
Abstract
Protein glycosylation has been described as the most abundant and complex post-translational modification occurring in nature. Recent studies have enhanced our view of how this modification occurs in bacteria highlighting the role of protein glycosylation in various processes such as biofilm formation, virulence and host-microbe interactions. We recently showed that the collagen- and laminin-binding adhesin Cnm of the dental pathogen Streptococcus mutans is post-translationally modified by the PgfS glycosyltransferase. Following this initial identification of Cnm as a glycoprotein, we have now identified additional genes (pgfM1, pgfE and pgfM2) that are also involved in the posttranslational modification of Cnm. Similar to the previously characterized ΔpgfS strain, inactivation of pgfM1, pgfE or pgfM2 directly impacts Cnm by altering its migration pattern, proteolytic stability and function. In addition, we identified the wall-associated protein A (WapA) as an additional substrate of Pgf-dependent modification. We conclude that the pgS-pgfM1-pgfE-pgfM2 operon encodes for a protein machinery that can modify, likely through the addition of glycans, both core and non-core gene products in S. mutans.
Similar content being viewed by others


Introduction
Protein glycosylation is considered the most abundant form of post-translational modification among living organisms and its broad distribution indicates that it is advantageous to the overall fitness of both prokaryotes and eukaryotes1,2. In bacteria, glycan decoration of surface-associated or secreted proteins has been shown to modulate an array of cellular processes, such as migration, communication and adhesion1,3,4,5,6,7. There are two major types of protein glycosylation; N-glycosylation and O-glycosylation whereby sugar moieties are directly attached to the amino terminal of an asparagine (N-glycosylation) or to the hydroxyl group of a serine or a threonine (O-glycosylation)1,8. First described in the gastrointestinal pathogen Campylobacter jejuni, bacterial N-glycosylation relies on the general Pgl glycosylation pathway, which is found in a large number of Gram-negative organisms9. Bacterial O-glycosylation, on the other hand, is far less characterized as the identification of specific O-glycosyltransferases and O-oligosaccharyltransferases is much more difficult due to their high level of heterogeneity compared to the canonical N-glycosylation machinery8,10. The complete characterization of O-glycosylated proteins is also more difficult as the system is generally more complex and often relies on stepwise attachment of glycoconjugates1. Given that bacterial glycoproteins are of increasing interest due to their abundance in nature and importance in health and disease, the analysis and characterization of glycosylation patterns is of great importance as it could represent new avenues of basic and clinical research11.
Sugar metabolism plays an essential role in the biology of the dental pathogen Streptococcus mutans contributing to energy generation, gene regulation, biofilm formation and virulence12,13,14,15. However, the use of sugar moieties to post-translationally modify proteins in S. mutans is poorly characterized. Recently, we showed that the collagen- and laminin-binding protein Cnm of S. mutans is post-translationally modified and that such modification requires the expression of a gene located downstream and co-transcribed with cnm that we named pgfS (for protein glycosyltransferase of S. mutans)6. Of note, the cnm gene is present in approximately 15% of S. mutans isolates and encodes an important virulence factor that mediates intracellular invasion and has been associated with systemic infections and caries severity16,17,18,19,20,21,22,23,24. Loss of PgfS-dependent modification of Cnm led to decreased collagen-binding activity and decreased resistance to protease degradation suggesting that Cnm glycosylation is required for both protein function and stability6. Immediately downstream and co-translated with pgfS is a gene encoding a putative integral membrane protein (smu2066c) that is followed by genes encoding an UDP-4-glucose epimerase (smu2065c) and a second membrane protein (smu2064c). Considering that bacterial protein glycosylation machineries often localize at the intracellular membrane interface1, the proximity of the glycosyltransferase pgfS gene with genes encoding a sugar epimerase and two membrane proteins suggested that together these four gene products represent an uncharacterized glycosylation system. Using Cnm as a model protein, we show herein that smu2064c, smu2065c and smu2066c are indeed required for the post-translational modification of Cnm. In many cases, strains lacking any one of these genes phenocopied the ΔpgfS strain although in the smu2064c deletion mutant Cnm appears to undergo partial modification. Interestingly, pgfS, smu2064c, smu2065c and smu2066c are found in all S. mutans strains sequenced to date and are therefore part of the S. mutans core genome. This observation led us to hypothesize that the role of these genes in protein decoration extends beyond Cnm glycosylation. In fact, we found that the surface adhesin WapA, encoded within the S. mutans core genome, is also modified by the Pgf system. Thus, we conclude that the pgfS-smu2064c-smu2065c-smu2066c gene cluster represents a novel posttranslational modification machinery, exerting its function likely through O-glycosylation, of S. mutans capable of modifying both core and non-core proteins.
Results
Transcriptional organization of the pgf genes
Previously, we demonstrated the requirement of pgfS, encoding a membrane-bound GT-A type glycosyltransferase, in the post-translational modification of Cnm6. Bioinformatics analysis of the available genome sequences of S. mutans revealed that pgfS is transcriptionally-coupled with smu2066c (herein pgfM1), which encodes a membrane protein of the O-mannosyltransferase (PMT)-2 subfamily containing 16 putative trans-membrane (TM) domains (Fig. 1A and B). While the protein product of S. mutans pgfM1 is not predicted to possess enzymatic activity, eukaryotic members of this family have been shown to catalyze the transfer of mannose from dolichyl phosphate-activated mannose (Dol-P-Man) to serine or threonine residues of secretory proteins25. Downstream of smu2066c and separated by 35 base pairs (bp) is smu2065c (herein pgfE), which encodes a UDP-4-glucose epimerase predicted to catalyze the interconversion of UDP-glucose and UDP-galactose. Further downstream and separated by 133 bp, smu2064c (herein pgfM2) encodes another membrane protein, with 10 putative TM domains, that shares 54% homology with PgfM1 (Fig. 1B).

Genetic organization of the pgf operon in S. mutans OMZ175 and conserved domains of Pgf proteins. (A) Despite the presence of putative Rho-independent transcriptional terminators (indicated by lollypop symbols) between cnm-pgfS and pgfE-pgfM2, RT-PCR analysis revealed that cnm, pgfS, pgfM1 and pgfE are co-transcribed. (B) Predicted domain organization of PgfS, PgfM1, PgfE and PgfM2: (GT) glycosyltransferase enzymatic domain, (TM) transmembrane domain, (E) epimerase enzymatic domain.
