
Abstract
Obesity is associated with a disturbed adipose tissue (AT) function characterized by adipocyte hypertrophy, an impaired lipolysis and pro-inflammatory phenotype, which contributes to insulin resistance (IR). We investigated whether AT phenotype in different AT depots of obese individuals with and without type 2 diabetes mellitus (T2DM) is associated with whole-body IR. Subcutaneous (SC) and visceral (V) AT biopsies from 18 lean, 17 obese and 8 obese T2DM men were collected. AT phenotype was characterized by ex vivo measurement of basal and stimulated lipolysis (mature adipocytes), adipocyte size distribution (AT tissue sections) and AT immune cells (flow cytometry). In VAT, mean adipocyte size, CD45+ leukocytes and M1 macrophages were significantly increased in both obese groups compared to lean individuals. In SCAT, despite adipocyte hypertrophy, no significant differences in immune cell populations between groups were found. In SCAT, multiple linear regression analysis showed that none of the AT phenotype markers independently contributed to HOMA-IR while in VAT, mean adipocyte size was significantly related to HOMA-IR. In conclusion, beside adipocyte hypertrophy in VAT, M1 macrophage- or B-cell-mediated inflammation, may contribute to IR, while inflammation in hypertrophic SCAT does not seem to play a major role in IR.
Similar content being viewed by others


Introduction
During the development of obesity, adipose tissue (AT) expansion frequently results in adipocyte hypertrophy (i.e. enlargement of the adipocyte), which is a known stressor for adipocytes1. Increases in AT mass and adipocyte volume result in a broad range of metabolic repercussions including, amongst others, a decreased insulin-mediated suppression2,3 and attenuated catecholamine or atrial natriuretic peptide (ANP)-mediated stimulation of AT lipolysis4,5,6,7. Especially in the postprandial state, this altered AT function is characterized by an impaired lipid buffering capacity8. Subsequently, this may lead to systemic lipid overflow and ectopic lipid accumulation in several insulin sensitive peripheral tissues like skeletal muscle, liver, pancreas, the heart and kidneys, which relates to the development of peripheral and systemic insulin resistance (IR)9.
Different immune cells are activated within hypertrophic AT (in case of resident immune cells) or attracted toward necrotic/apoptotic hypertrophic adipocytes, especially in the visceral AT (VAT), mainly based on rodent studies10,11. This VAT inflammation was proposed to be the strongest correlate of IR in human obesity12,13. In this regard, AT macrophages have gained much attention as important mediators of AT inflammation. In human obesity, AT macrophages display profound pro-inflammatory (M1) characteristics14,15 and are thereby thought to be the major source of pro-inflammatory cytokines and chemokines. This in turn may affect local AT lipolysis and further impede adipocyte function16 and reduce insulin sensitivity17,18,19. However, recent evidence indicates the presence of other innate and adaptive immune cells in the AT (reviewed by Mraz et al.)20. The cross-talk and dynamics of immune cells initiating and orchestrating AT inflammation and an impaired lipid metabolism in different AT depots are incompletely understood in the obese non-diabetic or type 2 diabetic state.
It is still an ongoing debate whether an impaired AT lipolysis or inflammation, or an interaction between both, is a cause or rather a consequence of IR in the obese state. Animal models propose a causative role of AT inflammation in the pathogenesis of IR21. In contrast, recent human studies indicated the development of hyperinsulinemia and dyslipidemia and (peripheral) IR upon overfeeding without affecting the inflammatory phenotype of the SCAT, suggesting that AT inflammation is not necessary to evoke peripheral IR in humans22,23. Of interest, healthy AT expansion and remodeling implies a certain degree of AT inflammation contrasting the general belief that inflammation negatively affects metabolism24.
Therefore, gaining better insights into the association between AT lipolysis, inflammation and IR is imperative to improve future development of therapeutic strategies to ameliorate the adverse metabolic consequences of obesity. In the present study, we studied subcutaneous and visceral AT morphology (adipocyte size), ex vivo lipolysis and immune cell populations (flow cytometry) in relation to whole-body IR in obese non-diabetic and type 2 diabetic (T2DM) men, compared with age-matched lean men.
