
Abstract
A novel blood-borne virus called the human hepegivirus 1 (HHpgV-1) was recently discovered in hemophilia patients. The present study aimed to investigate the presence of HHpgV-1 in hemophilia patients. A total of 436 serum samples were investigated for the presence of hepatitis C virus (HCV), human pegivirus-1 (HPgV-1), torque teno virus (TTV), and HHpgV-1. Out of the 436 patients, 163 (37.4%), 19 (4.4%), 76 (17.4%), and four (0.9%) patients were positive for HCV, HPgV-1, TTV, and HHpgV-1, respectively. HHpgV-1 patients had a mean viral load of 4.9 ± 0.3 log RNA copies/mL and were co-infected with HCV-1a, HPgV-1, and TTV. Moreover, three HHpgV-1-positive patients exhibited stage F0 liver fibrosis. HCV viral load in HHpgV-1-positive patients was lower than those of HHpgV-1-negative patients. Results also revealed that co-infection of HHpgV-1 with HPgV-1 and HCV may play a protective role in patients with chronic HCV. In conclusion, we detected a low frequency of HHpgV-1 infection in hemophilia patients, and results suggested that HHpgV-1 infection was correlated with the presence of other blood-borne viruses and is likely to also correlate with low HCV viral load and reduced severity of liver disease. Additional studies are required to further investigate the clinical importance of HHpgV-1.
Similar content being viewed by others


Introduction
Blood-derived products from patients with hemophilia, treated by factor VIII concentrates, are possible sources of transfusion-transmitted infections (TTI). TTI, especially viral infections, such as viral hepatitis, or infections by human immunodeficiency virus (HIV) and human pegivirus-1 (HPgV-1; formerly known as GBV-C or HGV), can potentially be transmitted via blood transfusions1.
These infections were major health problems in the 1980s, when 65–75% of hemophilia patients were infected with HIV-12.
This concern continued with the finding that approximately 80% of hemophilia patients treated with blood-derived products before 1992 were infected with hepatitis C virus (HCV)3.
Current screening practices and blood donor selection methods have improved TTI detection in blood-derived products, so that the residual risk of infection with hepatitis B virus (HBV), HCV, and HIV has decreased to approximately 1 or <1 million transfused units. However, despite this success, residual risk of infection remains1,4.
Kapoor et al. reported the discovery of human hepegivirus-1 (HHpgV-1), a new blood-borne virus that belongs to the Flaviviridae family and shares features with HCV and HPgV-1. HHpgV-1 was detected in serum specimens from two hemophilia patients and two blood transfusion recipients who received blood/plasma-derived products5.
Very little information is available on the prevalence of HHpgV-1. Accordingly, epidemiological studies from different parts of the world must be conducted to gain insight into the characteristics and disease mechanisms of HHpgV-1. Therefore, the present study aimed to investigate HHpgV-1, a new blood-borne virus, in Iranian hemophilia patients.
Results
Patient’s clinical characteristics
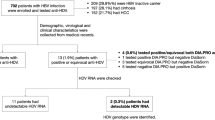
A total of 436 hemophilia patients were enrolled in this study. Of these patients, 288 (66.1%) and 148 (33.9%) were male and female, respectively. The mean age in all patients was 29.2 ± 8.3 years. In addition, mean ALT, AST, and ALK were 47.7 ± 29.9, 45.3 ± 25.4, and 175.2 ± 130.5, respectively (Table 1).
Detection of HCV, HPgV-1 and TTV in hemophilia patients
The frequency of HCV, HPgV-1, and TTV in hemophilia patients was 163 (37.4%), 19 (4.4%), and 76 (17.4%), respectively. After genotyping for HCV, the frequency of HCV-1a was 104 (63.8%) and that of HCV-3a was 59 (36.2%). Liver fibrosis stages in chronic HCV infection were 44 (27.0%) in F0, 85 (52.2%) in F1, 24 (14.7%) in F2, and 10 (6.1%) in F3. The mean HCV and HPgV-1 viral loads were 7.3 × 105 ± 2.8 × 105 IU/mL (range, 1.2 × 105–1.4 × 107 IU/mL) and 5.4 ± 0.4 log RNA copies/mL (range, 4.8–6.0 log RNA copies/mL), respectively (Table 1).