RT-PCR analyses revealed that pgfS, pgfM1 and pgfE are co-transcribed (Figs 1A and S1). These results are in agreement with in silico analysis that indicates that there are no putative promoters or transcriptional terminators in the 3′ UTR between pgfM1 and pgfE. On the other hand, the 133-bp intergenic region between pgfE and pgfM2 contains a putative Rho-independent transcriptional terminator located 4 bp downstream of the pgfE stop codon, which is followed by a putative σA-type promoter located 86 bp upstream of the pgfM2 start codon. However, RT-PCR analysis indicated that pgfE and pgfM2 are also co-transcribed possibly because transcription termination at pgfE is not 100% efficient, which is similar to the terminator located between cnm and pgfS6. Finally, RT-PCR analysis revealed that the gene downstream pgfM2, which encodes a putative ferrochelatase, is not co-transcribed with pgfM2 (data not shown).
All genes within the pgf operon contribute to modification and stability of Cnm
To assess the involvement of pgfM1, pgfE and pgfM2 in the post-translational modification of Cnm, strains bearing individual gene deletions were generated (ΔpgfM1, ΔpgfE and ΔpgfM2) as well as a quadruple mutant lacking all four pgf genes (Δpgf). All mutant strains were readily isolated and grew as well as the parent strain OMZ175 under balanced growth conditions (i.e., BHI at 37 °C in a 5% CO2 atmosphere; data not shown). However, Western blot analysis revealed that the mobility of Cnm was altered in all mutants compared to the parent strain OMZ175 (Fig. 2A), a trait that could be restored after reintroduction of each deleted pgf gene (Fig. 2B). Specifically, similar to the ΔpgfS strain6, deletion of pgfM1, pgfE or the entire pgf operon produced a ~90 kDa variant of Cnm that was substantially smaller than the 140 kDa product identified in OMZ175. On the other hand, deletion of pgfM2 yielded an intermediate-sized protein band of ~120 kDa. Finally, Cnm migration on SDS-PAGE was not altered in a strain lacking the ferrochetalase-encoding smu2063c gene, which lies downstream of the pgf operon (Fig. 2A). Because smu2063c is not co-transcribed with pgfM2 and does not modify Cnm, we did not include this strain in subsequent experiments. It is worth mentioning that we have previously indicated that the mature Cnm migrates at ~120 kDa6,19. However, increased resolution of migration on SDS-PAGE using 10% acrylamide gels identified Cnm migrating at ~140 kDa. Moreover, Cnm has a predicted molecular weight of ~54 kDa, indicating that Cnm has an abnormal electrophoretic migration pattern even in the absence of Pgf modification6. Whole protein analysis of purified native Cnm by quadruple time of flight (Q-TOF) mass spectrometry confirmed the predicted 54 kDa product (data not shown). Thus, the aberrant migration of Cnm in SDS-PAGE is likely due to the combination of Pgf-dependent glycosylation as well as the high negative charge density of the Cnm C-terminal domain as discussed previously6,19.

Modification of Cnm by the pgf operon. (A) Western blot analysis using anti-rCnm shows a band of ~140 kDa corresponding to the fully mature Cnm in OMZ175. Smaller variants of Cnm correspond to partially glycosylated (~120 kDa, ΔpgfM2) and unglycosylated Cnm (~90 kDa, ΔpgfS, ΔpgfM1, ΔpgfE and Δpgf). (B) In locus complementation of the single mutants restored the size of Cnm. (C) Lectin-binding analysis using biotinylated wheat germ agglutinin (WGA) shows that the mature Cnm produced by OMZ175 is recognized by WGA whereas the Cnm versions produced by the different pgf mutans are not. (D) Whole cells of strains OMZ175 and Δpgf derivatives were treated with increasing concentrations of proteinase K for 30 min and Cnm stability monitored by Western blotting. In all cases, duplicated gels stained with Coomasie blue were used to confirm that the same amounts of protein were loaded to the individual lanes.
In agreement with the notion that the Pgf system is responsible for Cnm glycosylation, reactivity to the WGA lectin (was lost in all mutants but restored in the complemented strains (Fig. 2C). Of note, we previously showed that Cnm interacts with WGA but not with other lectins such as PNA and ConA6. In addition, we showed that Cnm also interacts with succinylated WGA, which recognizes only N-acetylglucosamine and that this interaction is inhibited by N-acetylglucosamine in a concentration-dependent manner6. Collectively, these results strongly suggest that PgfM1, PgfE and PgfM2 act in concert with PgfS to glycosylate Cnm. In addition, the intermediate size of Cnm in the ΔpgfM2 strain indicates that Cnm can be modified in the absence of PgfM2. The lack of WGA reactivity in the ΔpgfM2 mutant suggests that PgfM2 is responsible for the addition of the N-acetylglucosamine, the sugar moiety recognized by WGA in the lection blot6.
In a previous study, we also showed that the unmodified Cnm version produced by the ΔpgfS strain was highly susceptible to protease degradation6. Thus, we also expected the Cnm versions produced by the ΔpgfE, ΔpgfM1 and Δpgf strains to be less stable than the mature Cnm version. In fact, inactivation of pgfM1, pgfE or the entire pgf operon increased the susceptibility of Cnm to proteinase K compared to the fully glycosylated Cnm produced by the parent strain OMZ175 (Fig. 2D). In particular, Cnm was completely degraded by as little as 0.3 μg ml−1 of proteinase K in the quadruple Δpgf mutant. Consistent with the idea that Cnm is partially glycosylated in the ΔpgfM2 mutant, the Cnm product observed in this strain was slightly more resistant to proteinase K degradation than the Cnm products of the other Δpgf strains. It was, however, still more susceptible than that produced by OMZ175 (Fig. 2D).
Cnm-dependent phenotypes are negatively affected in Δpgf mutants
Expression of Cnm has been linked to robust collagen binding, human coronary artery endothelial cell (HCAEC) invasion and increased virulence in the Galleria mellonella invertebrate model18,19. Based on the defects in collagen binding, HCAEC invasion and killing of G. mellonella observed for the ΔpgfS strain6, we asked if ΔpgfE, ΔpgfM1 and, perhaps, ΔpgfM2 can phenocopy the ΔpgfS strain. A small but significant decrease in collagen binding (~20%) was observed for each pgf mutant (Fig. 3A) and HCAEC invasion rates were significantly reduced in the different pgf mutant backgrounds, particularly in the ΔpgfE, ΔpgfM1 and Δpgf strains (~90%) (Fig. 3B). Finally, mortality rates of G. mellonella were significantly lower (p < 0.05) in larvae infected with the pgf mutant strains indicating that loss of Cnm posttranslational modification negatively impacts S. mutans virulence (Fig. 3C).