Methods
Subjects
For this study, lean and obese age-matched male individuals who were scheduled to undergo laparoscopic abdominal (inguinal hernia or gallbladder removal) or bariatric surgery were recruited, as described previously in more detail7. The lean control group consisted of 18 male individuals. The obese group was composed of 25 male individuals, including 17 individuals without type 2 diabetes and 8 individuals with type 2 diabetes. Presence of type 2 diabetes was based on known clinical diagnosis (on average 2.5 years of diagnosis, ranging from newly diagnosed to 6 years). Obese diabetic individuals had glycated haemoglobin (HbA1c) levels ≥6.5% (45 mmol/mol) or were on glucose lowering medication. Major exclusion criteria were the use of exogenous insulin, presence or history of heart, lung or kidney disease and/or presence of endocrine anomalies. The study protocol was approved by the Medical Ethical Committee Jessa hospital, Hasselt, and Hasselt University, Belgium, in accordance with the Declaration of Helsinki (2008), and all individuals gave their written informed consent before participating in the study.
Anthropometric measurements and blood sampling
Body weight, height, waist/hip circumference and blood pressure were determined at the morning of surgery. Fat and lean body mass were estimated by bio-electrical impedance analyses (Bodystat 1500; Bodystat Ltd., Isle of Man, UK). Fasting venous blood samples were collected after an overnight fast for measurement of plasma glucose, serum insulin and HbA1C. Insulin sensitivity was assessed by the homeostatic model assessment index for insulin resistance (HOMA-IR), calculated from fasting glucose and insulin, according to the formula: fasting insulin (mU/l)*fasting glucose (mmol/l)/22.525. Plasma glucose concentration was measured by the glucose oxidase method using an AU2700 analyser (Beckman Coulter, Brea, CA, USA). Serum insulin concentration was assessed by immunoassay (ADVIA Centaur Insulin IRI; Siemens Medical Solutions Diagnostics, Tarrytown, NY, USA). HbA1C was assessed by high performance liquid chromatography using a HA-8160 Hi-Auto A1C analyser (Menarini, Zaventem, Belgium).
Abdominal subcutaneous and visceral adipose tissue biopsies
After an overnight fast, adipose tissue biopsies were taken from the periumbilical subcutaneous adipose tissue depot (SCAT) and the distal portion of the omentum majus (VAT). The tissue samples were immediately placed in saline and transported on ice to the laboratory for further processing. One portion of fresh AT was used for histological sections whereas another portion was used for isolation of the stromal vascular fraction (SVF) and mature adipocytes for lipolysis measurements.
Adipocyte size
A small part of the AT samples was fixed overnight in 4% paraformaldehyde and embedded in paraffin. Histological sections (8 µm) were cut, mounted on microscope glass slides and dried overnight in an incubator (37 °C). Haematoxylin and eosin staining was used. Digital images were captured using a Leica DFC320 digital camera (Leica DM3000 microscope, Leica, Rijswijk, The Netherlands) at 20× magnification. Adipocyte size and distribution was performed in a blinded fashion (coefficient of variation <5%) using computerized morphometric analysis (Leica QWin V3, Cambridge, UK) of individual adipocytes (at least 400 adipocytes per sample), as described previously26.
Adipocyte isolation, stromal vascular preparation and lipolysis measurement
Mature adipocytes and stromal vascular cells were obtained, as described previously7. Briefly, after collagenase digestion of AT fragments in Dulbecco’s modified Eagle’s medium (DMEM)-Ham’s F12, the resulting suspension was filtered and mature adipocytes were diluted in DMEM-Ham’s F12 supplemented with 3% BSA for lipolysis assays and incubated with isoprenaline (ISO, a non-selective β-adrenergic agonist; 10−6 M) or human ANP (Bachem) (10−4 M) for 3 h at 37 °C. Following incubation, glycerol concentration in the medium (lipolysis index) was determined using the EnzyChromeTM Adipolysis assay kit (Gentaur) and expressed per cell number or as maximal responsiveness to adrenergic or ANP stimulation (compared with baseline).