Frequency of HHpgV-1 and characterization of HHpgV-1 patients
We detected HHpgV-1 in four (0.9%) patients using semi-nested PCR assay and analyzed the isolates via partial- and whole-genome sequencing. Patients were named IHP-16, IHP-234, IHP-343, and IHP-381. HHpgV-1 patients were all male and had a median age of 66.3 ± 4.0 years old. In addition, these patients were simultaneously co-infected with HCV, HPgV-1, and TTV. The liver fibrosis stage of three patients was F0, while one patient was in the F1 stage. The mean viral load in all HHpgV-1-positive patients was 4.9 ± 0.3 log RNA copies/mL (range, 4.6–5.4 log RNA copies/mL), and is similar to those reported by Wang et al. and Coller et al.11,13. HCV viral load was significantly lower in patients with HHpgV-1 (P = 0.010). All patients had the HCV genotype 1a. Results revealed significant associations of HHpgV-1 infection with age (P = 0.001), HCV genotype (P = 0.002), HPgV-1 viral load (P = <0.001), TTV positivity (P = 0.001), and Liver fibrosis stage (P = 0.001). In addition, ALT (P = 0.542), AST (P = 0.547), and ALK (P = 0.118) levels were not associated with HHpgV-1 positivity. These results reveal that HHpgV-1 viremia may not affect liver enzymes levels in patients with HCV infection (Table 2).
Sequence and phylogenetic analysis of HHpgV-1
Comparison of the partial genome sequences with previously published HHpgV-1 genomes showed that IHP-16 and IHP-234 shared 99% identity with KT439329.1 (AK-790), while IHP-343 and IHP-381 shared 99% identity with KX528231.1 and KX528230.1, respectively. Furthermore, comparison of the whole genome sequence with previously published HHpgV-1 genomes revealed that IHP-16 and IHP-234 showed 100% and 96% identity with KT439329.1 (AK-790) and IHP-343 and IHP-381 had 100% identity with KX528231.1 and KX528230.1, respectively (Fig. 1A,B).

Phylogenetic tree based on a partial helicase gene sequence (A) and whole genome sequence of HHPgV-1 (B). Only bootstrap values >80% are shown.
Discussion
This is a preliminary study to provide first evidence of the frequency of HHpgV-1 as new blood-borne virus in Iranian patients with hemophilia. Subsequent specific semi-nested PCR screening of hemophilia samples from 432 subjects failed to detect any HHpgV-1-positive samples. HHpgV-1 was also infrequently identified in four hemophilia patients, and subsequently, they were confirmed using Illumina sequencing. This result was in keeping with that of three previous studies, which detected low frequency of HHpgV-1 in hemophilia patients5,14,15.
In the current study, four positive samples had co-infection of HHpgV-1 with HCV, HPgV-1, and TTV. This result is completely consistent with data from four published reports, in which HHpgV-1 was mainly or only identified in serum containing HCV, HPgV-1 RNA, and other blood-borne virus. These results indicate the conclusion that HHpgV-1 circulates less frequently in humans but especially in patients with HCV infection is relatively frequent5,13,14,15.
Compared to HCV and HPgV-1, the low frequency of detection of HHpgV-1 in hemophilia patients as a high-risk group may be described by the fact that (I) HHpgV-1 is an uncommon viral infection in humans, (II) HHpgV-1 is less persistent, (III) due to the limited sample numbers and areas tested, or (IV) the PCR-based screening method was incapable to amplify sequences of more genetically divergent variants of HHpgV-15. Whole-genome sequencing has been demonstrated to have high accuracy and sensitivity for the detection of new viral RNA, such as HHpgV-1 RNA, in human blood, which can be highly divergent and indiscernible using conventional PCR techniques14,15. One limitation of the present study is that not all samples were subjected to whole-genome sequencing because of limited funding.