Phenotypic characterization of Δpgf strains. (A) Collagen binding of S. mutans OMZ175 and its Δpgf derivatives. (B) Antibiotic protection assay showing the percentage of HCAEC invasion for each strain of S. mutans after 5 h of incubation. *P ≤ 0.05. (C) Killing of G. mellonella larvae infected with S. mutans strains over a 96 h period. When compared to the parent OMZ175, virulence of all mutant strains was significantly attenuated (P ≤ 0.05).
Cnm modification does not contribute to collagen-binding activity
At this point, it remains unclear if posttranslational modification is directly important for Cnm function, or if the phenotypes observed are solely linked to the observed loss of protein stability. To begin to address this question we tested the effect of WGA, which is predicted to interact with the glycosylated threonine-rich region (TRR) of Cnm, on collagen-binding activity of OMZ175 and Δpgf strains. We found that WGA has a modest effect (∼20% inhibition) on the collagen-binding activity of either OMZ175 or ΔpgfS (Fig. 4A). However, WGA interfered with the collagen binding capacity of glycosylated (from OMZ175) or unglycosylated (from ΔpgfS) Cnm in a similar manner suggesting that the main function of Cnm modification is to increase protein stability. As seen before19, a polyclonal antibody against the Cnm collagen-binding domain (CBD) and upstream N-terminus, which are not predicted to undergo post-translational modification, nearly abolished the ability of OMZ175 and ΔpgfS to bind to collagen (Fig. 4A). Next, we used flow cytometry to evaluate the reactivity of anti-Cnm antibodies with the bacterial cell surface. When compared to OMZ175, the mean fluorescence intensity (MFI) of Cnm was significantly lower in the pgf mutants, except ΔpgfM2 (Figs 4B and S2). These results are consistent with the diminished stability of Cnm produced by these strains, although we cannot rule out that posttranslational modification of Cnm may facilitate protein translocation or surface localization. We also used surface plasmon resonance (SPR) to characterize the binding of different Cnm fragments to immobilized collagen. Three different truncated constructs (rCnm N1, rCnm N2, rCnm N1+2) (Fig. 4C and D) were designed from the full length (rCnm FL) and used for real time binding analyses with type I collagen. The results from the SPR analysis clearly show that the rcnm N2 domain plays a predominant role in adhering to collagen, whilst the rcnm N1 by itself does not adhere to collagen (Figs 4E and S3). Notably, the rCnm N1+2 fragment displayed a higher KD (∼3-fold higher) value when compared to the rCnm N1 fragment (8.17 × 10−7 versus 2.89 × 10−7, respectively) suggesting that the N1 domain stabilizes the interaction after the initial adherence to the N2 domain. Finally, the KD rates for rCnm FL and rCnm N1+2 were of a similar order of magnitude. These results are consistent with the known localization of collagen binding activity within the N2 domain and serves as further indication that the C-terminal threonine-rich domain predicted to undergo posttranslational modification does not have a direct contribution to the collagen-binding activity of Cnm. While we attempted to study the influence of glycosylation in collagen binding by SPR, we were not able to obtain signals due to the difficulty in purifying a sufficient quantity of native Cnm from S. mutans OMZ175. Taken together our results suggest that attenuation of Cnm-related phenotypes in the different pgf mutants is due primarily to the decrease in Cnm protein stability, rather than a direct influence of the posttranslational modification on Cnm functionality.

Cnm does not depend on posttranslational modification for collagen recognition. (A) Inhibition of collagen binding by anti-Cnm and WGA. (B) Expression of Cnm on the surface of OMZ175 and its Δpgf derivatives by flow cytometry expressed as mean fluorescence intensity (MFI). (C and D) Different Cnm constructs used in SPR analysis. The different Cnm domains shown in panel C are: (S) secretion signal, (N1) collagen-binding subdomain N1, (N2) collagen-binding subdomain N2, (TRR) threonine-rich region, and (W) LPxTG motif. (E) SPR analysis of the Cnm constructs. The constructs were injected at concentration of 0.125 μM, 0.25 μM, 0.5 μM, 1.0 μM and 2.0 μM collagen-prepared chip surfaces and the kinetics of association (KA) and dissociation (KD) rate constants (C) were deduced using the 1:1 Langmuir Kinetic model. All experiments were carried out at least in triplicate.
WapA is modified by the Pgf machinery
While cnm is present in ~15% of S. mutans strains, in silico analysis revealed that the pgfS, pgM1, pgfE and pgfM2 genes are part of the S. mutans core genome. We suspected, therefore, that additional proteins encoded by the S. mutans core genome might be subjected to Pgf-dependent modification. Using the NetOGlyc 4.0 glycoprotein prediction server (http://www.cbs.dtu.dk/services/NetOGlyc/), we identified a number of surface-localized and secreted proteins encoded by S. mutans core genes that are predicted to undergo O-glycosylation (Table 1). We focused on the WapA (wall-associated protein A) adhesin, which has been shown in previous studies to mediate collagen binding in vitro, to contribute to sucrose-independent biofilm formation, and to serve as a target of immune protection26,27,28. Of note, the predicted amino acid sequence of WapA is 100% conserved between UA159, a Cnm− non-invasive serotype c strain, and OMZ175 (Cnm+, serotype f) strains. While the predicted size of full-length WapA is ~45 kDa, Western blot analysis of WapA produced by the OMZ175 and UA159 strains revealed protein bands migrating at ~70 kDa, presumably corresponding to a modified form of WapA. A smaller band of ~29 kDa was also observed corresponding to the previously described proteolytic cleavage of WapA at amino acid residues 330 to 340 known as Antigen A or Antigen III29,30 (Fig. 5A). Consistent with the notion that the 70 kDa band represents a modified form of WapA, this band was not present in lysates of ΔpgfS and ΔpgfM1 that instead demonstrated a band, sometimes as a doublet, migrating close to the predicted molecular weight of WapA (~45 kDa). Interestingly, the band observed for the ΔpgfE mutant was slightly larger those of the ΔpgfS and ΔpgfM1 mutants while the WapA produced by ΔpgfM2 was larger than those of the other pfg mutants but still smaller than that produced by the parent OMZ175 strain (Fig. 5A). Similar results were observed in a pgfS deletion mutant created in the UA159 background strain indicating that the Pgf-dependent modification of WapA is conserved across different S. mutans strains (Fig. 5B). Finally, migration of the extracellular Antigen A/Antigen III cleavage product of WapA29,30 was not affected by deletion of genes encoding S. mutans Pgf glycosylation machinery components. The amino acids predicted to undergo O-glycosylation in WapA are located immediately downstream of the proteolytic cleavage site reported to result in the generation of Antigen A/Antigen III29.