Flow cytometry
Multicolour flow cytometry measurements were performed and analysed as described previously27. Briefly, isolated SVF cells, which were obtained following collagenase digestion and lysis of red blood cells, were stained for flow cytometry using two antibody cocktails. Cocktail 1 included CD45-PE-Cy7 (BD 557748), CD3-fitc (BD 561807), CD19-fitc (BD 555412), CD56-fitc (BD 562794), CD66b-fitc (BD 555724), CD11b-BV421 (Biologend 301324), and CD11c-APC-Cy7 (Biolegend 337218). Cocktail 2 included CD45-PE-Cy7 (BD 557748), CD3-V500 (BD 561416), CD4-PerCP (Biolegend 300528), CD8 APC-H7 (BD 641400), CD19-BV421 (Biolegend 302234), and CD56-APC (Biolegend 318310). Samples were measured with a FACS-Canto II (BD Biosciences). Results were analysed with FACSdiva (BD Biosciences) and FlowJo software. Since weight of the AT samples was unavailable, data are expressed as percentage of cells relative to total number of cells (based on forward and side scatter plot).
Statistical analyses
All data are presented as mean ± S.E.M. Shapiro-Wilk tests were used to examine normality and variables with a skewed distribution were ln-transformed prior to analysis. Clinical characteristics with a skewed distribution were reported as median (interquartile range). Group differences were analysed using one-way ANOVA. Post-hoc testing was performed using Bonferroni adjustment for multiple testing when one-way ANOVA showed a significant group difference. Correlation analysis was applied by Pearson’s correlation. Multiple linear regression analyses including age, total fat mass and the AT phenotype markers that were significantly correlated with HOMA-IR were used to investigate whether AT morphology, lipolysis and inflammation were independently related to whole-body IR (dependent variable, expressed as HOMA-IR). These analyses were performed for both AT depots separately. Statistical significance was set at p < 0.05 (two-tailed). Analyses were performed using SPSS 22.0 for Windows (SPSS).
Results
Subjects’ characteristics
By design, no differences in age between lean individuals and both obese groups were observed. As expected, lean individuals’ body composition and metabolic parameters, including insulin sensitivity (expressed as HOMA-IR) and glycaemic control (indicated by fasting plasma glucose and HbA1c levels) were significantly different compared to the obese groups (Table 1). BMI, body fat percentage and body fat distribution was not significantly different between both obese groups, while blood pressure measures were comparable between groups. Fasting plasma glucose levels, whole-body IR (HOMA-IR) and glycated haemoglobin (HbA1c) were significantly elevated in the obese T2DM individuals as compared to obese individuals (Table 1). Details with respect to medication use in both obese groups are summarized in Supplementary Table S1.
SCAT and VAT morphology
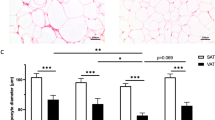
Mean adipocyte size was clearly elevated in both obese groups as compared to the lean group in both SCAT (p < 0.001 for obese individuals) and VAT (p < 0.001 for both obese and obese T2DM individuals) (Table 1). However, there was no significant difference in mean adipocyte size between the different AT depots within obese groups (Table 1). The difference in mean adipocyte size between lean and obese individuals was attributed by a shift toward a significantly higher proportion of large (70–89 µm) and very large (>90 µm) adipocytes, and a lower proportion of small (<50 µm) and medium size (50–69 µm) adipocytes in the obese individuals, in both the SCAT (Fig. 1A) and VAT (Fig. 1B). Detailed lipolysis data for the individuals in Table 1 have been reported previously7. Briefly, we observed an increased basal lipolysis in the SCAT and VAT of obese and obese diabetic individuals and an attenuated maximal ANP- and ISO-induced lipolysis in the SCAT of obese diabetic men as compared to lean individuals (with intermediate values for obese individuals).

