
Abstract
Bud dormancy transition is a vital developmental process for perennial plant survival. The process is precisely regulated by diverse endogenous genetic factors and environmental cues, but the mechanisms are not yet fully understood. Prunus mume is an ideal crop for bud dormancy analysis because of its early spring-flowering characteristics and small sequenced genome. Here, we analyzed the transcriptome profiles at the three endodormancy stages and natural flush stage using RNA sequencing combined with phytohormone and sugar content measurements. Significant alterations in hormone contents and carbohydrate metabolism have been observed, and α-amylases, Glucan Hydrolase Family 17 and diphosphate-glycosyltransferase family might play crucial roles in the interactions between hormones and sugars. The following hypothetical model for understanding the molecular mechanism of bud dormancy in Prunus mume is proposed: low temperatures exposure induces the significant up-regulation of eight C-repeat binding factor genes, which directly promotes all six dormancy-associated MADS-box genes, resulting in dormancy establishment. The prolonged cold and/or subsequently increasing temperature then decreases the expression levels of these two gene families, which alleviates the inhibition of FLOWERING LOCUS T and reopens the growth-promoting pathway, resulting in dormancy release and the initiation of the bud break process.
Similar content being viewed by others
Introduction
Perennial plant species face a multitude of abiotic stresses during their long lifespans. In winter, low temperature and short photoperiod are the most serious threats to most tree species in temperate zones1. Bud dormancy is a bet-hedging strategy for avoiding injury in unsuitable environments and for synchronizing their annual growth. Furthermore, bud dormancy is an agronomically important trait, influencing flower uniformity and fruit quality in the following growing season2. Lang et al.3 distinguished three types of bud dormancy: paradormancy, mainly promoted by other plant organs; endodormancy, maintained by signals internal to the bud; ecodormancy, induced by external environmental factors. Dormancy is regulated by complex endogenous signaling networks, such as hormone signaling and sink/source organ activity, and multiple environmental signals, such as temperature and day length, to ensure flowering at right time4. In recent years, dormancy regulation has garnered additional interest owing to global warming and unseasonal temperature fluctuations5.
Recently, increasing transcriptomic studies have been carried out to define gene regulatory networks in buds during the dormancy transition process, and several conserved factors been identified. An important breakthrough was the determination of the close relationship between dormancy-associated MADS-box (DAM) genes and dormancy phase transition, up-regulated during dormancy induction and down-regulated during release6. This seasonal expression patterns are reported in peach7,8,9,10, pear11, apple12,13, Japanese pear14 and Japanese apricot15. Recently, Niu et al.16 constructed a proposed DAM centered model in pear. The low temperatures in autumn promote the accumulation of C-repeat binding factor (CBF), which can directly promote DAM expression. Subsequently, the increased DAM inhibits FLOWERING LOCUS T (FT) expression, which can induce the endodormancy establishment. After enough chilling achieved, miR6390 promotes the degradation of DAMs mRNAs to promote endodormancy release. Although an increasing number of studies have focused on this field and achieved some significant achievements, the genetic factors underlying the control of dormancy are still not well understood.
The crucial roles of phytohormones in bud dormancy regulation are becoming more evident17. It is well known that abscisic acid (ABA) and gibberellins (GAs) play antagonistic roles in bud dormancy18,19,20. A high level of ABA increases the depth of dormancy, while a high level of GA increases the release of dormancy. ABA levels may increase in the autumn, resulting in the inhibition of cell proliferation and the induction of dormancy. The participation of GAs in breaking poplar bud dormancy has been well reported21. GA induces the up-regulation of 1,3-β-glucanases to hydrolyze callose at the plasmodesmata (PD), providing conduit for FT transport to promote dormancy release process22,23. In fact, not only endogenous ABA/GA individual concentrations, but also a dynamic balance of ABA/GA hormone-signaling network is thought to be central to dormancy induction and release20,24,25,26. In addition, recent findings demonstrate that other hormones, such as indole-3-acetic acid (IAA), ethylene (ET), cytokinin (CK), salicylic acid (SA), jasmonic acid (JA), and brassinosteroids (BR), are also critical for inducing and maintaining dormancy and, therefore, might act as key protectors of seed dormancy27. The interactions among these plant hormones controls the interconnected molecular process of bud dormancy, but their detailed roles remain to be discovered.
Tarancón et al.4 proposed that dormant buds are in carbon starvation situation triggered by environmental and endogenous cues. In the face of this carbon-limiting situation, the plants adjust carbohydrate metabolism between storage and soluble sugars to support essential maintenance functions28,29. For example, total soluble sugars increase at the onset of cold conditions, reaching their maximum at full cold hardiness and declining during de-acclimation30. Additionally, sucrose may be accumulated to protect against cold-induced damage under winter’s low-temperature conditions31. Recently, energy metabolism was demonstrated as being essential for dormancy transition, not only as energy substance but also as sugar-related signals32,33,34. While the role of sugar as an energy supply has been relatively well studied, the possible roles in mediating bud dormancy transition are still largely unknown.
Prunus mume, a rosaceous tree, is highly valued for its ornamental and economic importance. The most striking distinction of P. mume flowers is the early spring-flowering35. These flower buds continue to develop until late autumn, and then enter into dormant stage triggered by short daylength and low temperature. After passing a certain period of chilling temperature, flowers resume growth once environmental conditions are suitable36. During the long breeding time, the availability of diverse germplasm having different chilling requirements and flowering times provided excellent biomaterials for bud dormancy analyses. Previous transcript studies of dormancy release for several Prunus genus, such as P. avium37,38, P. armeniaca39 and P. persica40,41,42,43 have been performed, and some important regulators and signaling pathways have been identified. However, the gene regulatory networks are not well defined. Furthermore, the conservation of these identified genes throughout tree species is not well established4. Thus, understanding how P. mume perceives these seasonal changes to orchestrate the genetic regulation is critical, especially in the context of climate unseasonal change.
Endodormancy differs from the other types of dormancy. Endodormant buds cannot resume growth under favorable conditions, and require enough chilling accumulation for the transition to ecodormancy. Genes showing chilling-mediated differential expression patterns are candidates for internal factors controlling endodormancy. In this study, we further separate endodormancy into three stages (EDI, EDII and EDIII) according to chilling accumulation, and a natural flush (NF) stages were also taken into consideration during the analysis of the entire dormancy-activity cycle using RNA sequencing (RNA-Seq). Additionally, the sugars and hormones contents were measured using a spectrophotometer and high-performance liquid chromatography coupled to electrospray ionization–tandem mass spectrometry (HPLC–ESI–MS/MS). These novel transcriptomes offer comprehensive expression profiling data for a dynamic view of gene variations during bud dormancy in P. mume.
Results
Quantification and measurement of bud dormancy status in P. mume
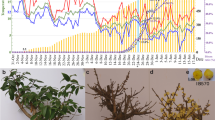
Until now, quantifying the exact dormancy status has been a prerequisite of dormancy studies, and no convincing method nor validated molecular marker have been available for demarcating the status18. In this study, we measured the dormancy level using repeated samplings of branch cuttings as detailed in the Methods section. Figure 1a shows the percentage of bud breaking after 10 d of controlled cultivation in a phytotron, and morphologic features at four representative sampling points. The chilling accumulation (number of hours below 7.2 °C) was recorded using a HOBO (Fig. 1b). On Nov. 6 [accumulated chilling hours (CH) = 0], leaves began to fall, and flower buds had no signs of flushing under controlled conditions. By Nov. 22, the accumulated chilling hours achieved 248 CHs, the one-year-old shoots still had no flush sign in the phytotron. Thus, we termed this time point as endodormancy I (EDI). From Nov. 29, the flower buds started to be released (~3%). However, the flush rate was negligible and unstable among the three replicates, and the so-called flushed buds were aborted immediately. On Dec. 6, with a 15% flush rate and 605 CHs, buds fluctuated depending on the cuttings. Moreover, the flushed buds were mainly at the middle position of the shoots, indicating different dormancy states even in the same shoot. By Dec. 14, there was a 45% flush rate and 748 CHs. The flush rate was stable. Therefore, we termed this time point as endodormancy II (EDII). From Dec. 14 to Jan. 6, bud-burst percentage continued to increase, and time to bud burst continued to decrease. By Jan. 6, the flower buds had completely flushed, and, more importantly, all of the flushed flowers were morphologically normal after a 6-d controlled cultivation in a phytotron. Therefore, we termed this time point as endodormancy III (EDIII). On Feb. 18, flower buds showed green tips under natural conditions, suggesting that dormancy had been completely released. Thus, based on morphology and chilling accumulation, we considered Nov. 22 as EDI, Dec. 14 as EDII, Jan. 6 as EDIII and Feb.18 as NF.