Modification of WapA by the pgf operon. (A,B) Western blot analysis using anti-WapA antibody shows a band of 70 kDa corresponding to the fully mature WapA in OMZ175 (A) and UA159 (B). Smaller variants of WapA correspond to partially (∼60 kDa ΔpgfM2) and unglycosylated WapA (∼50 kDa, ΔpgfS, ΔpgfM1, ΔpgfE and Δpgf). (C) Whole cells of strains OMZ175 and ΔpgfS derivatives were treated with increasing concentrations of proteinase K for 30 min and WapA stability monitored by Western blotting.
Next, we tested the stability of WapA produced by wild-type OMZ175 and ΔpgfS strains upon treatment with increasing concentrations of proteinase K. In agreement with the role of glycosylation in protein stability, the WapA product of the ΔpgfS strain was more sensitive to proteinase K degradation than the WapA produced by OMZ175 (Fig. 5C). Taken together, our results demonstrate that WapA and Cnm both serve as substrates of the Pgf system. We conclude, therefore, that the Pgf system can modify both core and non-core gene products in S. mutans.
Discussion
Initially thought to be a process unique to eukaryotic organisms, protein glycosylation is now recognized to occur in all three domains of life being the most abundant and complex posttranslational modification in nature1,2. Previously, we showed that PgfS, a GT-A type glycosyltransferase encoded by a gene located immediately downstream of cnm, is required for the glycosylation of Cnm6. In this report, we showed that the PgfS-dependent modification of Cnm is a cooperative multi-protein effort that includes at least two membrane proteins (PgfM1 and PgfM2) and an UDP-4-glucose epimerase (PgfE). However, unlike cnm, the pgfS-M1-E-M2 genes are present in all S. mutans strains. Here, we also showed that the Pgf system can also modify WapA, a surface-associated protein found to be encoded in the genomes of all S. mutans strains sequenced to date. Moreover, pgf homologs are found in other streptococcal species, usually next to genes coding for surface proteins with collagen-binding domains6,19. Thus, Pgf-mediated protein modification seems to be broad in scope and to be responsible for the posttranslational modification, likely through glycosylation, of both core and non-core proteins of S. mutans and, possibly, proteins of other Streptococci.
In Streptococcus parasanguinis and Streptococcus gordonii, O-glycosylation of the fimbrial protein Fap1 and the GspB adhesin, respectively, is stepwise and mediated by several proteins31,32,33. However, in other cases such as in Acinetobacter baumani, O-glycosylation occurs en bloc with the glycan transferred from a lipid carrier to the target protein via O-oligosaccharyltransferases (O-Otases)34. Based on the knowledge that protein glycosylation in Streptococci has been only shown to occur in a sequential manner and that conserved O-Otases are absent in S. mutans, we believe that Cnm and WapA are glycosylated by the Pgf system in a sequencial manner rather than en bloc. In support to this possibility, both Cnm and WapA appear to produce a partially gycosylated product in the ΔpgfM2 background, suggesting that PgfM2 participates in the final steps of both Cnm and WapA glycosylation (Fig. 6). Previously, we showed that the mature (glycosylated) Cnm reacts with WGA and that it specifically recognizes GlcNAc but not sialic acid6. The loss of Cnm recognition by WGA in the ΔpgfM2 mutant suggests that Cnm is probably modified with other sugars that are not recognized by this lectin prior to the addition to GlcNAc. Based on current evidence, we propose that PgfM2 coordinates the addition of GlcNAc as a final step in the protein glycosylation pathway. In contrast, loss of pgfM1 completely abolished glycosylation suggesting that PgfM1 cooperates with PgfS early in the process (Fig. 6). As part of our future efforts, functional and structural studies will be undertaken to conclusively demonstrate that the Pgf machinery add glycans sequentially.

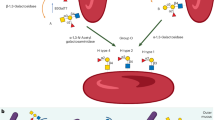
Proposed model of Pgf-mediated protein modification in S. mutans. Protein localization was predicted using the TMHMM 2.0 software and ribbon structure models were built using the PHYRE software. Both PgfM1 and PgfM2 are integral membrane proteins with an extracellular domain of unknown function. PgfS is membrane-bound with its N-terminal catalytic domain located intracellularly. PgfE is predicted to be intracellular, where it functions as a homodimer to direct the NAD+-dependent interconversion of UDP-galactose and UDP-glucose. (A) PgfS recognizes the protein target and initiates the process of glycosylation by adding sugars that were initially transported through PgfM1 and subsequently activated by the PgfE sugar epimerase. (B) Upon initial transfer, PgfS will add sugars that were transported by PgfM2 and activated by PgfE. Upon completion of the glycosylation process, the mature glycoprotein will be transported to the extracellular milieu and anchored at the cell wall surface via proteolytic cleavage of the C-terminal LPXTG motif.
Despite our best efforts, we have been unable to identify the glycan(s) attached to Cnm using mass spectometry (MS) analysis, possibly because of the heavily glycosylated nature of Cnm makes the protein refractory to enzymatic cleavage. Native Cnm purified from S. mutans OMZ175 was completely resistant to enzymatic deglycosylation using a cocktail containing five major glycosidases (PNGase F, sialidase A, O-glycanase, β(1-4)-galactosidase, of β-N-acetylglucosaminidase) and three different proteases (trypsin, Glu-C and pepsin) that failed to digest the threonine-rich B region predicted to undergo O-glycosylation6. While lectin binding analyses have been instrumental in confirming the glycosylated nature of Cnm, our future efforts will be directed toward engineering a Cnm variant that contains a cleavable site within the threonine-rich B-domain. Thus, upon protease digestion, glycopeptides containing far fewer repeats would be released, hopefully enabling glycan analysis. In addition, as the field of glycobiology continues to evolve, novel approaches in MS, nuclear magnetic resonance (NMR), analytical glycoscience and single-molecule force spectroscopy, among others, should also facilitate a more in-depth characterization of Pgf-dependent glycosylation and its substrates.
The discovery of the Pgf system and the identification of homologous systems in other streptococcal species open the possibility of using S. mutans as a model organism for glycoproteomics research. This possibility is particularly attractive considering that S. mutans is a genetically amenable organism with a large number of fast and reliable genetic tools available35. Thus, defining the individual functions of PgfS, PgfM1, PgfE and PgfM2 and their biochemical requirements during the glycosylation process will be of paramount importance if this machinery is to be employed in heterologous expression systems and/or in therapeutic targeting of glycosylated virulence factors36,37. While a thorough analysis of the Cnm glycosylation process has been difficult due to the technical constraints of its highly repetitive threonine-rich domain, the identification of WapA as an additional target of the Pgf system provides an alternate substrate for the characterization of this glycosylation pathway. In fact the magnitude of the band shift on SDS-PAGE, and the number of threonine and serine residues within the protein, suggest that WapA is not as heavily glycosylated as Cnm and therefore may be more amenable to MS analysis.