Adipocyte size distribution. The frequency of small (<50 µm), medium (50–69 µm), large (70–89 µm) and very large (>90 µm) adipocytes was determined in SCAT (A) and VAT (B) of lean (black bars), obese (white bars) and obese diabetic (white squared bars) individuals. Obese groups clearly had higher proportions of large and very large adipocytes in combination with smaller proportions of small and medium size adipocytes, resulting in a higher mean adipocytes size compared to lean individuals both in SCAT (p < 0.001 for obese individuals and p < 0.01 for obese diabetic individuals) and VAT (p < 0.001 for both obese groups) Data are means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Elevated inflammatory cell populations in the SVF of obese VAT
In SCAT, no significant differences were found in immune cell populations, expressed as percentage of total cells, between groups (Table 2). In contrast, the VAT of both obese groups showed a clear increase in proportion of leukocytes (CD45+) numbers, expressed as percentage of total cells, compared to lean controls (p < 0.001 for obese individuals and p < 0.01 for obese T2DM individuals). However, no significant differences were found between groups for the proportion of B-cells (CD19+), T-cell subsets (T-helper cells or cytotoxic T-cells) or NK cells (CD56+) (Table 2). For VAT macrophages, we observed an increased proportion of M1-macrophages (CD11c+; expressed as percentage of total cells) in obese individuals (p < 0.01) compared to lean controls. A trend toward a higher proportion of CD11c− M2 macrophages in obese VAT was observed (pgroup = 0.088) (Table 2). No group differences were observed when representing cell subsets as a fraction of total parent cells (either as % of CD45+ lymphocytes or as % of macrophages), thereby reflecting qualitative characteristics (Supplementary Table S2).
Determinants of whole-body insulin resistance
First, we investigated whether AT functional variables (adipocyte size, basal lipolysis and maximal lipolytic responsiveness to ISO or ANP) and AT immunophenotype were associated with whole-body IR, estimated using HOMA-IR (Table 3). In SCAT, basal lipolysis (expressed per cell number; p = 0.002) and mean adipocyte size (p = 0.004) were positively correlated with HOMA-IR. Furthermore, SCAT CD3+ T-cells (p = 0.018), CD8+ cytotoxic T-cells (p = 0.001) and CD56+ NK-cells (p = 0.005) correlated positively with HOMA-IR (Table 3). In VAT, mean adipocyte size correlated positively (p < 0.001) with HOMA-IR. Moreover, VAT CD45+ leukocytes (p = 0.001) and CD19+ B-cells (p = 0.034) were positively correlated with HOMA-IR, while VAT CD8+ cytotoxic T-cells tended to show a positive association with HOMA-IR (r = 0.29, p = 0.069) (Table 3). Of interest, AT functional variables showed some correlations with AT immunophenotype in the SCAT as well as in the VAT, respectively (Supplementary Table S3).
Subsequently, multiple linear regression analyses including age, total fat mass and the AT phenotype markers that were significantly correlated with HOMA-IR (indicated in Table 3) were used to investigate whether AT morphology, lipolysis and inflammation were independently related to whole-body IR (dependent variable, expressed as HOMA-IR). In SCAT, none of the AT phenotype markers were significantly associated to HOMA-IR (Table 4). In VAT, adipocyte size (β = 0.599, p = 0.002) was significantly associated to HOMA-IR, explaining about 17% the variance in HOMA-IR (pmodel < 0.001) (Table 4).
Discussion
To the best of our knowledge, this is the first study reporting detailed data on the relationship between depot-specific AT phenotype (i.e. morphology, lipolysis and inflammation) and whole-body IR (HOMA-IR) in male lean, obese non-diabetic individuals and obese humans with type 2 diabetes. As reported previously7, obese individuals have an attenuated basal lipolysis in both SCAT and VAT, and a decreased maximal ANP- and ISO-mediated lipolytic responsiveness in SCAT, compared to age-matched lean individuals. Here we demonstrate that this altered AT phenotype was further characterized by adipocyte hypertrophy and an increased immune cell infiltration, especially in the VAT. Of interest, in the VAT adipocyte size was positively related to whole-body IR. In contrast, none of the subcutaneous AT functional measures or immunophenotype were independently related to whole-body IR.