Characterization of flower bud dormancy status and seasonal temperature records. (a) Changes in bud dormancy status represented by bud-break percentages after 10 d phytotron cultivation and morphologic features at four representative time points. (b) Daily maximum, minimum and average temperatures and cumulative chilling hours during the sample collection periods.
Hormone contents in buds during dormancy transition
The levels of hormones were analyzed at four dormant points by HPLC–ESI–MS/MS (Fig. 2). The ABA content was high in EDI, decreased sharply by 44% from EDI to EDII and then by 27% from EDII to EDIII and 21% from EDIII to NF (Fig. 2a). The IAA content decreased by 48.5% from EDI to EDII but then increased sharply by 40% from EDII to EDIII and 130% from EDIII to NF (Fig. 2b). To our surprise, a consistent trend was observed for the IAA and GA contents. The GA3 content was relatively low during the EDI, EDII and EDIII dormancy stages but then increased sharply by 303.5% from the EDIII to NF stages (Fig. 2c). This trend was also observed for the GA1 and GA4 contents, although their levels were very low (Fig. 2d,e). Interestingly, the ABA/GA3 ratio was observed with little changes between EDI and EDII, and then decreased sharply by 64.1% from EDII to EDIII and 81.8% from EDIII to NF (Fig. 2f).

Phytohormone content in flower buds during the dormancy process in P. mume. (a) ABA; (b) IAA; (c) GA3; (d) GA1; (e) GA4; (f) ABA/GA3 ratio. Values are means of three replicates ± SE. Data are means with three biological replicates each, with error bars representing standard error. Non-overlapping letters (a–d) indicate significant differences between different bud stages, based on analysis of variance and multiple range test procedures with a confidence level of 95%.
Sugar contents in buds during dormancy transition
The contents of sugars and their catabolites were also analyzed at the four time points (Fig. 3). Starch and amylose levels were high at the beginning of bud dormancy, and then decreased significantly from EDI to NF stages (Fig. 3a,b). Soluble sugar, sucrose and glucose increased significantly from the EDI to EDII stage and then decreased sharply from the EDII to EDIII stage, peaking during the EDII stage (Fig. 3c,d and e). The contents of soluble sugars then increased by 138.0% from the EDIII to NF stage, suggesting that a considerable amount of energy was needed to support flower flush in early spring. The sucrose and glucose contents underwent similar change trends, increasing dramatically by 104.5% and 65.5%, respectively, from the EDIII to NF stage.

Sugar content in flower buds during the dormancy process in P. mume. (a) starch; (b) amylose; (c) total soluble sugar; (d) sucrose; (e) glucose. Values are means of three replicates ± SE. Data are means with three biological replicates each, with error bars representing standard error. Non-overlapping letters (a–d) indicate significant differences between different bud stages, based on analysis of variance and multiple range test procedures with a confidence level of 95%.
Global analysis of RNA-Seq Data
To obtain a genome-wide view of the transcriptome changes in the dormancy transition progression of P. mume, four developmental stages were constructed using RNA-Seq. After stringent quality checks and data trimming, a total of 438,914,500 high quality clean reads containing 65,837,175,000 nucleotides was obtained (Table 1). The percentages of Phred quality scores ≥20 at each base position were all greater than 98%. More than 84% of high quality reads from individual samples could be mapped on the reference genome, and 24,212 (out of 31,390 predicted genes) genes were found to be expressed (data not shown). More details are shown in Table 1. All of the raw read data were deposited in the Genome Sequence Archive with the project ID PRJCA000291.
Overall relatedness among the 12 transcriptomes
The degree of reproducibility was evaluated based on the square of the Pearson correlation coefficient, represented by the R2 value. A principal component analysis (PCA) was also performed to estimate the overall relatedness of transcriptomes. Overall, the expressions between all of the biological replicates were highly correlated, and this dataset could be used for further statistical analyses. Additionally, the R2 values of EDI vs EDII and EDII vs EDIII were high, and the R2 values between replicates of NFs were relatively low (data not shown). The PCA result was consistent with the analysis of the Pearson correlation coefficient (Supplementary Fig. S1). These data suggested that the dormancy release was a smooth transition process, and that the NF was a stage of dramatic changes with a more complex transcriptome.
Successive pairwise comparisons of DEG profiles
Because of the smooth dormancy transition, we compared transcriptomes of each successive stage with false discovery rates ≤0.05 and fold changes ≥1.5 and fold change ≤−1.5 (|log2Ratio| ≥0.58496). In total, 5,831 DEGs were found to be significantly changed during the dormancy transition process (Supplementary Table S1). Among these, 1,198 DEGs occurred between the EDI and EDII libraries, of which 515 were up-regulated and 683 were down-regulated. Between EDII and EDIII, there were 1,116 DEGs, with 403 up-regulated and 713 down-regulated. As expected, the largest number, 4,751 DEGs, was found between EDIII and NF (1,749 up- and 3,002 down-regulated), indicating that the transcript changed dramatically at these key stages. The numbers of DEGs in EDIII vs NF was greater than in EDI vs EDII or EDII vs EDIII, indicating the involvement of complex developmental events during NF. We also analyzed the overlap between the pairwise comparisons at both stages as shown in a Venn diagram (Supplementary Fig. S2). A total of 4,787 (82.1%) transcripts exhibited developmental stage-specific differential expressions, and only a small fraction of genes (190, 3.3%) were found to be common in all the comparisons. A large proportion of DEGs (392), overlapped between EDI vs EDII and EDII vs EDIII, suggesting a relatively smooth transition at the EDI-EDII-EDIII.
GO, KEGG and MapMan annotation analyses of all DEGs
DEGs were annotated by GO, KEGG and MapMan analyses to examine putative functional differences between different successive developmental stages. The significantly enriched GO terms (P-value < 0.05 FDR corrected) were involved in the cellular location, molecular function and biological processes (Supplementary Fig. S3). EDI vs EDII and EDII vs EDIII had far fewer GO terms than EDIII vs NF. Surprisingly, the GO term “microtubule-based process” was only identified in the EDI vs EDII cluster, and “hormone transport” was only identified in the EDII vs EDIII cluster. The terms “response to hormone” and “metabolism” were significantly enriched only in the EDIII vs NF comparison (Supplementary Table S2). The DEGs were then subjected to a KEGG pathway mapping, and the top 20 enriched pathways in each comparison are shown in Supplementary Fig. S4. KEGG showed the dramatic changes in the metabolism of various hormones and starch and sucrose. In addition, several other metabolic pathways, such as biosynthesis of secondary metabolites and secondary cell wall, were also significantly represented (Supplementary Table S3). The MapMan BINs analysis was also used to reveal a global view of changes in several important metabolic pathways and related functional groups during the dormancy transition process. Metabolic activities within the bud are significantly down-regulated during dormancy and then reactivated during release (Supplementary Fig. S5a–c). These effects and changes were mainly in “glycolysis”, “sucrose degradation”, “cell wall” and “lipid metabolism”. In corroboration with previous studies, we found that the DEGs participating in several hormone metabolism and signaling pathways, like ABA, GA, IAA, ET, JA, SA and BR were significantly changed (Supplementary Fig. S5d–f; Supplementary Table S4). Overall, the results were consistent with our enriched GO and KEGG pathways.