After discovering HCV in 1989, HPgV-1 in 1995, TTV in 1997, TTV-like minivirus (TLMV), and SEN virus (SENV) in 2000, no more transfusion-associated virus infections have been reported16. In addition, many cases of post-transfusion hepatitis with unknown etiology still occur among blood-transfusion recipients. However, chronic HCV infection is a major health problem and leads to liver cirrhosis, hepatocellular cancer, liver transplantation, and death17,18,19,20,21. In return, it was reported that most HPgV-1 infections were usually asymptomatic, transient, and self-limiting22.
Interestingly, several studies have also shown that co-infection with HIV and HPgV-1 can delay progression to acquired immune deficiency syndrome (AIDS), which can be potentially explained by reduced viral replication and/or impairment in the host immune system23,24,25. Moreover, previous studies have revealed that co-infection with Ebola and HPgV-1 viruses was correlated with improved patient survival26.
In the current study, the mean HCV viral load in HHpgV-1-positive patients was lower than that in HHpgV-1-negative patients and three patients were in the F0 liver disease stage. Results suggested that co-infection of HCV with HHpgV-1 and HPgV-1 caused a reduction in HCV viral load and also decreased the severity of liver damage in chronic HCV. Berzsenyi et al. showed that HPgV-1 infection in chronic HCV was correlated with reduced liver disease severity27. Moreover, Kandathil et al. showed similar results that is, no evidence that HHpgV-1 leads to liver disease15. HHpgV-1, like HPgV-1, is presumably not a significant cofactor in the development of HCV-related liver disease. These results give us with clues about the medical consequences of HHpgV-1 infection, raising the eventuality that HHPgV-1 infection may lead in limited or absence of pathogenicity13. However, further studies are required to verify the mechanisms underlying the interactions of HCV with HHpgV-1 and HPgV-1.
In a very recent study, Wang et al. reported the tight association HHpgV-1 with HCV. Also, in line with our study, HHpgV-1 seems not to worsen HCV-related liver fibrosis. As a result, they showed that HHpgV-1 could effect on the replication and pathogenesis of HCV13.
Whether HHpgV-1 plays an important role in the development of human hepatitis remains unclear. HHpgV-1 has the characteristics of both hepaciviruses and pegiviruses, thereby increasing the risk of chronic infections like HCV and HPgV-1 infection. Given that the effects of HHpgV-1 on human health have not been well characterized, a more precise evaluation of HHpgV-1 frequency in human populations is warranted28.
HHpgV-1 is likely to be infectious and can potentially be added to the list of viral infections that are investigated at Hemotherapy Services; further experiments can be conducted to prove this issue16. Historically, the prevalence of HCV infection was underestimated and was resolved only when the full range of the genetic variation of HCV was characterized. However, continued efforts to recognize more cases of HHpgV-1 infection are ongoing, for better understanding of the epidemiology, pathogenesis, and clinical relevance of HHpgV-111.
In conclusion, our results demonstrated that the low prevalence of HHpgV-1 infection in hemophilia patients may be correlated with ongoing HCV, HPgV-1, and TTV infection and may be associated with lower HCV viral load and reduced liver disease severity. However, additional studies are required to further characterize the clinical importance of HHpgV-1.
Patients and Methods
Study population
The current study was performed at the Pasteur Institute of Iran (PII) from October 2016 to August 2017 and conducted in accordance with the 1975 Declaration of Helsinki and relevant local regulations. The study was approved by the Ethical Committee of PII, and written informed consent was directly obtained from all participants. Serum samples from a total of 436 hemophilia patients were collected at the Iranian Comprehensive Hemophilia Care Center (ICHCC) and PII. Severity and staging of liver disease patients were extracted from the patient records. Patients were classified into the following five groups based on the severity of fibrosis: no fibrosis (F0), portal fibrosis alone (F1), portal fibrosis with rare septae (F2), portal fibrosis with many septae (F3), and cirrhosis (F4)6. Aspartate Aminotransferase (AST), alkaline phosphatase (ALK), and Alanine Aminotransferase (ALT) were detected according to laboratory protocols. All patients were negative for both hepatitis B surface antigen (Radim SPA, Pomezia-Rome, Italy) and HIV antibodies (Radim SPA, Pomezia-Rome, Italy).