The present findings indicate that adipocyte hypertrophy in the obese SCAT and VAT is a major determinant of whole-body IR, which is in line with previous studies28,29,30. In addition, VAT adipocyte hypertrophy was independently and positively associated (β = 0.599, p = 0.002) with whole-body IR, also after adjustment for age and total fat mass. Although adipocyte hypertrophy is the preferential expansion mechanism within the AT, in the chronic state of obesity SCAT adipogenesis is commonly impaired31, possibly resulting in lipid spill-over to other tissues (the VAT as well) and thus the development of metabolic complications32. The latter may explain our finding that adipocyte hypertrophy in VAT was an independent determinant of whole-body IR. Importantly, abdominal subcutaneous adipocyte hypertrophy may thus substantially contribute to lipid overflow, ectopic lipid deposition and expansion of VAT, which in turn results in worsening of whole-body IR.
The presence of a chronic pro-inflammatory AT microenvironment may limit adipocyte hypertrophy, by activating fibrotic signalling and impairing adipocyte functionality33, thereby contributing to metabolic alterations frequently observed in the obese state34,35. In addition to adipocyte hypertrophy, low-grade AT inflammation is essential for healthy AT expansion and remodelling, as some cells are implicated in the clearance of cell debris released by necrotic adipocytes36, act as a local buffer against increased lipid content37, and promote angiogenesis38. In this regard, our data show a significantly increased proportion of CD45+ leukocytes and CD11c+ M1 macrophages in the VAT of obese and obese diabetic humans. In contrast, an inflammatory phenotype was less pronounced in the SCAT of obese individuals compared to lean or obese T2DM individuals, where we only observed a trend toward a higher proportion of CD11c+ M1 (pro-inflammatory) macrophages in the obese SCAT (Table 2). Additionally, no immune-related markers in SCAT were significantly related to HOMA-IR, when adjusting for total fat mass and age. Indeed, the VAT is believed to have a more pronounced inflammatory phenotype than the SCAT39, which may possibly be ascribed to an increased production of inflammatory mediators by adipocytes themselves and by cells in the SVF40. Concerning the latter, one should be aware that SCAT is more important in quantitative terms, since SCAT accounts for approximately 80% of total AT mass. This may also explain why total fat mass rather than adipocyte morphology or the SCAT function was the strongest independent determinant of whole-body IR.
Therefore, in the VAT, the potential involvement of the elevated immune cells (i.e. CD45+ leukocytes and CD11c+ M1 macrophages) in the development of AT inflammation and IR in obese humans is likely, indicated by a strong positive correlation between these CD45+ leukocytes and HOMA-IR (r = 0.514; p = 0.001) and a tendency for a positive correlation between CD8+ cytotoxic T-cells and HOMA-IR (r = 0.290; p = 0.069) (Table 3). Notably, VAT CD19+ B (r = 0.332, p = 0.034) cells were positively correlated with HOMA-IR. B lymphocytes are known to be elevated in obesity and IR, at least in diet-induced obese mice41. In epididymal AT, by orchestrating T cell populations and their activity (i.e. production of pro-inflammatory cytokines) and the release of pathogenic antibodies, B cells can decrease glucose tolerance41. Recently, the pro-inflammatory features of B cells in a high-fat diet setting were confirmed in obese mice42. These data, together with the present findings, suggest that B cells exert pro-inflammatory effects in obese AT and may contribute to IR. However, the precise mechanisms via which these cells impair insulin sensitivity require further investigation, especially in human obesity.