Cluster analysis of hormone-regulated DEGs during bud dormancy transition
DEGs encoding hormone metabolism and signaling were further hierarchically clustered. As shown in Fig. 4, four main gene clusters were determined. Cluster A included 51 genes, some exhibiting their highest levels at EDI and some at EDII, and then gradually decreased as dormancy release progressed. ABA signaling genes, such as ABF2, ABF3, ABF4 and HAI1, GA associated genes, such as GA2OX1, GA2OX6, GA1 and RGL1, as well as with IAA-related genes, such as ATB2, WAG1, GH3.1, GH3.17, PIN1, PIN6 and DFL2, all grouped within this cluster. UDP-glycosyltransferases 71B6 (UGT71B6), UGT73C1 and UGT74E2, encoding UDP-glucosyl transferases that preferentially glucosylates ABA, CK and SA respectively, exhibited this expression pattern. Cluster B (21 genes) showed high expression level at EDI stage, and dramatically decreased at EDII, and then maintained relatively low level at EDIII and NF stages. Among them, NCED3 and NCED5 are key ABA biosynthesis genes, and GA2OX2 is key GA catabolism gene, which are in agreement with high ABA and low GAs levels at EDI stage. Several ethylene response factors (ERFs), including ERF2, ERF4, ERF5, ERF9, ERF105, involved in ET-activated signaling pathway, also displayed this expression pattern. Cluster C contained 12 genes, which showed low expression levels at the EDI and EDII stages, then sharply increased at EDIII, and dramatically decreased at NF. GA3OX3 and ACO5, keys genes involved in GA and ET biosynthetic process, and BRS1, involved in BR signaling pathway, belonged to this cluster. PIN8, encoding an auxin transporter, also displayed this expression pattern. Genes in Cluster D (66 genes) typically showed almost constant low expression levels from EDI to EDIII, and then sharply increased at the NF stage, when the dormancy was completely released. Among them, some key genes that respond to ABA (such as NCED4, AREB3), GA (GA5, RGA2, GIDC), IAA (PIN3, ILL6), BR (BAS1, EXL5), ET (ERF7, EAT1) and JA (AOC4, LOX2) were grouped within this cluster. UGT74B1, encoding a UDP-glucosyl transferase that preferentially glucosylates IAA, belonged to this cluster. More details can be seen in Supplementary Table S5.

Hierarchical cluster analysis of hormone-related DEGs during the dormancy process in P. mume. Red indicates high relative gene expression and green indicates low relative gene expression. Letters assigned to major clusters are indicated on the dendrogram. Some key genes were highlighted with triangular symbol.
Cluster analysis of sugar-related DEGs during bud dormancy transition
Sugar-related DEGs, including those involved in carbohydrate biosynthesis and metabolism, transportation and signaling, were grouped into three major clusters as shown in Fig. 5 and listed in Supplementary Table S6. In general, genes in Cluster A were expressed at high levels in at least one of the dormancy stages and at significantly lower levels at the NF stage. The high expression levels of BMY3, BMY4, BAM1, pseudo-response regulator 5 (PRR5), PRR7, PRR9 and starch excess 1 in this cluster, the key genes in the starch metabolic process, are consistent with the decline in the starch contents. In addition, sucrose-proton symporter 2 (SUC2), a major sucrose transporter, exhibited the same expression pattern. The genes in Cluster B (4 genes) exhibited a significant increase between the EDI and EDII stages and then decreased between the EDII and EDIII stages. Among them, major facilitator superfamily protein is involved in sugar transport, and SUS3 is involved in sucrose biosynthesis. Group C, with the largest DEGs (33 genes), exhibited completely opposite expression profile with Cluster A, with a sharply increasing expression pattern during the dormancy release process. Among them, beta-amylase 5 (BYM5), BMY2, alpha-amylase-like (AMY1), AMY3 and heteroglycan glucosidase 1 are involved in starch metabolic processes, and sedoheptulose-bisphosphatase and NDH dependent flow 6 are involved in starch biosynthetic processes. Moreover, many of them are related to sucrose and glucose biosynthetic and metabolic processes, such as sucrose synthase 6 (SUS6), sucrose beta-fructofuranosidase, glucose insensitive 2 (GIN2), and trehalose-phosphatase/synthase 7 (TPS7). In addition, 1,3-β-glucanase, UDP-glc 4-epimerase 1 and maternal effect embryo arrest 31, which involved in carbohydrate biosynthetic and metabolic process, also belong to this cluster.

Hierarchical cluster analysis of sugar-related DEGs during the dormancy process in P. mume. Red indicates high relative gene expression and green indicates low relative gene expression. Letters assigned to major clusters are indicated on the dendrogram. Some key genes were highlighted with triangular symbol.
Cluster analysis of dormancy candidate genes during bud dormancy transition
In 1960, Chouard hypothesized that vernalization and dormancy development share similar signaling components and mechanisms44. Thereafter, evidence from various herbaceous and perennial species has directly or indirectly supported this hypothesis. Thus, 78 dormancy candidate genes (CGs) conserved in Arabidopsis4, and 79 CGs in the regulation of dormancy and flowering date in Prunus species, blackcurrant, poplar and Arabidopsis, were collected45. After strict homolog identification, 146 P. mume putative homologs were identified and 117 genes met the DEG criteria. Thus, only the 117 P. mume putative dormancy CGs were used for further analyses (Supplementary Table S7). Based on their expression profiles, the dormancy genes were analyzed using hierarchical clustering, and four major expression profiles were determined (Fig. 6). In general, genes in Cluster A displayed high expression levels in at least one of the dormancy stages and then sharply decreased at the re-active stage. Among these, 8 putative PmCBFs (named as PmCBF1–8 according physic locus) showed this expression pattern and all grouped together. MADS-box transcription factors, including PmDAM1–6, short vegetative phase (SVP), suppressor of overexpression of CO 1 (SOC1), flowering locuc C (FLC), which are proven components in the dormancy process, exhibited this expression profile. Glucan Hydrolase Family 17 (GH17) and GH17-101 regulating callose degradation, belonged to this cluster. Seven genes in Cluster B exhibited low expression levels at the EDI and EDII stages, and then sharply increased in the EDIII and NF stages. Of these genes, FT has been hypothesized as the essential component of the flowering network and many genes and signaling pathways converge on FT. SWEET11 and sugar transporter 14 (STP14), involved in carbohydrate transport, are grouped in this cluster. Cluster C (three genes) exhibited low expression levels at EDI and increased sharply at true endo-dormancy stage, and then decreased again at the EDIII and NF stages. GH17-61 and STP1, close paralogs of GH17-101 and STP14 respectively, belonged to this cluster. Genes in Cluster D displayed consistent low levels in dormancy stages and much higher levels at the active stage. Among them, KINβ1, a subunit of the SnRK1 kinase, plays an important role in bud dormancy development by promoting sugar metabolism and cell division.

Hierarchical cluster analysis of 117 dormancy CGs during the dormancy process in P. mume. Red indicates high relative gene expression and green indicates low relative gene expression. Letters assigned to major clusters are indicated on the dendrogram. Some key genes were highlighted with triangular symbol.
Validation of RNA-Seq using qPCR
The total RNAs performed for RNA sequencing libraries were used as template for qPCR to validate the RNA-seq data. The qPCR of 12 hormone- and sugar-related genes at four dormancy stages showed the same trends as the RNA-Seq results, despite some differences in magnitude, suggesting that the RNA-Seq data in this study are reliable (Supplementary Fig. S6; Supplementary Table S8).
Discussion
Bud dormancy is a complex programmed strategy that allows perennials to survive extreme low temperature in winter. As the chilling accumulation increases and favorable environmental conditions occur, the external signals are perceived by receptors, resulting in the generation of many secondary signaling molecules. Recently, expression profiles have been performed on several members of the Prunus genus. In this paper, to draw a complete dormancy-activity cycle, we reported a comprehensive transcriptome study at four critical developmental stages. We particularly focused on the MapMan BINs “major carbohydrate metabolism” and “hormone metabolism”, as well as their interaction.
The importance of hormone homeostasis in bud dormancy has been well reviewed18. ABA and GA are the core hormones that antagonistically regulate bud dormancy status. ABA and GA might interact and have both cause: ABA participates in the suppression of GA biogenesis19, and GA also negatively regulates ABA biogenesis during seed germination20. In the present investigation, the ABA content steadily decreased from EDI to EDIII, and even to NF (Fig. 2a). The GAs levels declined from EDI to EDII and then steadily increased (Fig. 2c–e). The ABA/GA ratio steadily decreased during the dormancy release process in our paper (Fig. 2f). The findings in this study regarding endogenous ABA/GA are consistent with those in grapes24, sweet cherry25 and tree peony26.