Adhesion is a critical first step for infection initiation within a host and involves bacterial surface molecules. Many virulence factors associated with initial colonization, and/or invasion of host cells have been demonstrated to undergo posttranslational modification through glycosylation1. The importance of protein glycosylation in virulence has been demonstrated in many bacteria including Gram-positive pathogens1,38,39,40,41. In S. agalactiae, for example, the O-glycosylated serine-rich surface protein Srr1 is an important virulence factor that contributes to adherence to lung epithelial cell lines and to virulence in a rat model of neonatal sepsis41. Inactivation of the glycosylation machinery (GtfCDEFGH) impacted the ability of the mutant strains to display full virulence indicating that glycosylation of Srr1 contributes to bacterium-host cell interactions and resistance to proteolysis41. Here, we showed that Cnm modification contributes to invasion of host cells and virulence in the G. mellonella model. Thus our findings also serve to strengthen the growing literature that posttranslational modification through glycosylation of surface proteins plays an important and still underestimated role in streptococcal virulence.
Taken altogether, this study strongly indicates that the pgfS-M1-E-M2 operon encodes for a glycosylation pathway responsible for the posttranslational modification of at least two S. mutans surface proteins. We showed that this posttranslational modification increases the proteolytic stability of Cnm and WapA, a desirable trait for surface exposed adhesins that are constantly challenged by host and bacteria-derived proteases. Thus, the Pgf system could represent an attractive new target for the development of therapeutic approaches against S. mutans and, possibly, other Streptococci. In addition, a better understanding of how the Pgf system functions can potentially lead to the utilization of this system as a new tool in synthetic biology for the modification of recombinant proteins used for therapeutic and non-therapeutic purposes1,11,36,42.
Methods
Bacterial strains and culture conditions
Strains of S. mutans used in this study are listed in Table 2. Strains were routinely cultured in brain heart infusion (BHI) medium at 37 °C in a humidified 5% CO2 atmosphere. When required, 1 mg ml−1 kanamycin or 10 μg ml−1 erythromycin was added to BHI broth or plates. Primary HCAEC were purchased form Lonza Laboratories and cultured in endothelial cell basal medium 2 (EBM-2; Lonza) supplemented with EGM-2MV single-use aliquots (Lonza), as suggested by the supplier.
Genetic manipulation of S. mutans
Mutation of pgfS (smu2067c) in S. mutans OMZ175 and UA159 was previously described along with its complemented counterparts6. The genes smu2066c (pgfM1), smu2065c (pgfE) and smu2064c (pgfM2) were individually replaced with a non-polar kanamycin cassette via allelic exchange. A quadruple mutant was also generated by replacing all pgf genes (pgfS-M1-E-M2) with the same non-polar kanamycin cassette used to create the single mutant strains. Briefly, PstI sites were introduced to DNA fragments containing the 5′ and 3′ regions of pgfM1, pgfE or pgfM2 using the primers listed in Table 3. For the quadruple mutant construction (Δpgf), DNA fragments containing the 5′ region of pgfS and 3′ region of pgfM2 were generated using primers pgfS F1 and pgfM2 R2. Upon amplification, PCR products were digested with PstI and then ligated to a PstI-digested non-polar kanamycin cassette. The wapA gene was inactivated in OMZ175 using a non-polar kanamycin cassette. Briefly, KpnI sites were introduced to DNA fragments containing the 5′ and 3′ regions of wapA using the primers listed in Table 3. Upon amplification, PCR products were digested with KpnI and then ligated to a KpnI -digested non-polar kanamycin cassette. Transformation of S. mutans OMZ175 was carried out by growing bacterial cells to an OD600 of 0.1 followed by the addition of 250 ng of DNA along with 100 nM of synthetic competence stimulating peptide (CSP)43. Cultures were grown for 4 hours and transformants selected on plates containing kanamycin. All mutations were confirmed by PCR and sequencing of the insertion site.
Genetic complementation of the pgf single mutants was achieved by restoring the original sequence at the exact same location as detailed elsewhere6. Briefly, a PCR product containing the intact and flanking region of each pgf gene was amplified from OMZ175 using primers listed in Table 3. Cultures were grown overnight in chemically defined medium containing 0.5% glucose (CDMG)44, diluted 1:20 in fresh CDMG and grown to OD600 0.1. Cultures of ΔpgfM1, ΔpgfE or ΔpgfM2 mutants were then transformed with 250 ng of their respective gene fragment in the presence of 100 nM of ComX-inducing peptide (XIP)45, along with 50 ng of pCJK9646, which confers resistance to erythromycin, and allowed for the selection of naturally competent cells. After growing cells for 4 h, positive transformants were selected on plates containing erythromycin. Resulting colonies were patched on plates containing erythromycin or kanamycin and colonies that grew in erythromycin but not in kanamycin were screened by PCR. Reintroduction of the pgfM1, pgfE and pgfM2 genes was confirmed by sequencing each gene and respective flanking regions.
In silico analysis
The Prokaryotic Promoter Predictor software (http://bioinformatics.biol.rug.nl/websoftware/ppp/) was used to determine putative promoters driving transcription of pgfS, pgfM1, pgfE and pgfM2, and ARNold software (http://rna.igmors.u-psud.fr/toolbox/arnold/) was used to determine putative transcriptional terminators. The FGENESB bacterial operon and gene prediction software (http://www.softberry.com/berry.phtml?topic = fgenesb&group = programs&subgroup = gfindb) was used to predict the co-transcription of pgfS, pgfM1, pgfE and pgfM2.
RT-PCR analysis
For RT-PCR, RNA was extracted from S. mutans OMZ175 cultures grown to mid-exponential phase (OD600 ~ 0.5) as previously described47. cDNA from 0.5 μg of RNA was synthesized using a high-capacity cDNA reverse transcriptase kit containing random primers (Applied Biosystems). Primers specific for coding and intergenic regions of cnm, pgfS, pgfM1, pgfE and pgfM2 (Table 3) were used to determine the transcriptional organization of these genes.
Western blot analysis
Overnight cultures were used to prepare whole cell protein lysates of S. mutans OMZ175, UA159 and derivatives. Briefly, cells were suspended in PBS hand homogenized in the presence of 0.1 mm glass beads using a bead-beater (Biospec) in three intervals of 30 seconds. Protein lysates (10 mg ml−1) were separated on 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). Cnm detection was performed using rabbit anti-rCnmA polyclonal antibody19 diluted 1:2000 in PBS + 0.1% Tween 20. WapA detection was performed using rabbit anti-WapA polyclonal antibody48 diluted 1:2000 in PBS + 0.1% Tween 20. To visualize protein bands, HRP-labeled anti-rabbit horseradish peroxidase secondary antibody (Sigma-Aldrich) diluted 1:2000 was used followed by detection with an enhanced chemiluminescent (ECL) Western blotting detection kit (GE Healthcare).
