
Statistical analysis
The statistical analysis of the difference between means of paired samples was performed using the paired t test. Multiple comparisons were performed using One-way ANOVA with post-hoc Tukey HSD test. The results are given as the confidence interval (*p < 0.05, **p < 0.01, ***p < 0.005). All the experiments described in the figures with a quantitative analysis have been performed at least three times in duplicate. Other experiments were performed three times with similar results. We used actin as a loading control and the histograms represent the ratio (value of protein of interest)/(value of actin).
References
Willems, P. H., Rossignol, R., Dieteren, C. E., Murphy, M. P. & Koopman, W. J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab 22, 207–218 (2015).
Kensler, T. W. & Wakabayashi, N. Nrf2: friend or foe for chemoprevention? Carcinogenesis 31, 90–99 (2010).
Krajka-Kuzniak, V., Paluszczak, J. & Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacological reports: PR 69, 393–402 (2017).
Scarpulla, R. C. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem 97, 673–683 (2006).
Kovac, S. et al. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta 1850, 794–801 (2015).
Dinkova-Kostova, A. T. & Abramov, A. Y. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88, 179–188 (2015).
Strom, J., Xu, B., Tian, X. & Chen, Q. M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J 30, 66–80 (2016).
Muthusamy, V. R. et al. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic Biol Med 52, 366–376 (2012).
Wang, P., Li, C. G., Qi, Z., Cui, D. & Ding, S. Acute exercise stress promotes Ref1/Nrf2 signalling and increases mitochondrial antioxidant activity in skeletal muscle. Experimental physiology 101, 410–420 (2016).
Santacatterina, F. et al. Down-regulation of oxidative phosphorylation in the liver by expression of the ATPase inhibitory factor 1 induces a tumor-promoter metabolic state. Oncotarget 7, 490–508 (2016).
Khan, A. U. et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine 3, 43–53 (2016).
Zhao, J. et al. Big mitogen-activated protein kinase 1 protects cultured rat aortic smooth muscle cells from oxidative damage. Journal of pharmacological sciences 116, 173–180 (2011).
Charni, S. et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J Immunol 185, 3498–3503 (2010).
Lopez-Royuela, N. et al. Extracellular-signal-regulated kinase 5 modulates the antioxidant response by transcriptionally controlling Sirtuin 1 expression in leukemic cells. Int J Biochem Cell Biol 53, 253–261 (2014).
Abe, J., Kusuhara, M., Ulevitch, R. J., Berk, B. C. & Lee, J. D. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem 271, 16586–16590 (1996).
Kim, M. et al. Laminar flow activation of ERK5 protein in vascular endothelium leads to atheroprotective effect via NF-E2-related factor 2 (Nrf2) activation. J Biol Chem 287, 40722–40731 (2012).
Nigro, P., Abe, J. & Berk, B. C. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal 15, 1405–1414 (2011).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Mysickova, A. & Vingron, M. Detection of interacting transcription factors in human tissues using predicted DNA binding affinity. BMC genomics 13(Suppl 1), S2 (2012).
Nagar, S. et al. MEF2D haploinsufficiency downregulates the NRF2 pathway and renders photoreceptors susceptible to light-induced oxidative stress. Proc Natl Acad Sci USA 114, E4048–E4056 (2017).
Villalba, M. et al. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol 45, 106–113 (2013).
Villalba, M. et al. Chemical metabolic inhibitors for the treatment of blood-borne cancers. Anti-cancer agents in medicinal chemistry 14, 223–232 (2014).
Catalán E, et al. MHC-I modulation due to metabolic changes regulates tumor sensitivity to CTL and NK cells. Oncoimmunology 4, (2015).
Rathore, M. G. et al. The NF-kappaB member p65 controls glutamine metabolism through miR-23a. Int J Biochem Cell Biol 44, 1448–1456 (2012).
Reitzer, L. J., Wice, B. M. & Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 254, 2669–2676 (1979).
Rossignol, R. et al. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 64, 985–993 (2004).
Allende-Vega, N. et al. The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. Oncotarget 6, 19228–19245 (2015).
Catalán, E. et al. MHC-I modulation due to metabolic changes regulates tumor sensitivity to CTL and NK cells. Oncoimmunology 4, e985924 (2015).
Khan, A. U. H. et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis Through the ERK5/MEF2 Pathway. Scientific reports 7, 10654 (2017).
Rushworth, S. A. et al. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 120, 5188–5198 (2012).
Moreno-Loshuertos, R. et al. Evolution meets disease: penetrance and functional epistasis of mitochondrial tRNA mutations. PLoS genetics 7, e1001379 (2011).
Speijer, D. Oxygen radicals shaping evolution: why fatty acid catabolism leads to peroxisomes while neurons do without it: FADH(2)/NADH flux ratios determining mitochondrial radical formation were crucial for the eukaryotic invention of peroxisomes and catabolic tissue differentiation. Bioessays 33, 88–94 (2011).