Inflammatory cells may have a modulatory function in chronically increased lipolysis. Indeed, beside the presence of inflammation additional mechanisms that may be (in)directly related to inflammation may be of importance in worsening of IR in obesity, as previously suggested43. Of interest, Kosteli et al.37 suggested that AT macrophages may buffer excess intracellular fatty acids upon increased lipolysis during fasting. The chronic elevation and uptake of lipids by macrophages may alter their phenotype44, promoting the progression to an inflammatory (CD11c+) M1 polarisation and AT inflammation45. However, in the present study no association between SCAT or VAT immunophenotype and measures of lipolysis were observed, suggesting that adipocyte lipolysis does not activate inflammatory pathways in human AT macrophages, as shown recently46. In line, only in the VAT a correlation between M1 and M2 macrophages and adipocyte size was found (Supplementary Table S3), respectively, suggesting that different immunological processes or alternative stimuli for AT macrophages (e.g. concentration of saturated fatty acids or ratio of monounsaturated to saturated fatty acids) could be present between abdominal SCAT and VAT depots in humans. Moreover, altered AT oxygen tension may have a putative role in AT dysfunction in obese insulin resistant conditions, as recently reviewed47. However, further studies are warranted to obtain more insight into the triggers of AT dysfunction within different AT depots.
Future studies should include human mechanistic experiments to strengthen these findings by focussing on intra-AT relationships and to address causality in a larger sample size. Our study was also performed in male individuals and it is not known whether the outcomes can be extrapolated to females, since it is evident that there are sex-specific differences in fat metabolism and white AT function48. In addition, it needs to be mentioned that a significant proportion of the obese patients in the present study received several forms of glucose lowering or lipid lowering medication (Supplementary Table S1), which may impact the AT phenotype and have cofounded the present results49,50. Furthermore, the use of HOMA-IR as a surrogate markers to assess whole-body IR and bioimpedance measurements to determine body composition may be two limitations of this study. It would be of interest to relate AT functional characteristics to tissue-specific (e.g. muscle, AT or liver) IR, which need to be determined in future studies using for example a two-step hyperinsulinemic-euglycemic clamp combined with a glucose tracer.
In conclusion, this study is the first to characterize the SCAT and VAT phenotype in detail in male lean, obese and obese type 2 diabetic subjects. In VAT, adipocyte hypertrophy independently contributes to IR, possibly via pro-inflammatory M1 macrophage- and/or B cell-mediated inflammation. In SCAT, AT function or immunophenotype were only of minor importance with respect to whole-body IR. These alterations in AT mass and phenotypes likely impact cardiometabolic risk and may contribute to the development of type 2 diabetes.
References
Khan, T. et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29, 1575–1591 (2009).
Jocken, J. W. et al. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J Clin Endocrinol Metab 92, 2292–2299 (2007).
Bickerton, A. S. et al. Adipose tissue fatty acid metabolism in insulin-resistant men. Diabetologia 51, 466–1474 (2008).
Jocken, J. W., Blaak, E. E., van der Kallen, C. J., van Baak, M. A. & Saris, W. H. Blunted beta-adrenoceptor-mediated fat oxidation in overweight subjects: a role for the hormone-sensitive lipase gene. Metabolism 57, 326–332 (2008).
Nielsen, T. S., Jessen, N., JØrgensen, J. O., MØller, N. & Lund, S. Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 52, R199–R222 (2014).
Rydén, M. et al. Impaired atrial natriuretic peptide-mediated lipolysis in obesity. Int J Obes (Lond) 40, 714–720 (2015).
Verboven, K. et al. Attenuated atrial natriuretic peptide-mediated lipolysis in subcutaneous adipocytes of obese type 2 diabetic men. Clin Sci (Lond) 130, 1105–1114 (2016).
McQuaid, S. E. et al. Downregulation of adipose tissue fatty acid trafficking in obesity: a driver for ectopic fat deposition? Diabetes 60, 47–55 (2011).