DNA and RNA extraction
Total RNA was extracted using the High Pure Viral RNA Kit (Roche Diagnostics Deutschland GmbH, Mannheim, Germany) and cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics Deutschland GmbH, Mannheim, Germany), according to the manufacturer’s instructions. TTV DNA was extracted using High Pure PCR Template Preparation Kit (Roche Diagnostics Deutschland GmbH, Mannheim, Germany), according to the manufacturer’s instructions.
HCV detection and genotyping
A nested PCR assay was prepared for detection of HCV using the primer pair HCV-251F (5′-AGCGTCTAGCCATGGCGT-3′) and HCV-22R (5′-GCACGGTCTACGAGACCT-3′) during in the first round of PCR (264 bp) and HCV-FN-F2 (5′-GTGGTCTGCGGAACCGG-3′) and HCV-RN-R2 (5′-GGGCACTCGCAAGCACCC-3′) during the second round of PCR (174 bp). PCR conditions for the first round were 95 °C for 5 min, followed by 30 cycles of 94 °C for 45 s, 59 °C for 45 s, and 72 °C for 45 s, and 72 °C for 5 min as a final extension. PCR conditions for the second round were to the same as the first, but annealing temperature (62 °C) was different. PCR-restriction fragment length polymorphism assay (PCR-RFLP) was used to detect HCV genotypes. HCV-genotyping by RFLP assay has also been confirmed by sequencing of the 5′ non-coding region fragments7.
HPgV-1 and Torque Teno Virus (TTV) detection
The primer pairs for detection of HPgV-1 were: HPg-F1 (5′-CCTTGGACCCAGGTICCNACIGA-3′) and HPg-R1 (5′-CCTGGTGGGGTRGCIGTNGC-3′) in the first round of PCR (242 bp) and HPg-F2 (5′-GGACCCAGGTGCCIACGGAYGC-3′) and HPg-R2 (5′-CCTGGTGGGGTRGCGGTNGCRTA-3′) in the second round of PCR (208 bp)5. The reaction involved an initial denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 45 s, 59 °C for 45 s, and 72 °C for 45 s, and a final extension at 72 °C for 7 min. Identical cycling conditions were applied during the second round, with an annealing temperature of 59 °C for the remaining 40 cycles.
Semi-nested PCR was used to detect the presence of TTV DNA. Three specific primers (NG059, NG061, and NG063) were used to amplify the viral DNA as previously described8. The 386 bp (first round) and 271 bp (second round) PCR products were analyzed by electrophoresis on a 2% agarose gel.
Screening method of HHpgV-1
We used a semi-nested PCR method with helicase gene primers for the detection of HHpgV-1. The PCR primers HHpgV-ak1 (5′-GTTGTATTCGCCACAGCCAC-3′) and HHpgVak2 (5′-TCAAAGTTTCCTGTGTAGCCTGT-3′) were used in the first round and HHpgV-ak3 (5′-GTATTCGCCACAGCCACTCC-3′) and HHpgV-ak2 were used in the second round of semi-nested PCR5. The PCR conditions for the first round included 5 min of denaturation at 94 °C, followed by 35 cycles of 94 °C for 45 s, 57 °C for 45 min, and 72 °C for 45 min, and a final extension at 72 °C for 7 min. The PCR conditions for the second round were 5 min of denaturation at 94 °C, followed by 35 cycles of 94 °C for 45 s, 59 °C for 45 s, and 72 °C for 45 s, and a final extension at 72 °C for 10 min. The amplification products of the first and second PCR round were 317 bp and 314 bp, respectively.
