
Abstract
Synaptic long-term depression (LTD) is believed to underlie critical mnemonic processes in the adult hippocampus. The roles of the metabotropic and ionotropic actions of glutamate in the induction of synaptic LTD by electrical low-frequency stimulation (LFS) in the living adult animal is poorly understood. Here we examined the requirement for metabotropic glutamate (mGlu) and NMDA receptors in LTD induction in anaesthetized adult rats. LTD induction was primarily dependent on NMDA receptors and required the involvement of both the ion channel function and GluN2B subunit of the receptor. Endogenous mGlu5 receptor activation necessitated the local application of relatively high doses of either competitive or non-competitive NMDA receptor antagonists to block LTD induction. Moreover, boosting endogenous glutamate activation of mGlu5 receptors with a positive allosteric modulator lowered the threshold for NMDA receptor-dependent LTD induction by weak LFS. The present data provide support in the living animal that NMDA receptor-dependent LTD is boosted by endogenously released glutamate activation of mGlu5 receptors. Given the predominant perisynaptic location of mGlu5 receptors, the present findings emphasize the need to further evaluate the contribution and mechanisms of these receptors in NMDA receptor-dependent synaptic plasticity in the adult hippocampus in vivo.
Similar content being viewed by others

Introduction
Elucidation of the mechanisms of long-term potentiation (LTP) and long-term depression (LTD) provide a means of understanding synaptic plasticity processes that are believed to underlie information storage processes in learning and memory1,2,3. In the case of CA3-to-CA1 synapses in the hippocampus, LTD induction has been divided into NMDA receptor (NMDAR) and metabotropic glutamate receptor (mGluR) –dependent forms. Until recently, the ability of the ion channel of the NMDAR to conduct Ca2+ currents over prolonged periods was considered fundamental for the induction of NMDAR-dependent LTD by the standard low-frequency conditioning stimulation protocol (LFS, 1 Hz). Controversially, this view has been challenged recently by evidence that non-canonical, metabotropic, roles of the NMDAR mediate this form of LTD induction4,5,6. A major other form of synaptically evoked hippocampal LTD, usually induced by paired-pulse LFS, is mGluR-dependent and has been reported to operate via distinct signaling pathways7,8,9. However, this apparent dichotomy may only be valid under conditions where either one or other of these glutamate receptors is activated alone9,10. Indeed, it has been known for some time that the magnitude of exogenous mGlu5R agonist-induced LTD is dependent on the level of background NMDAR tone11. Since mGlu5Rs are predominantly located outside the synapse, especially perisynaptically within ~60 nm of the synapse edge12,13,14,15 and form part of an extensive signaling interactome with NMDARs16, if sufficient glutamate concentration accumulates at perisynaptic sites during LTD induction, significant cross-talk between these two receptors and their downstream signaling mechanisms is likely.
Presumably because of the known difficulty of inducing LTD in the intact hippocampus in vivo of adult animals17,18 most research on this topic has been performed in brain slices from young animals. Recently, we reported that high-intensity electrical LFS (LFS) reliably induced robust LTD in the hippocampus of anaesthetized rats19. In contrast to most previous in vitro studies (e.g. see10), the induction of this in vivo LTD was resistant to block by standard doses of either NMDAR or mGlu5R antagonists. Because LFS-evoked synaptically released glutamate will spillover to activate peri- and extra-synaptic glutamate receptors20,21, and therefore is likely to co-activate both mGlu5R and NMDARs, we wondered if an interaction between these receptors shaped the induction of LTD.
Therefore, we decided to revisit the glutamate receptor requirements for the induction of synaptic LTD in vivo. We found that relatively high doses of locally-injected NMDAR antagonists were required to block LFS-induced LTD. We show that mGlu5R activation by endogenously released glutamate facilitated the NMDAR-dependent LTD and made it resistant to block with standard doses of NMDAR antagonists. Moreover, the GluN2B subunit and a functional ion channel in NMDARs were obligatory for such LTD induction in vivo.
Results
Role of NMDA and mGlu5 receptors in the induction of hippocampal synaptic LTD by low-frequency conditioning stimulation in vivo
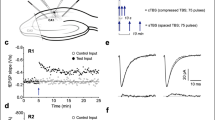
Because we had previously found that the induction of LTD of electrically evoked field excitatory postsynaptic potentials (fEPSPs) by LFS, consisting of 900 high-intensity pulses at 1 Hz, was not blocked by standard systemic doses of either NMDAR or mGlu5R antagonists when applied alone19, we wondered if both types of receptors needed to be blocked simultaneously. Consistent with our previous finding19, using a dose (10 mg/kg, i.p.) of the competitive NMDAR antagonist CPP known to completely inhibit synaptic LTP in the CA1 area22, the application of LFS after injection of CPP induced robust and stable synaptic LTD similar in magnitude to that induced in control, vehicle-injected rats (Fig. 1a,b). Again, confirming our previous report19, application of LFS in MTEP-treated rats induced robust and stable LTD. Initially we tested the standard dose of MTEP (3 mg/kg, i.p., n = 5) which is known to non-competitively block mGlu5Rs, with a rat hippocampal receptor occupancy of >90%23. When this dose proved ineffective we doubled the dose (n = 4) with similar results, so we pooled the data for the two doses (Fig. 1c,d). Intriguingly, even though neither of these treatments on their own affected LTD, combined systemic injection of these antagonists completely abrogated electrical induction of synaptic LTD (Fig. 1e,f).