Stinkens, R., Goossens, G. H., Jocken, J. W. & Blaak, E. E. Targeting fatty acid metabolism to improve glucose metabolism. Obes Rev 16, 715–757 (2015).
Osborn, O. & Olefsky, J. M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 18, 363–374 (2012).
Mathis, D. Immunological goings-on in visceral adipose tissue. Cell Metab 17, 851–859 (2013).
Klöting, N. et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab 299, E506–E515 (2010).
Hardy, O. T. et al. BMI-independent inflammation in omental adipose tissue associated with insulin resistance in morbid obesity. Surg Obes Relat Dis 7, 60–67 (2011).
Lumeng, C. N., Bodzin, J. L. & Saltiel, A. R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117, 175–184 (2007).
Lumeng, C. N., DelProposto, J. B., Westcott, D. J. & Saltiel, A. R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57, 3239–3246 (2008).
Langin, D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res 53, 482–491 (2006).
Olefsky, J. M. & Glass, C. K. Macrophages, inflammation and insulin resistance. Annu Rev Physiol 72, 219–246 (2010).
Ouchi, N., Parker, J. L., Lugus, J. J. & Walsh, K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11, 85–97 (2011).
Glass, C. K. & Olefsky, J. M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab 15, 635–645 (2012).
Mraz, M. & Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J Endocrinol 222, R113–127 (2014).
Xu, H. et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112, 1821–1830 (2003).
Tam, C. S. et al. Short-term overfeeding may induce peripheral insulin resistance without altering subcutaneous adipose tissue macrophages in humans. Diabetes 59, 2164–2170 (2010).
Johannsen, D. L. et al. Effect of 8 weeks of overfeeding on ectopic fat deposition and insulin sensitivity: testing the adipose tissue expandability hypothesis. Diabetes Care 37, 2789–2797 (2014).
Blüher, M. Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clin Sci (Lond) 130, 1603–1614 (2016).
Matthews, D. R. et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentration in man. Diabetologia 28, 412–419 (1985).
Goossens, G. H. et al. Increased adipose tissue oxygen tension in obese compared with lean men is accompanied by insulin resistance, impaired adipose tissue capillarization, and inflammation. Circulation 124, 67–76 (2011).
Wouters, K. et al. Circulating classical monocytes are associated with CD11c+ macrophages in human visceral adipose tissue. Sci Rep 7, 42665 (2017).
Weyer, C., Foley, J. E., Bogardus, C., Tataranni, P. A. & Pratley, R. E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type 2 diabetes independent of insulin resistance. Diabetologia 43, 1498–1506 (2000).
Lönn, M., Mechlig, K., Bengtsson, C. & Lissner, L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J 24, 326–331 (2010).
Acosta, J. R. et al. Increased fat cell size: a major phenotype of subcutaneous white adipose tissue in non-obese individuals with type 2 diabetes. Diabetologia 59, 560–570 (2016).
Kim, S. M. et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab 20, 1049–1058 (2014).
Pellegrinelli, V., Carobbio, S. & Vidal-Puig, A. Adipose tissue plasticity: how fat depots respond differently to pathophysiological cues. Diabetologia 59, 1075–1088 (2016).
Abdennour, M. et al. Association of adipose tissue and liver fibrosis with tissue stiffness in morbid obesity: links with diabetes and BMI loss after gastric bypass. J Clin Endocrinol Metab 99, 898–907 (2014).
Strissel, K. J. et al. Adipocyte death, adipose tissue remodeling and obesity complications. Diabetes 56, 2910–2918 (2007).
Pellegrinelli, V. et al. Human adipocyte function is impacted by mechanical cues. J Pathol 233, 183–195 (2014).
Cinti, S. et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46, 2347–2355 (2005).
Kosteli, A. et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest 120, 3466–3479 (2010).
Cao, Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab 18, 478–489 (2013).
Hamdy, O., Porramatikul, S. & Al-Ozairi, E. Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev 2, 367–373 (2006).
Fain, J. N. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells – a review. Mediators Inflamm. https://doi.org/10.1155/2010/513948 (2010).