HCV, HPgV-1 and HHpgV-1 RNA quantification
HCV RNA viral load was measured using Amplicor Monitor HCV 2.0 (Roche Diagnostics Deutschland GmbH, Mannheim, Germany), according to the manufacturer’s instructions, respectively. A quantitative PCR (qPCR) method using primers and probes directed against the HHpgV-1 5′-untranslated region (UTR) and nonstructural proteins 2/3 (NS2/3) genes and 5′ UTR of the HPgV-1 was used to determine for the presence of HHpgV-1 and HPgV-1 viral RNAs9,10,11.
Partial- and whole-genome sequencing and phylogenetic analysis
A partial helicase gene (nucleotide 4488 to 5021) of HHpgV-1 was sequenced. In brief, for purification of PCR products, we used the High Pure PCR Product Purification Kit (Roche Diagnostics Deutschland GmbH, Mannheim, Germany). Subsequently, purified PCR products were sequenced using an ABI Automated Sequencer (Applied Biosystems, Foster City, CA, USA).
All datasets were obtained using the Illumina HiSeq. 2500 platform (Illumina Inc., San Diego, CA, USA), yielding an average of 150 million reads per sequencing lane. All HHpgV-1 genome sequences in the NCBI GenBank Database were compared using MEGA6 using the neighbor-joining model and the maximum-likelihood algorithm, with 1000 bootstrap replicates applied to compute branch strength12. In addition, the outgroup sequence for the analysis was HCV (AB559564.1).
Nucleotide sequence accession number
The HHpgV-1 complete genome has been submitted to GenBank under accession number MF775734, MG210577, MG210578, and MG210579 for IHP-16, IHP-234, IHP-343, and IHP-381, respectively.
Statistical analysis
Statistical analyses were carried out using the IBM SPSS Statistics for Windows v.22.0 (IBM Corp. Armonk, New York). All statistical tests were two-tailed, and P < 0.05 was considered statistically significant. Fisher’s exact test/χ2 was performed to detect significant associations among qualitative variables, while the Mann-Whitney U-test was performed for continuous variables.
References
Di Minno, G. et al. Current concepts in the prevention of pathogen transmission via blood/plasma-derived products for bleeding disorders. Blood Rev 30, 35–48 (2016).
Franchini, M. & Mannucci, P. M. Past, present and future of hemophilia: a narrative review. Orphanet J Rare Dis 7, 24 (2012).
Plug, I. et al. Mortality and causes of death in patients with hemophilia, 1992–2001: a prospective cohort study. J Thromb Haemost 4, 510–516 (2006).
Stramer, S. L. & Dodd, R. Y. Transfusion‐transmitted emerging infectious diseases: 30 years of challenges and progress. Transfusion 53, 2375–2383 (2013).
Kapoor, A. et al. Virome analysis of transfusion recipients reveals a novel human virus that shares genomic features with hepaciviruses and pegiviruses. MBio 6, e01466–01415 (2015).
Kalantari, H., Hoseini, H., Babak, A. & Yaran, M. Validation of hepascore as a predictor of liver fibrosis in patients with chronic hepatitis C infection. Hepat Res Treat 2011 (2011).
Pohjanpelto, P., Lappalainen, M., Widell, A., Asikainen, K. & Paunio, M. Hepatitis C genotypes in Finland determined by RFLP. Clin Diagn Virol 7, 7–16 (1996).
Fateh, A. et al. Study on Prevalence of TTV among Cirrhotic patients due to Hepatitis B & C in Ahwaz University Hospitals during the Years 2004–2005. Iran J Virol 3, 29–33 (2009).
George, S. L., Xiang, J. & Stapleton, J. T. Clinical isolates of GB virus type C vary in their ability to persist and replicate in peripheral blood mononuclear cell cultures. Virology 316, 191–201 (2003).
Rydze, R. T., Bhattarai, N. & Stapleton, J. T. GB virus C infection is associated with a reduced rate of reactivation of latent HIV and protection against activation-induced T-cell death. Antivir Ther 17, 1271 (2012).