The decline in the ABA content might contribute to the decreased ABA biosynthesis genes and the increase of ABA catalysis genes. Pm011164 and Pm010425, homologs of ATNCED3 and ATNCED5, respectively, exhibited high expression levels during EDI and then sharply decreased during EDII (Fig. 4). Pm031212 (homolog of ATCYP707A1) exhibited a high expression level at the EDI stage and then gradually decreased during the EDII stage, as confirmed by qPCR (Fig. 4 and Supplementary Fig. S6). Interestingly, Pm014836 (homolog of ATUGT71B6) which can preferentially glucosylate ABA, was expressed at a high level during the EDII and EDIII stages, as confirmed by RNA-Seq and qPCR (Fig. 4 and Supplementary Fig. S6). Our results indicated that at the EDI stage, ABA might be hydrolyzed by CYP707A1 into phaseic acid and/or dihydrophaseic acid. However, at the EDII and EDIII stages, active ABA was glucosylated by ABA glucociltransferase into ABA glucocil ester (ABA-GE), and stored in the flower buds and/or stems. Moreover, ABA-GE could be degraded by β-glucosidases (BGs) in only one step, if necessary46. To our surprise, BGs homologs in P. mume exhibited low expression levels (data not shown), suggesting that ABA-GE was not degraded by BGs during the bud dormancy release process in P. mume.
In the early stage of dormancy formation, the decrease in active GA levels was closely related to growth cessation. After enough chilling accumulation, these contents then increased47. In our study, the GA3 content at the EDI stage were slightly greater than at the EDII stage, and then, they sharply increased from the EDII to EDIII stage (Fig. 2c). This might contribute to the increased transcript abundance of GA3 biosynthesis genes and decreased GA3 catabolic genes. Pm027461, homologs of ATGA3OX3, respectively, a key GA biosynthesis gene, exhibited low expression levels at the EDI stage and then were sharply up-regulated at the EDII and EDIII stages (Fig. 4). GA2OXs, such as Pm010412 and Pm011163, which deactivate bioactive GA, showed relatively high expression levels at the EDI stage and significantly low levels at the EDII stage (Fig. 4). These results were consistent with the decline in the GA contents from EDI to EDII and with the increase from EDII to EDIII.
The balance in ABA/GA signaling is also crucial for bud development. Shu et al.20 reported that ABSCISIC ACID-INSENSITIVE 4 (ABI4) is a key factor that regulates seed dormancy by mediating the ABA/GA balance. ABI4 positively regulates ABA catabolism by binding directly to the promoters of CYP707A1 and CYP707A2, but no direct targeting of GA metabolism genes by ABI4 has been detected. Nevertheless, in sorghum, SbABI4 and SbABI5 can directly bind to the promoter of SbGA2ox3, likely activating its expression and affecting seed dormancy48. ABI3, as the major downstream component of ABA signaling, is another main regulator of seed dormancy and germination49. Transgenic poplar overexpressing ABI3, do not form terminal buds, suggesting a role for ABA in bud dormancy establishment50. In our present study, the expression level of the ABI3 homolog Pm020417 was very low, suggesting that the ABA-signaling pathway was blocked during the dormancy release process (Fig. 4). Thus, the ABA/GA balance during biogenesis and/or their signaling levels may be essential for the bud dormancy–activity cycle in P. mume.
IAA is an essential hormone that is involved in almost all aspects of plant development and in processing environmental cues. For bud dormancy, the auxin-related mechanism remains largely unknown. Recently, Qiu et al.51 reported an increase in the IAA level and several enriched auxin-signaling pathways in the vascular cambium during the transition from dormancy to active growth in Chinese fir. El-Yazal et al.52 discovered that exogenous IAA could alter endogenous hormones and hasten bud break from dormancy in apple trees. Here, the IAA content was up-regulated from the dormancy to reactivity stages (Fig. 2b). Auxin response factors (ARFs), as transcriptional activators, play crucial roles in auxin metabolic pathways during bud dormancy in hybrid aspen53. The expression profiles of ARFs observed here indicated that Auxin signaling pathways might involve in the bud dormancy process of P. mume. Consistent with previous studies, there were up- and down-regulated ARFs during the same comparative periods, suggesting their potential roles are blurred.
In general, dormant buds are considered to be in an inactive state with limited metabolic activities, and the dormancy–activity transition process involved the extensive reconfiguration of carbohydrate metabolism54. In the present investigation, significant changes in sugar-related genes and content levels suggested that sugar might participate in and play important roles in the bud dormancy development of P. mume. Short day lengths and low temperatures during autumn triggered starch accumulations in stems and buds, and subsequently the stored starch was degraded to soluble sugars in response to freezing temperatures during winter28. An energy flow from source to metabolic sink was observed in our study. Starch and amylopectin levels were high in buds at the beginning of dormancy and then declined gradually until dormancy was completely released (Fig. 3a,b). Soluble sugar, sucrose and glucose levels showed the opposite dynamic pattern (Fig. 3c–e). These results are in agreement with those of leafy spurge crown buds during the dormancy transition process29. The high concentrations of sucrose and glucose improved the freezing tolerance by lowering the freezing point of free water and disrupting the formation of ice crystals30. This enables buds to survive winter. The hydrolysis of starch might also provide a carbon skeleton for the synthesis of needed amino acids, lipids and metabolites associated with bud growth as shown in Supplementary Fig. S5 and Supplementary Table S4.
In bud dormancy, the sucrose could promote bud release, not only as an energy supply, but also as a sugar signal that was independent from auxin signaling32. In the present research, the transcriptome data indicated that sugar metabolism and signaling are involved in bud dormancy development. The expression profiles of key genes involved in starch metabolism, and sucrose and glucose biosynthesis, such as BMY5, BYM1, AMY1, AMY3, SUS3, SUS6 and TPS7, were consistent with a decrease in the starch and an increase in sucrose and glucose (Fig. 5). The genes involved in sugar transport, such as GTL1, SUC2, PMT5, SAG29 and MISS1, as well as sugar signaling, such as GIN2, PCAP1 and BETAFRUCT4, also increased with the dormancy release process. Similar results have been reported in other species, such as Arabidopsis33, poplar55 and grapevine34. Thus, these genes may be required for energy reconfiguration to acclimate to the external environment and survive cold winters.
Sugar alone, or through interactions with hormones, can induce or suppress many growth-related genes56. Additionally, crosstalk has been reported between sugar and hormones, including GA, ABA, IAA and SA. The α-amylase family is a relatively well-characterized example of interactions between hormone and sugar in dormancy process. α-Amylases act as key enzymes in starch degradation to generate soluble sugars, playing critical roles in the dormancy process57. They are tightly regulated by GA and ABA. In general, GA promotes the production of α-amylases by GAMYB, and ABA blocks the expression of α-amylases by an ABA-induced protein kinase (PKABA1) and two ABA-inducible WRKY proteins. PKABA1 and WRKY proteins suppress the GA-induced α-amylase’s expression by strongly inhibiting the expression of GAMYB and by competing with GAMYB, respectively58,59,60. In addition, SA might inhibit α-amylase production not by inhibiting α-amylase enzyme activities but by suppressing a GA-induced low pI α-amylase gene expression57. Moreover, SA and ABA promote HvWRKY38 expression through an independent pathway and consequently, suppress the GA-inducible α-amylase further during seed dormancy process in rice57. Meanwhile, the application of exogenous glucose affects GA-mediated α-amylase expression and activity levels through complex pathways61. During the leafy spurge bud dormancy process, GA promotes the synthesis and activity levels of α-amylases, suggesting the potential for GA involvement in the breakdown of starch62.
Another example of sugar and hormone interactions involves the GA-inducible GH17 family. During bud dormancy establishment, PD is blocked by callose, resulting in the inhibition of cell-to-cell communication22. Then, low temperatures induce the accumulation of 1,3-β-glucanases, which can hydrolyze callose. GH17 is a relatively larger family, with 50 members in Arabidopsis, and groups into three clades (α, β and γ). Most members of α-clade localize to PD, while the γ-clade’s members localize to lipid bodies. Both of them can remove callose at PD and play crucial roles in the seed dormancy process23. In poplar, over 100 members were identified, and both α- and γ-clade members potentially target the PD’s callose21. In addition, 10 members are GA responsive and well investigated in relation to bud dormancy21. More importantly, three (GH17-44, GH17-61 and GH17) were mapped as CGs involved in bud dormancy and flowering time on different linkage groups constructed from two progenies of sweet cherry45. In our study, the high expression levels of GH17s (Pm027564, Pm025785 and Pm001330 homologs of GH17, GH17-101 and GH17-61, respectively) and high contents of GAs suggest that they play crucial roles during the bud dormancy process of P. mume (Fig. 5).