Proteinase K susceptibility assay
Susceptibility of Cnm and WapA to protease degradation was determined as previously described6. Briefly, cells from overnight cultures of the indicated S. mutans strains were pelleted by centrifugation and resuspended in 1 × PBS pH 7.2 containing increasing amounts of proteinase K (Sigma-Aldrich). After 30 min incubation on ice, protease activity was neutralized by addition of a protease inhibitor cocktail for 5 min (Thermo Scientific). Bacterial cells were washed once with PBS and Cnm stability was analyzed by Western blot.
Collagen binding and human coronary artery endothelial cells (HCAEC) invasion
In vitro assays for collagen binding and HCAEC invasion were performed as previously described18,19. Briefly, for collagen binding assays, 100 μl of PBS-washed bacterial suspensions containing approximately 1 × 109 CFU ml−1 were added to each well of a microtiter plate containing immobilized type I collagen from rat tail (Sigma-Aldrich). Adherent cells were stained with 0.05% crystal violet (CV) solution and OD575 was measured. To determine the contribution of Cnm glycosylation to collagen binding, experiments were performed as described above but bacteria were pre-incubated with either 500 ng of anti-rCnmA rabbit antiserum or with 1 µg of wheat germ agglutinin (WGA) for 30 min. Samples were then washed with PBS and 100 µl of the cell suspensions were then added to corresponding wells. For HCAEC invasion, 1 ml of 2% FBS-EBM-2 medium containing 1 × 107 CFU ml−1 of S. mutans was used to infect HCAEC-containing wells [multiplicity of infection (MOI) of 100:1], for 2 h in the absence of antibiotics followed by 3 h incubation in 1 ml of 2%FBS-EBM-2 medium containing 300 μg ml−1 gentamicin and 50 μg ml−1 penicillin G to kill extracellular bacteria. After incubation with antibiotics, HCAECs were lysed with 1 ml of sterile water and the mixture of lysed HCAEC and S. mutans plated onto TSA agar to determine the number of intracellular bacteria. The percentage of invasion for each strain was calculated based on the initial inoculum and the intracellular bacteria recovered from HCAEC lysates. All experiments were performed at least in triplicate. A one-way ANOVA was performed to verify the significance of binding and invasion between parent and mutant strains. P values ≤ 0.05 were considered significant.
Lectin-binding analysis
Whole cell lysates of S. mutans strains were separated by SDS-PAGE and transferred to a PVDF membrane as described above. After blocking with 5% bovine serum albumin (BSA) for 1 h at room temperature, the membranes were incubated with 20 μg ml−1 of biotinylated wheat germ agglutinin (WGA) (Vector Laboratories) in PBS containing 0.5% BSA for 1 h at room temperature. Membranes were washed three times with PBS containing 0.1% Tween 20, followed by incubation with HRP-conjugated streptavidin (Cell Signaling Technology). Bound lectins were visualized using the ECL detection kit (GE Healthcare).
Galleria mellonella infection
For the G. mellonella infection model, 5 μl aliquots containing 1 × 108 CFU ml−1 in sterile saline of overnight-grown cultures of S. mutans were injected into the hemocoel of each larva via the last left proleg as detailed elsewhere47. Larvae injected with heat-inactivated S. mutans strains (30 min at 80 °C) were used as controls. After injection, larvae were kept in the dark at 37 °C, and survival was recorded at selected intervals. Experiments were performed in triplicate. Kaplan-Meier killing curves were plotted and estimations of differences in survival were compared using the log-rank test. P values ≤ 0.05 were considered significant.
Expression and purification of recombinant Cnm
To generate full-length recombinant Cnm (rCnm-FL), a derivative of the cnm gene encoding amino acids 32–465 was amplified from OMZ175 using the primers listed in Table 3. The amino acids for the N-terminal secretion signal (1–31) were excluded to avoid toxic effects in E. coli. The amplified PCR product and the expression vector pET16b (Clontech) were each digested with NdeI and BamHI, ligated and transformed into E. coli BL21 (DE3). Cells harboring the pET16b::rCnm-FL were grown in LB containing ampicillin to OD600 0.5 and expression was achieved by the addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 18 h at 24 °C. Three additional constructs (rcnm N1, rcnm N2, rcnm N1+2) were designed from rCnm FL using primers listed in Table 3. The PCR products and the pET23d expression vector were then digested with XhoI and NcoI, ligated and transformed into E. coli BL21 (DE3). Cells bearing the recombinant plasmids were grown to OD600 0.6 and expression was achieved by the addition of 1 mM IPTG at 37 °C for 4 h. For protein purification, cells were harvested by centrifugation, lysed, filtered and then loaded onto His-Prep affinity columns (GE Healthcare). Non-specific proteins were eluted with 50 mM imidazole in binding buffer, while proteins of interest were eluted with a 50–300 mM imidazole gradient of the same buffer. After confirming expression and purification of the different fragments by SDS-PAGE, the respective fractions were pooled and dialyzed against 20 mM HEPES, pH 7.4 for subsequent analysis.
Surface plasmon resonance
Real-time binding analyses of Cnm proteins (rCnm N1, rCnm N2 rCnm N1+2 and rCnm FL) with type I collagen (BD-Biosciences, Franklin Lakes, NJ) were carried out using the BIAcore 2000 system (GE Healthcare) (Fig. 4C). A CM5 chip was labeled with collagen ligand as previously described49, using the amine coupling kit (GE Healthcare) followed by blocking of both control and experimental surfaces using 1 M ethanolamine. Various concentrations (0.125 µM to 2.5 µM) of rCnm analytes were injected over the prepared chip surfaces and dissociation was measured for 8 to 10 min at a flow rate of 20 μl min−1 of binding buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 2.5 mM CaCl2) at 25 °C. The regeneration of the surface between experiments was accomplished using 1 M NaCl, 20 mM EDTA, pH 7.2. All experiments were carried out in triplicate and the kinetics of the association (KA) and dissociation (KD) rate constants were deduced using the 1:1 Langmuir Kinetic model on the BIA-Evaluation software (GE Healthcare).