Coller, K. E. et al. Antibodies to the Novel Human Pegivirus 2 Are Associated with Active and Resolved Infections. J Clin Microbiol 54, 2023–2030 (2016).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30, 2725–2729 (2013).
Wang, H. et al. A Novel Human Pegivirus, HPgV-2 (HHpgV-1), Is Tightly Associated With Hepatitis C Virus (HCV) Infection and HCV/Human Immunodeficiency Virus Type 1 Coinfection. Clin Infect Dis 66, 29–35 (2018).
Bonsall, D. et al. Evaluation of viremia frequencies of a novel human pegivirus by using bioinformatic screening and PCR. Emerg Infect Dis 22, 671 (2016).
Kandathil, A. J. et al. Presence of Human Hepegivirus-1 in a Cohort of People Who Inject Drugs. Ann Intern Med 167, 1–7 (2017).
Cuestas, M. L. New virus discovered in blood supply: Human hepegivirus-1 (HHpgV-1). Rev Argent Microbiol 48, 180–181 (2016).
Asnavandi, M. et al. EGFR rs11506105 and IFNL3 SNPs but not rs8099917 are strongly associated with treatment responses in Iranian patients with chronic hepatitis C. Genes Immun 18, 144–151 (2017).
Sakhaee, F. et al. The impact of genetic variation in IL28B, IFNL4 and HLA genes on treatment responses against chronic hepatitis C virus infection. Infect Genet Evol 54, 330–337 (2017).
Fateh, A. et al. Comparison of three different methods for detection of il28 rs12979860 polymorphisms as a predictor of treatment outcome in patients with hepatitis C virus. Osong Public Health Res Perspect 7, 83–89 (2016).
Fateh, A. et al. High resolution melting curve assay for detecting rs12979860 IL28B polymorphisms involved in response of Iranian patients to chronic hepatitis C treatment. Asian Pac J Cancer Prev 16, 1873–1880 (2015).
Sadeghi, S. et al. Effect of IL15 rs10833 and SCARB1 rs10846744 on virologic responses in chronic hepatitis C patients treated with pegylated interferon-α and ribavirin. Gene 630, 28–34 (2017).
Mushahwar, I., Erker, J., Dille, B. & Desai, S. Recently discovered blood-borne viruses. In: Forum (Genoa, Italy). 98–122 (2001).
Mohr, E. L. & Stapleton, J. T. GB virus type C interactions with HIV: the role of envelope glycoproteins. J Viral Hepat 16, 757–768 (2009).
Bhattarai, N. & Stapleton, J. T. GB virus C: the good boy virus? Trends Microbiol 20, 124–130 (2012).
Tillmann, H. L. & Manns, M. P. GB virus-C infection in patients infected with the human immunodeficiency virus. Antiviral Res 52, 83–90 (2001).
Lauck, M. et al. GB virus C coinfections in West African Ebola patients. J Virol 89, 2425–2429 (2015).
Berzsenyi, M. D., Bowden, D. S., Roberts, S. K. & Revill, P. A. GB virus C genotype 2 predominance in a hepatitis C virus/HIV infected population associated with reduced liver disease. J Gastroenterol Hepatol 24, 1407–1410 (2009).
Rasmussen, A. L. Probing the Viromic Frontiers. MBio 6, e01767–01715 (2015).
Acknowledgements
We would like to thank all of the patients who participated in the study.
Author information
Authors and Affiliations
Contributions
Y.B. and F.S.: performed the experiments; A.A.S. and P.P.: clinical sample and data acquisition; F.V. and F.R.J.: analyzed data, interpreted data; M.R.A. and S.D.S.: performed the experiments, read and approved final manuscript; A.F.: designed and supervised clinical study, interpreted data, read and approved manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bijvand, Y., Aghasadeghi, M.R., Sakhaee, F. et al. First detection of human hepegivirus-1 (HHpgV-1) in Iranian patients with hemophilia. Sci Rep 8, 5036 (2018). https://doi.org/10.1038/s41598-018-23490-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23490-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