Sugars can also affect hormone metabolism, and one mechanism is through UDP-glucose conjugation. UGTs are the most common enzymes that catalyze this process. UGTs can transfer UDP-glucose biosynthesized from sucrose through sucrose synthase to free hormones, resulting in conjugated inactive hormones63. At present, 107 UGTs have been identified in Arabidopsis64. Among them, eight UGTs were characterized to have ABA-associated UGT activity65, and five UGTs showed UGT activity for CK66. Free bioactive ABA in plant cells can be lowered in two ways, hydroxylation and conjugation. CYP707A1 hydroxylates ABA at the C-8´position and converts to phaseic acid and dihydrophaseic acid; on the other hand, UGT71B6 preferentially glucosylates ABA to form ABA-GE67. ABA-GE, as a stable storage form, contributes to ABA homeostasis because ABA-GE can be hydrolyzed through BGs in one step46. However, to our knowledge, the exact UGTs that can preferentially glucosylate GAs have not been identified. In our study, the dynamic patterns of several UGTs, such as Pm014836 (UGT71B6, associated with ABA), Pm030035 (UGT74B1, IAA), Pm014886 (UGT85A1, CK) and Pm026307 (UGT73C1, CK), suggested that hormone conjugation plays important roles during the dormancy process of P. mume (Fig. 4).
Recently, Niu et al.16 constructed a proposed CBF-DAM-FT model in pear. A hypothetical model on low temperature-induced CBF’s increasing freezing tolerance was proposed68. In brief, low temperature-induced CBF expression results in increased GA20X expression and the accumulation of DELLA proteins, which can reduce the GA content and block the GA signaling pathway, respectively. The overexpression of CBF genes can result in increased cold hardiness in several perennials, including poplar69, apple70,71 and grape72. Horvath et al.73 reported that CBF controlled cold-responsive EeDAM1 by binding CBF sites in its promoter. In peach, CBF sites were found in the promoters of PpDAM5 and PpDAM6, which were well-characterized within the dormancy process; however, no CBF sites were found in PpDAM1, PpDAM2 or PpDAM3, which have not been associated with dormancy8,74,75. There is also some evidence that DAM regulates dormancy by inhibiting the flower-promoting FT genes73. The overexpression of DAM1 in Arabidopsis resulted in decreased FT expression and delayed flowering time73. Chromatin immunoprecipitation assays indicated that DAM could bind to the CArG boxes in the promoter regions of FT76. Yeast one-hybrid and transient expression analyses demonstrated that PpDAM1 could bind to the PpFT2 promoter and inhibit its expression16.
In the P. mume genome, 13 putative PmCBF homologs and six tandemly arrayed PmDAMs were identified35. Among them, 8 genes were significantly changed during dormancy development. Surprisingly, all (PmCBF1-8) decreased during the dormancy release process, displaying the same pattern and grouped together as shown in Fig. 6. Moreover, all six PmDAMs were up-regulated more than twofold, and displayed similar dynamic patterns and grouped together, peaking at the EDI stage (Fig. 6). These results were consistent with other transcriptomic dormancy studies on peach8,10 and pear11,77. In the upstream regions of PmDAM4, 5 and 6, more CBF-binding sites were found than in peach. In PmDAM1 and 6, one and two novel CBF sites were found, respectively, while PmDAM2 and 3 had no CBF sites. Remarkably, the expression levels of these PmDAMs correlated with the number of CBF sites. PmDAM4, 5 and 6 (with several CBF sites) showed the highest levels, followed by PmDAM1 (with one novel CBF sites) and then PmDAM2 (with no CBF sites) (Fig. 6; Supplementary Table S7). Thus, dormancy-associated DAMs may be controlled by CBFs binding to the CBF sites in the upstream regions. In the present study, Given that DAM5 and DAM6 were well characterized as having dormancy-associated expressions, it is reasonable to hypothesize that these DAM genes might function in dormancy development in P. mume. In addition, other MADS-box genes, such as SVP (Pm002166), SOC1 (Pm018089) and FLC (Pm016141), that are involved in bud dormancy displayed the same expression patterns as the DAMs and belonged to the same cluster (Fig. 6). These results suggested that MADS-box genes might be closely associated with the dormancy transition process in P. mume.
Conclusions
By combining the physiological and transcriptomic analyses, a hypothetical model was proposed for understanding the interactions between hormones and sugars (Fig. 7). During the four crucial dormancy stages, significant alterations in hormone contents and carbohydrate metabolism were observed, and α-amylase, GH17 and UGT families may play crucial roles in the interactions between hormones and sugars. In autumn, low temperatures exposure activated the significant up-regulation of eight PmCBFs, and then, the PmCBFs blocked the GA-signaling pathway by accumulating DELLA proteins. In addition, the increased levels of PmCBFs activated the up-regulation of all six PmDAMs, resulting in growth cessation and dormancy establishment. Prolonged chilling and/or subsequently increasing temperature then reduced PmCBF expression levels, resulting in high levels of bioactive GAs and the reopening of GA pathways. The down-regulated PmDAMs removed the inhibition to FT. The high level of FT expression and the reopened growth-promoting GA-pathway promoted the dormancy release process and bud break under appropriate conditions.

A hypothetical model for understanding the molecular mechanism of bud dormancy in P. mume. α-Amylase, GH17 and UGT families might play crucial roles in the interactions between hormones and sugars. Positive and negative regulatory actions are indicated by arrows and lines with bars, respectively.
Methods
Plant materials and dormancy status of flower buds
P. mume cultivar ‘Lve’ grown in the Jiufeng International Plum Blossom Garden, Beijing, China (40°07′N, 116° 11′ E) was used in this study. The temperature was recorded by HOBO U23-003 (Massachusetts, United States) every 30 minutes. A typical P. mume leaf axil has three separate buds, a single vegetative bud and two flower buds, and only the flower buds were sampled for further study. Flower buds were collected approximately every 7 d between 12 and 14 a.m. from 6 November 2015 to 8 March 2016 according to our multi-year field observation. Additionally, at every time point, 20 one-year-old shoots were randomly collected, and they were cultivated in water under the following phytotron conditions: 16/8 h 25/16 °C day/night at 70% relative humidity. The water was changed and the basal ends of the shoots were cut every 2 d. After 10 d, the dormancy status was evaluated as percentage bud break, which is the showing of a green tip. Flower buds on the cuttings were collected and stored at −80 °C until use.
RNA extraction, Illumina sequencing, and quality control
For 12 bud samples, total RNA were extracted using the NanoDrop® 2000 (Thermo, CA, USA), and ~5 μg RNA was used for the RNA preparations. Sequencing libraries were generated using NEBNext®Ultra™ RNA Library Prep Kit for Illumina®(NEB, USA) and index codes were added to attribute sequences to each sample. The library preparations were sequenced on an Illumina Hiseq. 2500 platform and paired-end reads were generated following manufacturer’s recommendations. The sequencing reads were filtered using FastQC to remove primer/adapter and low-quality reads. The observed clean data with high quality were aligned to the P. mume genome, and then normalized into Reads Per Kilobase of transcript per million mapped reads (RPKM) values. DESeq R package (1.18.0) was used to analysis differential expression of two successive stages. The resulting p-value was corrected by Benjamini and Hochberg for controlling the false discovery rate (FDR). Differentially expressed genes (DEGs) were defined by FDR ≤0.05 and absolute fold change ≥1.5 (|log2Ratio| ≥0.58496) in successive stages.
PCA, Venn and Heatmap diagrams
Principal component analysis (PCA) was performed using the online OmicShare tools (www.omicshare.com/tools). A Venn diagram of distribution of DEGs was constructed using the R package “VennDiagram”, and the Heatmap was constructed using the R package “pheatmap”.
GO, KEGG and MapMan analyses
For all DEGs, functional annotation by gene ontology (Go) terms was analyzed using the Blast2GO program, with p-value ≤ 0.05 as significantly enriched. To analyze the main biological functions, we mapped the DEGs to terms in the KEGG (Kyoto encyclopedia of genes and genomes) pathways using R software. Mapman version 3.5.1 (http://mapman.gabipd.org/web/guest) was also used to identify specific enriched pathways. TAIR10 version (http://www.arabidopsis.org/) was used as reference with a P-value cut-off of ≤0.05 as ontology.