Combined administration of antagonists of NMDA and mGlu5 receptors, when given systemically, is necessary to prevent the induction of hippocampal synaptic LTD by low-frequency conditioning stimulation in vivo. (a) The competitive NMDAR antagonist CPP (10 mg/kg, i.p.), injected alone 2.25 h prior to electrical LFS (900 high-intensity pulses at 1 Hz) (see also Supplementary Fig. S1), had no significant effect on LTD induction (CPP: 64.4 ± 10%, mean ± SEM fEPSP amplitude expressed as a % of the pre-LFS baseline, n = 10; controls: 66.2 ± 7.1%, n = 10; P < 0.05 both compared with respective pre-LFS baselines, P > 0.05 between groups; two-way ANOVA RM-Sidak). (b) Summary of the mean EPSP amplitude data in (a) before (pre) and 1 h after (post) application of LFS. (c) Injection of the mGlu5R negative allosteric modulator MTEP (3 or 6 mg/kg, i.p., hash) alone did not significantly alter the induction of LTD by LFS 1 h later (MTEP: 75.0 ± 3.8%, n = 9; controls: 64.3 ± 7.7%, n = 9; P < 0.05 both compared with baselines, P > 0.05 between groups, two-way ANOVA RM-Sidak). (d) Summary of the mean EPSP amplitude data in (c). (e) Combined administration of CPP (10 mg/kg, i.p., 2.25 h pre-LFS) with MTEP (3 mg/kg, i.p., 1 h pre-LFS, hash), blocked induction of LTD (CPP + MTEP: 96.7 ± 4.9%, n = 8, P > 0.05 compared with pre-LFS baseline; Veh: 60.6 ± 3.8%, n = 9, P < 0.05 compared with baseline and between groups; two-way ANOVA RM-Sidak). (f) Summary of the mean EPSP amplitude data in (e). *P < 0.05. Insets show typical fEPSP traces at the times indicated. Calibration bars: vertical, 1 mV; horizontal, 10 ms.
These findings indicate that LFS increases glutamate concentration sufficiently to activate mGlu5Rs, which are found predominantly outside, especially on the edge of, the synapse12,13,14,15. Co-activation of mGlu5Rs with NMDARs located in this vicinity is known to strongly enhance the function of the NMDARs24,25,26. Group II mGluRs also are located perisynaptically27, can enhance NMDA-evoked currents28,29, and have been implicated in the induction/maintenance of LTD30,31,32. However, unlike mGlu5Rs, they do not appear to have a role during LTD induction by LFS in vivo. Thus, the high potency mGlu2/3 R antagonist LY341495 (3 mg/kg, i.p.) failed to significantly affect the magnitude of LTD when administered alone (Fig. 2a,b), or when co-administered with CPP (Fig. 2c,d).