Lenaz, G. et al. Mitochondrial Complex I: structural and functional aspects. Biochim Biophys Acta 1757, 1406–1420 (2006).
Scialo, F. et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab 23, 725–734 (2016).
Lapuente-Brun, E. et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 (2013).
Guaras, A. et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell reports 15, 197–209 (2016).
Young, A. et al. Flow activation of AMP-activated protein kinase in vascular endothelium leads to Kruppel-like factor 2 expression. Arteriosclerosis, thrombosis, and vascular biology 29, 1902–1908 (2009).
Wang, Q. et al. Dimethyl Fumarate Protects Neural Stem/Progenitor Cells and Neurons from Oxidative Damage through Nrf2-ERK1/2 MAPK Pathway. Int J Mol Sci 16, 13885–13907 (2015).
Kamakura, S., Moriguchi, T. & Nishida, E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. Journal of Biological Chemistry 274, 26563–26571 (1999).
Ternette, N. et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell reports 3, 689–700 (2013).
Ooi, A. et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20, 511–523 (2011).
Garaude, J. et al. ERK5 activates NF-kappaB in leukemic T cells and is essential for their growth in vivo. J Immunol 177, 7607–7617 (2006).
Schweppe, R. E., Cheung, T. H. & Ahn, N. G. Global gene expression analysis of ERK5 and ERK1/2 signaling reveals a role for HIF-1 in ERK5-mediated responses. J Biol Chem 281, 20993–21003 (2006).
Arias-Gonzalez, L. et al. ERK5/BMK1 is a novel target of the tumor suppressor VHL: implication in clear cell renal carcinoma. Neoplasia 15, 649–659 (2013).
Smolkova, K. et al. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol 43, 950–968 (2011).
Kato, Y. et al. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO Journal 16, 7054–7066 (1997).
Kato, Y. et al. Big mitogen-activated kinase regulates multiple members of the MEF2 protein family. Journal of Biological Chemistry 275, 18534–18540 (2000).
Kasler, H. G., Victoria, J., Duramad, O. & Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol 20, 8382–8389 (2000).
Yang, C. C., Ornatsky, O. I., McDermott, J. C., Cruz, T. F. & Prody, C. A. Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids Res 26, 4771–4777 (1998).
Stockwin, L. H. et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127, 2510–2519 (2010).
Pichard, L. et al. Human hepatocyte culture. Methods Mol Biol 320, 283–293 (2006).
Charni, S. et al. ERK5 Knockdown generates mouse leukemia cells with low MHC class i levels that activate NK cells and block tumorigenesis. J Immunol 182, 3398–3405 (2009).
Garaude, J. et al. SUMOylation regulates the transcriptional activity of JunB in T lymphocytes. J Immunol 180, 5983–5990 (2008).
Garaude, J., Kaminski, S., Cherni, S., Hipskind, R. A. & Villalba, M. The Role of ERK5 in T-Cell Signalling. Scand J Immunol 62, 515–520 (2005).
Acknowledgements
We thank Dr. Robert A. Hipskind for his helpful comments regarding this manuscript. FACs analysis and microscopy were performed at the platform Montpellier Rio Imaging (MRI). The collection of clincial data and samples (HEMODIAG_2020) at the CHRU Montpellier was supported by funding from Région Languedoc Roussillon. All our funders are public or charitable organizations. This work was supported by a fellowship from the Higher Education Commission, Pakistan (AK) and fellowships from the Ministère de l’Enseignement Supérieur et de la Recherche (MESR) (DNV).
Author information
Authors and Affiliations
Contributions
A.K., N.A.-V., D.G., S.G., C.G., D.-N.V., S.O., G.T., perform experiments. J.G., M.D., C.D., F.B., A.A., J.-M.C., J.-A.E., G.C., C.-H.L., J.H. and M.V. wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, A.U.H., Allende-Vega, N., Gitenay, D. et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci Rep 8, 7420 (2018). https://doi.org/10.1038/s41598-018-23884-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23884-4
This article is cited by
-
Mitochondrial stress: a key role of neuroinflammation in stroke
Journal of Neuroinflammation (2024)
-
The metabolism of cells regulates their sensitivity to NK cells depending on p53 status
Scientific Reports (2022)
-
Simultaneous augmentation of muscle and bone by locomomimetism through calcium-PGC-1α signaling
Bone Research (2022)
-
Mitochondrial fission links ECM mechanotransduction to metabolic redox homeostasis and metastatic chemotherapy resistance
Nature Cell Biology (2022)
-
MEKK3-MEK5-ERK5 signaling promotes mitochondrial degradation
Cell Death Discovery (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