Winer, D. A. et al. B lymphocytes promote insulin resistance through modulation of T lymphocytes and production of pathogenic IgG antibody. Nat Med 17, 610–617 (2011).
DeFuria, J. et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA 110, 5133–5138 (2013).
Barbarroja, N. et al. Progression from high insulin resistance to type 2 diabetes does not entail additional visceral adipose tissue inflammation. PLoS One 7, e48155 (2012).
Klein-Wieringa, I. R. et al. Adipocytes modulate the phenotype of human macrophages through secreted lipids. J Immunol 191, 1356–1363 (2013).
Prieur, X. et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes 60, 797–809 (2011).
Caspar-Bauguil, S. et al. Fatty acids from fat cell lipolysis do not activate an inflammatory response but are stored as triacylglycerols in adipose tissue macrophages. Diabetologia 58, 2627–2636 (2015).
Goossens, G. H. & Blaak, E. E. Adipose tissue oxygen tension: implications for chronic metabolic and inflammatory diseases. Curr Opin Clin Nutr Metab Care 15, 539–546 (2012).
Newell-Fugate, A. The role of sex steroids in white adipocyte function. Reproduction 153, R133–R149 (2017).
Kothari, V., Galdo, J. A. & Mathews, S. T. Hypoglycemic agents and potential anti-inflammatory activity. J Inflamm Res 9, 27–38 (2016).
Welty, F. K., Alfaddagh, A. & Elajami, T. K. Targeting inflammation in metabolic syndrome. Transl Res 167, 257–280 (2016).
Acknowledgements
We thank all abdominal surgeons from the Jessa Hospital (Hasselt, Belgium) and Hospital East-Limburg (Genk, Belgium) for their contributions to subject recruitment. We thank University Biobank Limburg (UBiLim, Jessa Hospital, Belgium) for their technical support in sample handling and storage. Yvonne Essers, Nicole Hoebers, Maria Vroomen and José van de Gaar are greatly acknowledged for their technical support. This work was supported by the Internal resources from Hasselt University and Maastricht University. Part of the study was financed by The Netherlands Organization for Scientific Research (NWO) (Veni 916.12.056), The Netherlands Heart Foundation (2013T143) and a Seventh Framework Program (FP7) Grant (CIG 322070) to KW. The funding sources were not involved in any decision making with respect to this manuscript.
Author information
Authors and Affiliations
Contributions
K.V. recruited subjects, collected samples and performed lipolysis and adipocyte morphology measurements, analysed the data and wrote the manuscript. K.W. and K.G. designed, performed and analysed flow cytometry measurements. D.H. designed the study and revised the manuscript. M.B. performed measurements and analysed flow cytometry data. S.W. performed flow cytometry measurements. C.D.S. supervised statistical and flow cytometry analyses and revised the manuscript. G.G. supervised adipocyte morphology and lipolysis measurements and revised the manuscript. C.G.S. supervised flow cytometry analyses and revised the manuscript. E.B. designed the study, contributed to data interpretation and revised the manuscript. J.J. designed the study, supervised lipolysis experiments and contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verboven, K., Wouters, K., Gaens, K. et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci Rep 8, 4677 (2018). https://doi.org/10.1038/s41598-018-22962-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22962-x
This article is cited by
-
Saturated fatty acid concentrations are predictive of insulin sensitivity and beta cell compensation in dogs
Scientific Reports (2024)
-
Adipose tissue as a linchpin of organismal ageing
Nature Metabolism (2024)
-
White adipocyte dysfunction and obesity-associated pathologies in humans
Nature Reviews Molecular Cell Biology (2024)
-
Adipose Cells Induce Escape from an Engineered Human Breast Microtumor Independently of their Obesity Status
Cellular and Molecular Bioengineering (2023)
-
Adipose Tissue and Plasma Markers Associated with HbA1c Pre- and Post-bariatric Surgery: a Cross-sectional and Cohort Study
Obesity Surgery (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