Evaluation of cell-surface localized Cnm by flow cytometry
Strains were grown overnight in BHI, diluted 1:40 in fresh medium and grown to OD600 of 0.5. Cultures were then washed three times in PBS by centrifugation at 13,000 RPM for 3 min and 500 μl of cells (approximately 1 × 108 cfu ml−1) were incubated with 2.5 μg of purified anti-rCnm rabbit IgG in 500 μl PBS for 1 h at room temperature with continuous rotation. Cells were then washed three times in PBS followed by incubation with 5 μg of Alexa-488 anti-rabbit in 500 μl PBS for 1 h at room temperature with continuous rotation. Finally, cells were washed three more times in PBS, resuspended in 500 μl PBS and surface-localized Cnm was quantified using the Accuri C6 (BD-Biosciences) Flow Cytometer at the University of Florida Cytometry Core.
References
Nothaft, H. & Szymanski, C. M. Protein glycosylation in bacteria: sweeter than ever. Nat. Rev. Microbiol. 8, 765–778, https://doi.org/10.1038/nrmicro2383 (2010).
Abu-Qarn, M., Eichler, J. & Sharon, N. Not just for Eukarya anymore: protein glycosylation in Bacteria and Archaea. Curr. Opin. Struct. Biol. 18, 544–550, https://doi.org/10.1016/j.sbi.2008.06.010 (2008).
Egge-Jacobsen, W. et al. O-linked glycosylation of the PilA pilin protein of Francisella tularensis: identification of the endogenous protein-targeting oligosaccharyltransferase and characterization of the native oligosaccharide. J. Bacteriol. 193, 5487–5497, https://doi.org/10.1128/JB.00383-11 (2011).
Tang, G., Ruiz, T. & Mintz, K. P. O-polysaccharide glycosylation is required for stability and function of the collagen adhesin EmaA of Aggregatibacter actinomycetemcomitans. Infect. Immun. 80, 2868–2877, https://doi.org/10.1128/IAI.00372-12 (2012).
Bouche, L. et al. TheType B Flagellin of Hypervirulent Clostridium difficile Is Modified with Novel Sulfonated Peptidylamido-glycans. J. Biol. Chem. 291, 25439–25449, https://doi.org/10.1074/jbc.M116.749481 (2016).
Aviles-Reyes, A. et al. Modification of Streptococcus mutans Cnm by PgfS contributes to adhesion, endothelial cell invasion, and virulence. J. Bacterio.l 196, 2789–2797, https://doi.org/10.1128/JB.01783-14 (2014).
Lees-Miller, R. G. et al. A common pathway for O-linked protein-glycosylation and synthesis of capsule in Acinetobacter baumannii. Mol. Microbiol. 89, 816–830, https://doi.org/10.1111/mmi.12300 (2013).
Iwashkiw, J. A., Vozza, N. F., Kinsella, R. L. & Feldman, M. F. Pour some sugar on it: the expanding world of bacterial protein O-linked glycosylation. Mo.l Microbiol. 89, 14–28, https://doi.org/10.1111/mmi.12265 (2013).
Szymanski, C. M. & Wren, B. W. Protein glycosylation in bacterial mucosal pathogens. Nat. Rev. Microbiol. 3, 225–237, https://doi.org/10.1038/nrmicro1100 (2005).
Faridmoayer, A. et al. Extreme substrate promiscuity of the Neisseria oligosaccharyl transferase involved in protein O-glycosylation. J. Bio.l Chem. 283, 34596–34604, https://doi.org/10.1074/jbc.M807113200 (2008).
Fulton, K. M., Smith, J. C. & Twine, S. M. Clinical applications of bacterial glycoproteins. Expert Rev. Proteomics 13, 345–353, https://doi.org/10.1586/14789450.2016.1166054 (2016).
Moye, Z. D., Zeng, L. & Burne, R. A. Fueling the caries process: carbohydrate metabolism and gene regulation by Streptococcus mutans. J. Oral Microbiol. 6, https://doi.org/10.3402/jom.v6.24878 (2014).
Bowen, W. H. & Koo, H. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 45, 69–86, https://doi.org/10.1159/000324598 (2011).
Burne, R. A. et al. Opportunities for disrupting cariogenic biofilms. Adv. Dent. Res. 21, 17–20, https://doi.org/10.1177/0895937409335593 (2009).
Lemos, J. A., Abranches, J. & Burne, R. A. Responses of cariogenic streptococci to environmental stresses. Curr. Issues Mol. Biol. 7, 95–107 (2005).
Miller, J. H. et al. The collagen binding protein Cnm contributes to oral colonization and cariogenicity of Streptococcus mutans OMZ175. Infect. Immun. 83, 2001–2010, https://doi.org/10.1128/IAI.03022-14 (2015).
Freires, I. A. et al. Heterologous expression of Streptococcus mutans Cnm in Lactococcus lactis promotes intracellular invasion, adhesion to human cardiac tissues and virulence. Virulence 8, 18–29, https://doi.org/10.1080/21505594.2016.1195538 (2017).
Abranches, J. et al. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect. Immun. 79, 2277–2284, https://doi.org/10.1128/IAI.00767-10 (2011).
Aviles-Reyes, A., Miller, J. H., Simpson-Haidaris, P. J., Lemos, J. A. & Abranches, J. Cnm is a major virulence factor of invasive Streptococcus mutans and part of a conserved three-gene locus. Mol. Oral Microbiol. 29, 11–23, https://doi.org/10.1111/mom.12041 (2014).
Tonomura, S. et al. Intracerebral hemorrhage and deep microbleeds associated with cnm-positive Streptococcus mutans; a hospital cohort study. Sci Rep 6, 20074, https://doi.org/10.1038/srep20074 (2016).
Misaki, T. et al. Presence of Streptococcus mutans strains harbouring the cnm gene correlates with dental caries status and IgA nephropathy conditions. Sci Rep 6, 36455, https://doi.org/10.1038/srep36455 (2016).
Nomura, R. et al. Potential involvement of collagen-binding proteins of Streptococcus mutans in infective endocarditis. Oral Dis. 19, 387–393, https://doi.org/10.1111/odi.12016 (2013).
Nomura, R., Ogaya, Y. & Nakano, K. Contribution of the Collagen-Binding Proteins of Streptococcus mutans to Bacterial Colonization of Inflamed Dental Pulp. PLoS One 11, e0159613, https://doi.org/10.1371/journal.pone.0159613 (2016).
Watanabe, I. et al. Oral Cnm-positive Streptococcus mutans Expressing Collagen Binding Activity is a Risk Factor for Cerebral Microbleeds and Cognitive Impairment. Sci Rep 6, 38561, https://doi.org/10.1038/srep38561 (2016).
Loibl, M. & Strahl, S. Protein O-mannosylation: what we have learned from baker’s yeast. Biochim. Biophys. Acta 1833, 2438–2446, https://doi.org/10.1016/j.bbamcr.2013.02.008 (2013).