RNA-Seq validation using real-time quantitative-PCR (qPCR)
The cDNA was synthesized from ~2 μg of total RNA for each sample. The gene-specific primers were designed at the website (http://sg.idtdna.com/primerquest/Home/Index), and the specificity was tested using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome). Protein phosphatase 2A (PP2A, Pm006362) was used as the internal reference control. The analysis was performed with three biological replicates. Fold change was calculated using standard 2−ΔΔCT method. Twelve hormone-related and sugar-related genes qPCR primers are listed in Supplementary Table S8.
Measurements of sugar and hormone contents
Dry and/or fresh buds at four developmental stages were used for sugar and hormones extraction and determination. Dry buds (~0.3 mg) were used for starch and amylose extractions. The starch contents were measured as described by Rosa et al.78, and the amylose contents were determined with an amylose measurement kit (A152-1) from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). For soluble sugar, sucrose and glucose extractions, ~0.5 g fresh buds were used, and they were quantified using a soluble sugar kit (A145), sucrose measurement kit (A099-1) and glucose assay kit (F006), according to the manufacturer’s instructions, respectively. Meanwhile, ~1 g fresh buds were used for ABA, IAA, GA1, GA3 and GA4 extractions with three biological replicates. The hormonal quantification was carried out using HPLC–ESI–MS/MS with a standard measure as described by Dobrev and Vankova79 and Djilianov et al.80.
Bud candidate gene selection and the identification of P. mume orthologs
Based on available dormancy-related data in herbaceous and woody species, 78 dormancy candidate genes conserved in Arabidopsis published by Tarancón et al.4, and 79 CGs conserved in Prunus species, blackcurrant, poplar and Arabidopsis published by Castède et al.42 were selected (Supplementary Table S8). For each CG, the peach ortholog was identified from Arabidopsis sequence in Phytozome v12 (https://phytozome.jgi.doe.gov/pz/portal.html) and other published studies. The putative P. mume orthologs were identified by BLASP (E-value < 1e-10, identity >30% and coverage >70%) using Arabidopsis and peach sequences as query, and genes with the highest E-values were selected. As the well-characterized strong CGs, all 13 putative PmCBFs and 6 PmDAMs identified in P. mume genome19 were included for further analysis. Furthermore, a Pfam domain analysis (http://pfam.xfam.org/) was performed for each CG to ensure the accuracy of the orthologs in three species. In addition, phylogenetic trees were built using MEGA7.1 (Maximum-likelihood method, 1,000 bootstrap replicates) to identify the most closely related orthologs.
Statistical analysis
For hormone, sugar and qPCR results, statistical analysis was performed using SPSS version 19 (SPSS, Chicago, IL, USA) with ANOVA and Duncan’s test. Data are means with three biological replicates each, with error bar representing standard error. Non-overlapping letters (a–d) indicate significant differences between different bud stages, based on analysis of variance and multiple range test procedures with a confidence level of 95%.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files) and also are available from the corresponding author on reasonable request.
References
Hänninen, H. & Tanino, K. Tree seasonality in a warming climate. Trends in Plant Science 16, 412–416, https://doi.org/10.1016/j.tplants.2011.05.001 (2011).
Footitt, S., Clay, H. A., Dent, K. & Finch-Savage, W. E. Environment sensing in spring-dispersed seeds of a winter annual Arabidopsis influences the regulation of dormancy to align germination potential with seasonal changes. The New phytologist 202, 929–939, https://doi.org/10.1111/nph.12694 (2014).
Lang, G. A., Early, J. D., Martin, G. C. & Darnell, R. L. Endo-, para-, and ecodormancy: physiological terminology and classification for dormancy research. Hortscience 22, 371–377 (1987).
Tarancón, C., González-Grandío, E., Oliveros, J. C., Nicolas, M. & Cubas, P. A Conserved Carbon Starvation Response Underlies Bud Dormancy in Woody and Herbaceous Species. Frontiers in Plant Science 8, 788, https://doi.org/10.3389/fpls.2017.00788 (2017).
Abbott, A. G., Zhebentyayeva, T., Barakat, A. & Liu, Z. The Genetic Control of Bud-Break in Trees. Advances in Botanical Research 74, 201–228 (2015).
Böhlenius, H. et al. CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science 312, 1040–1043, https://doi.org/10.1126/science.1126038 (2006).
Bielenberg, D. G. et al. Sequencing and annotation of the evergrowing locus in peach [Prunus persica (L.) Batsch] reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genetics & Genomes 4, 495–507, https://doi.org/10.1007/s11295-007-0126-9 (2008).
Li, Z., Reighard, G. L., Abbott, A. G. & Bielenberg, D. G. Dormancy-associated MADS genes from the EVG locus of peach [Prunus persica (L.) Batsch] have distinct seasonal and photoperiodic expression patterns. Journal of Experimental Botany 60, 3521–3530, https://doi.org/10.1093/jxb/erp195 (2009).
Leida, C. et al. Identification of genes associated with bud dormancy release in Prunus persica by suppression subtractive hybridization. Tree physiology 30, 655–666, https://doi.org/10.1093/treephys/tpq008 (2010).
Leida, C., Conesa, A., Llacer, G., Badenes, M. L. & Rios, G. Histone modifications and expression of DAM6 gene in peach are modulated during bud dormancy release in a cultivar-dependent manner. The New phytologist 193, 67–80, https://doi.org/10.1111/j.1469-8137.2011.03863.x (2012).
Liu, G. et al. Transcriptomic analysis of ‘Suli’pear (Pyrus pyrifolia white pear group) buds during the dormancy by RNA-Seq. BMC genomics 13, 1, https://doi.org/10.1186/1471-2164-13-700 (2012).
Mimida, N. et al. Expression of DORMANCY-ASSOCIATED MADS-BOX (DAM)-like genes in apple. Biologia Plantarum 59, 237–244, https://doi.org/10.1007/s10535-015-0503-4 (2015).
Kumar, G., Rattan, U. K. & Singh, A. K. Chilling-Mediated DNA Methylation Changes during Dormancy and Its Release Reveal the Importance of Epigenetic Regulation during Winter Dormancy in Apple (Malus x domestica Borkh.). PloS one 11, e0149934, https://doi.org/10.1371/journal.pone.0149934 (2016).
Ubi, B. E. et al. Molecular cloning of dormancy-associated MADS-box gene homologs and their characterization during seasonal endodormancy transitional phases of Japanese pear. Journal of the American Society for Horticultural Science 135, 174–182 (2010).
Sasaki, R. et al. Functional and Expressional Analyses of PmDAM Genes Associated with Endodormancy in Japanese Apricot. Plant Physiology 157, 485–497, https://doi.org/10.1104/pp.111.181982 (2011).
Niu, Q. F. et al. Dormancy-associated MADS-box genes and microRNAs jointly control dormancy transition in pear (Pyrus pyrifolia white pear group) flower bud. Journal of Experimental Botany 67, 239–257, https://doi.org/10.1093/jxb/erv454 (2016).
Singh, R. K., Svystun, T., AlDahmash, B., Jonsson, A. M. & Bhalerao, R. P. Photoperiod- and temperature-mediated control of phenology in trees - a molecular perspective. The New phytologist 213, 511–524, https://doi.org/10.1111/nph.14346 (2017).
Cooke, J. E. K., Eriksson, M. E. & Junttila, O. The dynamic nature of bud dormancy in trees: environmental control and molecular mechanisms. Plant Cell and Environment 35, 1707–1728, https://doi.org/10.1111/j.1365-3040.2012.02552.x (2012).
Seo, M., Jikumaru, Y. & Kamiya, Y. Profiling of hormones and related metabolites in seed dormancy and germination studies. Methods in molecular biology (Clifton, N.J.) 773, 99–111, https://doi.org/10.1007/978-1-61779-231-1_7 (2011).
Shu, K. et al. ABI4 regulates primary seed dormancy by regulating the biogenesis of abscisic acid and gibberellins in Arabidopsis. PLoS Genet 9, e1003577, https://doi.org/10.1371/journal.pgen.1003577 (2013).
Rinne, P. L. et al. Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-beta-glucanases to reopen signal conduits and release dormancy in Populus. Plant Cell 23, 130–146, https://doi.org/10.1105/tpc.110.081307 (2011).
Rinne, P. & van der Schoot, C. Symplasmic fields in the tunica of the shoot apical meristem coordinate morphogenetic events. Development 125, 1477–1485 (1998).
Doxey, A. C., Yaish, M. W., Moffatt, B. A., Griffith, M. & McConkey, B. J. Functional divergence in the Arabidopsis beta-1,3-glucanase gene family inferred by phylogenetic reconstruction of expression states. Molecular biology and evolution 24, 1045–1055, https://doi.org/10.1093/molbev/msm024 (2007).
Zheng, C. et al. Abscisic acid (ABA) regulates grape bud dormancy, and dormancy release stimuli may act through modification of ABA metabolism. Journal of experimental botany 66, 1527–1542, https://doi.org/10.1093/jxb/eru519 (2015).
Duan, C., Li, X., Gao, D., Liu, H. & Li, M. Studies on regulations of endogenous ABA and GA3 in sweet cherry flower buds on dormancy. Acta Hortic. Sinica 31, 149–154, https://doi.org/10.3321/j.issn:0513-353X.2004.02.002 (2004).
Mornya, P. M. P. & Cheng, F. Seasonal changes in endogenous hormone and sugar contents during bud dormancy in tree peony. Journal of Applied Horticulture 15, 159–165 (2013).
Shu, K., Liu, X.-d, Xie, Q. & He, Z.-h. Two faces of one seed: hormonal regulation of dormancy and germination. Molecular plant 9, 34–45, https://doi.org/10.1016/j.molp.2015.08.010 (2016).
Ito, A., Sugiura, T., Sakamoto, D. & Moriguchi, T. Effects of dormancy progression and low-temperature response on changes in the sorbitol concentration in xylem sap of Japanese pear during winter season. Tree Physiology 33, 398–408, https://doi.org/10.1093/treephys/tpt021 (2013).
Anderson, J. V., Gesch, R. W., Jia, Y., Chao, W. S. & Horvath, D. P. Seasonal shifts in dormancy status, carbohydrate metabolism, and related gene expression in crown buds of leafy spurge. Plant, Cell & Environment 28, 1567–1578, https://doi.org/10.1111/j.1365-3040.2005.01393.x (2005).
Ning, D.-L. et al. Label-free quantitative proteomics analysis of dormant terminal buds of poplar. Molecular biology reports 40, 4529–4542, https://doi.org/10.1007/s11033-013-2548-9 (2013).
El Kayal, W. et al. Molecular events of apical bud formation in white spruce, Picea glauca. Plant, cell & environment 34, 480–500, https://doi.org/10.1111/j.1365-3040.2010.02257.x (2011).
Mason, M. G., Ross, J. J., Babst, B. A., Wienclaw, B. N. & Beveridge, C. A. Sugar demand, not auxin, is the initial regulator of apical dominance. Proceedings of the National Academy of Sciences 111, 6092–6097, https://doi.org/10.1073/pnas.1322045111 (2014).
Gonzalez-Grandio, E. & Cubas, P. Identification of gene functions associated to active and dormant buds in Arabidopsis. Plant signaling & behavior 9, e27994, https://doi.org/10.4161/psb.27994 (2014).
Díaz-Riquelme, J., Grimplet, J., Martínez-Zapater, J. M. & Carmona, M. J. Transcriptome variation along bud development in grapevine (Vitis vinifera L.). BMC plant biology 12, 181, https://doi.org/10.1186/1471-2229-12-181 (2012).
Zhang, Q. X. et al. The genome of Prunus mume. Nature Communications 3, https://doi.org/10.1038/ncomms2290 (2012).
Yamane, H. Regulation of Bud Dormancy and Bud Break in Japanese Apricot (Prunus mume Siebold & Zucc.) and Peach Prunus persica (L.) Batsch: A Summary of Recent Studies. Journal of the Japanese Society for Horticultural Science 83, 187–202, https://doi.org/10.2503/jjshs1.CH-Rev4 (2014).
Alburquerque, N., García-Montiel, F., Carrillo, A. & Burgos, L. Chilling and heat requirements of sweet cherry cultivars and the relationship between altitude and the probability of satisfying the chill requirements. Environmental & Experimental Botany 64, 162–170, https://doi.org/10.1016/j.envexpbot.2008.01.003 (2008).
Castède, S. et al. Genetic determinism of phenological traits highly affected by climate change in Prunus avium: flowering date dissected into chilling and heat requirements. New Phytologist 202, 703–715, https://doi.org/10.1111/nph.12658 (2014).
Campoy, J. A., Ruiz, D., Allderman, L., Cook, N. & Egea, J. The fulfilment of chilling requirements and the adaptation of apricot (Prunus armeniaca L.) in warm winter climates: An approach in Murcia (Spain) and the Western Cape (South Africa). European Journal of Agronomy 37, 43–55, https://doi.org/10.1016/j.eja.2011.10.004 (2012).
Okie, W. R. & Blackburn, B. Increasing chilling reduces heat requirement for floral budbreak in peach. HortScience 46, 245–252 (2011).
Habu, T. et al. 454-Pyrosequencing of the Transcriptome in Leaf and Flower Buds of Japanese Apricot (Prunus mume Sieb. et Zucc.) at Different Dormant Stages. Journal of the Japanese Society for Horticultural Science 81, 239–250 (2012).
Habu, T. et al. Custom Microarray Analysis for Transcript Profiling of Dormant Vegetative Buds of Japanese Apricot during Prolonged Chilling Exposure. (2014).
Zhong, W. et al. Genome-wide expression profiles of seasonal bud dormancy at four critical stages in Japanese apricot. Plant molecular biology 83, 247–264 (2013).
Chouard, P. Vernalization and its relations to dormancy. Annual Review of Plant Physiology 11, 191–238 (1960).
Castède, S. et al. Mapping of Candidate Genes Involved in Bud Dormancy and Flowering Time in Sweet Cherry (Prunus avium). PloS one 10, e0143250, https://doi.org/10.1371/journal.pone.0143250 (2015).
Baron, K. N., Schroeder, D. F. & Stasolla, C. Transcriptional response of abscisic acid (ABA) metabolism and transport to cold and heat stress applied at the reproductive stage of development in Arabidopsis thaliana. Plant Science An International Journal of Experimental Plant Biology s 188–189, 48–59, https://doi.org/10.1016/j.plantsci.2012.03.001 (2012).
Hoffman, D. E. Changes in the Transcriptome and Metabolome during the Initiation of Growth Cessation in Hybrid Aspens. Acta Universitatis Agriculturae Sueciae (2011).
Cantoro, R., Crocco, C. D., Benech-Arnold, R. L. & Rodriguez, M. V. In vitro binding of Sorghum bicolor transcription factors ABI4 and ABI5 to a conserved region of a GA 2-OXIDASE promoter: possible role of this interaction in the expression of seed dormancy. Journal of experimental botany 64, 5721–5735, https://doi.org/10.1093/jxb/ert347 (2013).
Bentsink, L. & Koornneef, M. Seed dormancy and germination. The Arabidopsis Book 6, (e0119 (2008).
Rohde, A. et al. PtABI3 impinges on the growth and differentiation of embryonic leaves during bud set in poplar. Plant Cell 14, 1885–1901 (2002).
Qiu, Z. B. et al. The regulation of cambial activity in Chinese fir (Cunninghamia lanceolata) involves extensive transcriptome remodeling. New Phytologist 199, 708–719, https://doi.org/10.1111/Nph.12301 (2013).
El-Yazal, M. A. S., El-Yazal, S. A. S. & Rady, M. M. Exogenous dormancy-breaking substances positively change endogenous phytohormones and amino acids during dormancy release in ‘Anna’ apple trees. Plant Growth Regulation 72, 211–220, https://doi.org/10.1007/s10725-013-9852-1 (2014).
Baba, K. et al. Activity-dormancy transition in the cambial meristem involves stage-specific modulation of auxin response in hybrid aspen. Proceedings of the National Academy of Sciences of the United States of America 108, 3418–3423, https://doi.org/10.1073/pnas.1011506108 (2011).
Rohde, A. & Bhalerao, R. P. Plant dormancy in the perennial context. Trends in Plant Science 12, 217–223, https://doi.org/10.1016/j.tplants.2007.03.012 (2007).
Ruttink, T. et al. A molecular timetable for apical bud formation and dormancy induction in poplar. Plant Cell 19, 2370–2390, https://doi.org/10.1105/tpc.107.052811 (2007).
Gibson, S. I. Sugar and phytohormone response pathways: navigating a signalling network. Journal of experimental botany 55, 253–264, https://doi.org/10.1093/jxb/erh048 (2004).