Li, H. & Wang, D. Streptococcus mutans wall-associated protein A promotes TLR4-induced dendritic cell maturation. Scand. J. Immunol. 80, 121–126, https://doi.org/10.1111/sji.12194 (2014).
Zhu, L. et al. Functional characterization of cell-wall-associated protein WapA in Streptococcus mutans. Microbiology 152, 2395–2404, https://doi.org/10.1099/mic.0.28883-0 (2006).
Qian, H. & Dao, M. L. Inactivation of the Streptococcus mutans wall-associated protein A gene (wapA) results in a decrease in sucrose-dependent adherence and aggregation. Infect. Immun. 61, 5021–5028 (1993).
Ferretti, J. J., Russell, R. R. & Dao, M. L. Sequence analysis of the wall-associated protein precursor of Streptococcus mutans antigen A. Mol. Microbiol. 3, 469–478 (1989).
Russell, M. W., Harrington, D. J. & Russell, R. R. Identity of Streptococcus mutans surface protein antigen III and wall-associated protein antigen A. Infect. Immun. 63, 733–735 (1995).
Zhu, F. et al. Engineering and dissecting the glycosylation pathway of a streptococcal serine-rich repeat adhesin. J. Biol. Chem.. https://doi.org/10.1074/jbc.M116.752998 (2016).
Bensing, B. A. et al. Novel aspects of sialoglycan recognition by the Siglec-like domains of streptococcal SRR glycoproteins. Glycobiology 26, 1222–1234, https://doi.org/10.1093/glycob/cww042 (2016).
Chen, Y., Seepersaud, R., Bensing, B. A., Sullam, P. M. & Rapoport, T. A. Mechanism of a cytosolic O-glycosyltransferase essential for the synthesis of a bacterial adhesion protein. Proc. Natl. Acad. Sci. USA 113, E1190–1199, https://doi.org/10.1073/pnas.1600494113 (2016).
Harding, C. M. et al. Acinetobacter strains carry two functional oligosaccharyltransferases, one devoted exclusively to type IV pilin, and the other one dedicated to O-glycosylation of multiple proteins. Mol. Microbiol. 96, 1023–1041, https://doi.org/10.1111/mmi.12986 (2015).
Lemos, J. A., Quivey, R. G. Jr., Koo, H. & Abranches, J. Streptococcus mutans: a new Gram-positive paradigm? Microbiology 159, 436–445, https://doi.org/10.1099/mic.0.066134-0 (2013).
Gross, B. J., Swoboda, J. G. & Walker, S. A strategy to discover inhibitors of O-linked glycosylation. J. Am. Chem. Soc. 130, 440–441, https://doi.org/10.1021/ja078125s (2008).
Dwek, R. A., Butters, T. D., Platt, F. M. & Zitzmann, N. Targeting glycosylation as a therapeutic approach. Nat. Rev. Drug Discov. 1, 65–75, https://doi.org/10.1038/nrd708 (2002).
Bleiziffer, I. et al. The Plasmin-Sensitive Protein Pls in Methicillin-Resistant Staphylococcus aureus (MRSA) Is a Glycoprotein. PLoS Pathog. 13, e1006110, https://doi.org/10.1371/journal.ppat.1006110 (2017).
Deng, L. et al. Oral streptococci utilize a Siglec-like domain of serine-rich repeat adhesins to preferentially target platelet sialoglycans in human blood. PLoS Pathog. 10, e1004540, https://doi.org/10.1371/journal.ppat.1004540 (2014).
Wang, N. Y. et al. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J. Infect. Dis. 210, 982–991, https://doi.org/10.1093/infdis/jiu151 (2014).
Mistou, M. Y., Dramsi, S., Brega, S., Poyart, C. & Trieu-Cuot, P. Molecular dissection of the secA2 locus of group B Streptococcus reveals that glycosylation of the Srr1 LPXTG protein is required for full virulence. J. Bacteriol. 191, 4195–4206, https://doi.org/10.1128/JB.01673-08 (2009).
Price, N. L. et al. Glycoengineered Outer Membrane Vesicles: A Novel Platform for BacterialVaccines. Sci. Rep. 6, 24931, https://doi.org/10.1038/srep24931 (2016).
Lau, P. C., Sung, C. K., Lee, J. H., Morrison, D. A. & Cvitkovitch, D. G. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J. Microbiol. Methods 49, 193–205 (2002).
Desai, K., Mashburn-Warren, L., Federle, M. J. & Morrison, D. A. Development of competence for genetic transformation of Streptococcus mutans in a chemically defined medium. J. Bacteriol. 194, 3774–3780, https://doi.org/10.1128/JB.00337-12 (2012).
Mashburn-Warren, L., Morrison, D. A. & Federle, M. J. A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol. Microbiol. 78, 589–606, https://doi.org/10.1111/j.1365-2958.2010.07361.x (2010).
Kristich, C. J., Wells, C. L. & Dunny, G. M. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. USA 104, 3508–3513, https://doi.org/10.1073/pnas.0608742104 (2007).
Kajfasz, J. K. et al. Two Spx proteins modulate stress tolerance, survival, and virulence in Streptococcus mutans. J. Bacteriol. 192, 2546–2556, https://doi.org/10.1128/JB.00028-10 (2010).
Besingi, R. N. et al. Functional amyloids in Streptococcus mutans, their use as targets of biofilm inhibition and initial characterization of SMU_63c. Microbiology 163, 488–501, https://doi.org/10.1099/mic.0.000443 (2017).
Oli, M. W., McArthur, W. P. & Brady, L. J. A whole cell BIAcore assay to evaluate P1-mediated adherence of Streptococcus mutans to human salivary agglutinin and inhibition by specific antibodies. J. Microbiol. Methods 65, 503–511, https://doi.org/10.1016/j.mimet.2005.09.011 (2006).
Acknowledgements
This work was supported by NIDCR R01 DE022559, NIDCR R01 DE021789, NIDCR T90 DE21990 and NHLBI F31 HL124952.
Author information
Authors and Affiliations
Contributions
A.A.R., C.D., L.J.B., J.A.L., J.A. conceived the experiments; A.A.R., I.A.F., S.P., C.D. and R.B. performed the experiments; A.A.R., L.J.B., J.A. and J.A.L. co-wrote the paper. All authors discussed the results and revised the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Avilés-Reyes, A., Freires, I.A., Besingi, R. et al. Characterization of the pgf operon involved in the posttranslational modification of Streptococcus mutans surface proteins. Sci Rep 8, 4705 (2018). https://doi.org/10.1038/s41598-018-23170-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23170-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