Xie, Z., Zhang, Z.-L., Hanzlik, S., Cook, E. & Shen, Q. J. Salicylic acid inhibits gibberellin-induced alpha-amylase expression and seed germination via a pathway involving an abscisic-acid-inducible WRKY gene. Plant molecular biology 64, 293–303, https://doi.org/10.1007/s11103-007-9152-0 (2007).
Gómez-Cadenas, A. et al. An abscisic acid-induced protein kinase, PKABA1, mediates abscisic acid-suppressed gene expression in barley aleurone layers. Proceedings of the National Academy of Sciences 96, 1767–1772, https://doi.org/10.1073/pnas.96.4.1767 (1999).
Gomez-Cadenas, A., Zentella, R., Walker-Simmons, M. K. & Ho, T. H. Gibberellin/abscisic acid antagonism in barley aleurone cells: site of action of the protein kinase PKABA1 in relation to gibberellin signaling molecules. Plant Cell 13, 667–679, https://doi.org/10.1105/tpc.13.3.667 (2001).
Xie, Z. et al. Interactions of two abscisic‐acid induced WRKY genes in repressing gibberellin signaling in aleurone cells. The Plant Journal 46, 231–242, https://doi.org/10.1111/j.1365-313X.2006.02694.x (2006).
Lu, C.-A., Ho, T.-h.D., Ho, S.-L. & Yu, S.-M. Three novel MYB proteins with one DNA binding repeat mediate sugar and hormone regulation of α-amylase gene expression. The Plant Cell 14, 1963–1980, https://doi.org/10.1105/tpc.001735 (2002).
Chao, W. S., Serpe, M. D., Anderson, J. V., Gesch, R. W. & Horvath, D. P. Sugars, hormones, and environment affect the dormancy status in underground adventitious buds of leafy spurge (Euphorbia esula). Weed Science 54, 59–68, https://doi.org/10.1614/WS-05-088R.1 (2006).
Yonekura-Sakakibara, K. Functional genomics of family 1 glycosyltransferases in Arabidopsis. Plant Biotechnology 26, 267–274 (2009).
Harborne, J. B. & Williams, C. A. Advances in flavonoid research since 1992. Phytochemistry 55, 481–504, https://doi.org/10.1016/S0031-9422(00)00235-1 (2000).
Lim, E.-K. et al. Resolution of (+)-abscisic acid using an Arabidopsis glycosyltransferase. Tetrahedron: Asymmetry 16, 143–147, https://doi.org/10.1016/j.tetasy.2004.11.062 (2005).
Hou, B., Lim, E. K., Higgins, G. S. & Bowles, D. J. N-glucosylation of cytokinins by glycosyltransferases of Arabidopsis thaliana. Journal of Biological Chemistry 279, 47822–47832, https://doi.org/10.1074/jbc.M409569200 (2004).
Xu, Z.-J., Nakajima, M., Suzuki, Y. & Yamaguchi, I. Cloning and characterization of the abscisic acid-specific glucosyltransferase gene from adzuki bean seedlings. Plant Physiology 129, 1285–1295 (2002).
Achard, P. et al. The cold-inducible CBF1 factor–dependent signaling pathway modulates the accumulation of the growth-repressing DELLA proteins via its effect on gibberellin metabolism. The Plant Cell 20, 2117–2129, https://doi.org/10.1105/tpc.108.058941 (2008).
Benedict, C. et al. The CBF1‐dependent low temperature signalling pathway, regulon and increase in freeze tolerance are conserved in Populus spp. Plant, Cell & Environment 29, 1259–1272, https://doi.org/10.1111/j.1365-3040.2006.01505.x (2006).
Wisniewski, M., Norelli, J., Bassett, C., Artlip, T. & Macarisin, D. Ectopic expression of a novel peach (Prunus persica) CBF transcription factor in apple (Malus × domestica) results in short-day induced dormancy and increased cold hardiness. Planta 233, 971–983, https://doi.org/10.1007/s00425-011-1358-3 (2011).
Wisniewski, M., Norelli, J. & Artlip, T. Overexpression of a peach CBF gene in apple: a model for understanding the integration of growth, dormancy, and cold hardiness in woody plants. Frontiers in plant science 6, https://doi.org/10.3389/fpls.2015.00085 (2015).
Tillett, R. L. et al. The Vitis vinifera C‐repeat binding protein 4 (VvCBF4) transcriptional factor enhances freezing tolerance in wine grape. Plant biotechnology journal 10, 105–124, https://doi.org/10.1111/j.1467-7652.2011.00648.x (2012).
Horvath, D. P., Sung, S., Kim, D., Chao, W. & Anderson, J. Characterization, expression and function of DORMANCY ASSOCIATED MADS-BOX genes from leafy spurge. Plant molecular biology 73, 169–179, https://doi.org/10.1007/s11103-009-9596-5 (2010).
Jiménez, S., Reighard, G. & Bielenberg, D. Gene expression of DAM5 and DAM6 is suppressed by chilling temperatures and inversely correlated with bud break rate. Plant molecular biology 73, 157–167, https://doi.org/10.1007/s11103-010-9608-5 (2010).
Yamane, H., Ooka, T., Jotatsu, H., Sasaki, R. & Tao, R. Expression analysis of PpDAM5 and PpDAM6 during flower bud development in peach (Prunus persica). Scientia Horticulturae 129, 844–848, https://doi.org/10.1093/jxb/err028 (2011).
Hao, X., Chao, W., Yang, Y. & Horvath, D. Coordinated expression of FLOWERING LOCUS T and DORMANCY ASSOCIATED MADS-BOX-like genes in leafy spurge. PloS one 10, e0126030, https://doi.org/10.1371/journal.pone.0126030 (2015).
Bai, S. L. et al. Transcriptome Analysis of Japanese Pear (Pyrus pyrifolia Nakai) Flower Buds Transitioning Through Endodormancy. Plant Cell Physiol 54, 121132–121151, https://doi.org/10.1093/Pcp/Pct067 (2013).
Rosa, M., Hilal, M., Gonzalez, J. A. & Prado, F. E. Low-temperature effect on enzyme activities involved in sucrose-starch partitioning in salt-stressed and salt-acclimated cotyledons of quinoa (Chenopodium quinoa Willd.) seedlings. Plant physiology and biochemistry 47, 300–307, https://doi.org/10.1016/j.plaphy.2008.12.001 (2009).
Dobrev, P. I. & Vankova, R. Quantification of abscisic acid, cytokinin, and auxin content in salt-stressed plant tissues. Plant salt tolerance: methods and protocols, 251–261 (2012).
Djilianov, D. L. et al. Dynamics of endogenous phytohormones during desiccation and recovery of the resurrection plant species Haberlea rhodopensis. Journal of Plant Growth Regulation 32, 564, https://doi.org/10.1007/s00344-013-9323-y (2013).
Acknowledgements
The research was supported by the Fundamental Research Funds for the Central Universities (No. 2016ZCQ02), the program for Science and Technology of Beijing (NO.Z171100002217005), Special Fund for Beijing Common Construction Project and the Fundamental Research Funds for the Central Universities (NO.BLYJ201613).
Author information
Authors and Affiliations
Contributions
Z.Z. conceived and designed the experiments. Z.X. and Z.K. performed the experiments and analyzed the data. Z.T., H.Y. and Y.C. provided technical support and theoretical support to this work. Z.Z. wrote the paper. Z.Q. supervised the project. Z.Z. and Z.Q. together with other authors revised and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Zhuo, X., Zhao, K. et al. Transcriptome Profiles Reveal the Crucial Roles of Hormone and Sugar in the Bud Dormancy of Prunus mume. Sci Rep 8, 5090 (2018). https://doi.org/10.1038/s41598-018-23108-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23108-9
This article is cited by
-
Unravelling the cascade of differentials (ABA and GA) in corm dormancy release of gladiolus (Gladiolus grandiflorus L.)
Plant Physiology Reports (2024)
-
Comparative transcriptomic analysis of transcription factors and hormones during flower bud differentiation in ‘Red Globe’ grape under red‒blue light
Scientific Reports (2023)
-
An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in peach reproductive tissues
Plant Methods (2022)
-
GA3 is superior to GA4 in promoting bud endodormancy release in tree peony (Paeonia suffruticosa) and their potential working mechanism
BMC Plant Biology (2021)
-
Elucidation of molecular and hormonal background of early growth cessation and endodormancy induction in two contrasting Populus hybrid cultivars
BMC Plant Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