Group II mGluR or mGlu1R antagonists alone or in combination with NMDAR blockade fails to inhibit synaptic LTD in vivo. (a) The potent group II mGluR antagonist LY341495 (3 mg/kg, i.p.) did not significantly affect the induction of LTD (LY341495: 74.8 ± 5.4%, n = 11; Veh: 68.2 ± 2.9%, n = 10; P < 0.05 both compared with baselines, P > 0.05 between groups; two-way ANOVA RM-Sidak) (see also Supplementary Fig. S1). (b) Summary of the mean EPSP amplitude data in (a). (c) Intraperitoneal administration of CPP (10 mg/kg, 2.25 h pre-LFS) combined with LY341495 (3 mg/kg, 1 h pre LFS), had no effect on LTD (CPP + LY341495: 61.2 ± 3.7%, n = 5; Veh: 64.9 ± 2.4%, n = 7, P < 0.05 compared to respective baselines and P > 0.05 between groups; two-way ANOVA RM-Sidak). (d) Summary of the mean EPSP amplitude data in (c). (e) Neither injection of the selective mGlu1R antagonist JNJ16259685 (0.5 mg/kg, s.c., asterisk) alone, nor when combined with CPP (10 mg/kg, i.p., 2.25 h pre-LFS) significantly affected the induction of LTD (JNJ16259685: 63.1 ± 1.8%, n = 6; CPP + JNJ16259685: 66.6 ± 4.0%, n = 6; P < 0.05 both compared with baselines, P > 0.05 between groups; two-way ANOVA RM-Sidak). (f) Summary of the mean EPSP amplitude data in (e). *P < 0.05. Calibration bars: vertical, 1 mV; horizontal, 10 ms.
Like mGlu5Rs, mGlu1Rs have been implicated in LTD and are located peri- and extra-synaptically7,33,34. The selective mGlu1R antagonist JNJ16259685 (0.5 mg/kg, s.c.), when injected alone or in combination with CPP (10 mg/kg, i.p.), failed to significantly affect the magnitude of LTD (Fig. 2e,f).
Whereas group II mGluRs enhance GluN2A subunit containing NMDAR function28, mGlu5R enhancement is likely achieved at least partly by promoting the phosphorylation of GluN2B subunits with consequent increased membrane stabilization of NMDARs35. Therefore, we tested the ability of a non-competitive GluN2B-selective antagonist, the negative allosteric modulator Ro 25-6981, to prevent LTD induction in the presence and absence of MTEP. Consistent with the CPP results, whereas Ro 25-6981 (12 mg/kg, i.p) alone failed to significantly alter LTD (Fig. 3a,b) the same dose of Ro 25-6981 given together with MTEP, greatly reduced the magnitude of LTD (Fig. 3c,d).

GluN2B-subunit-selective antagonist, systemically administered with an mGlu5R antagonist, prevents LFS induction of LTD. (a) The non-competitive GluN2B subtype selective NMDAR antagonist Ro 25-6981 (12 mg/kg, i.p., 1 h pre-LFS, hash) alone had no effect on LTD induction (Ro 25-6981: 63.1 ± 4.6%, n = 5; Veh: 64.3 ± 4.2%, n = 6; P < 0.05 both compared with baselines, P > 0.05 between groups; two-way ANOVA RM-Sidak). (b) Summary of the mean EPSP amplitude data in (a). (c) In contrast, a combination of Ro 25-6981 (12 mg/kg, i.p., first hash) with MTEP (3 mg/kg, second hash) strongly attenuated LTD (Ro 25-6981 + MTEP: 91.7 ± 2.1%, n = 6; Veh: 64.2 ± 1.4%, n = 7; P < 0.05 compared with respective baselines and between groups; two-way ANOVA RM-Sidak). (d) Summary of the mean EPSP amplitude data in (c). *P < 0.05. Calibration bars: vertical, 1 mV; horizontal, 10 ms.
These findings are consistent with the supposition that the interaction between mGlu5R and GluN2B subunit-containing NMDARs facilitates the induction of NMDAR-dependent LTD. However, an alternative potential explanation of these data is that the activation of either mGlu5Rs or NMDARs by LFS can independently mediate the induction of LTD in vivo.
Absolute requirement for GluN2B-subunit-containing NMDARs during LTD induction by LFS in vivo
We hypothesized that, if endogenously released glutamate during LFS activates mGlu5Rs to increase the number of functional NMDARs available for LTD induction, then the dose of NMDAR antagonist required to block LTD should be greater when administered alone, relative to when co-administered with an mGlu5R antagonist. In order to assess this possibility, rather than inject higher doses systemically which lead to increased risk of respiratory depression, we injected relatively high doses of NMDAR antagonists locally near the hippocampus, via the intracerebroventricular (i.c.v.) route, thereby minimizing the likelihood of significant systemic toxicity. We started by testing the effect of i.c.v. injection of the competitive antagonist D-AP5. Whereas a dose (100 nmol) that inhibits LTP induction36 was without significant effect, double this dose partially attenuated the magnitude of LTD (Fig. 4a,b). As an additional control, we tested the same dose (200 nmol) of L-AP5, the AP5 stereoisomer that is relatively inactive at NMDARs37, and found that it failed to affect LTD, consistent with a crucial role for NMDARs in LFS induction of LTD.

Relatively high doses of locally-injected NMDAR antagonists prevent LTD induction by LFS in vivo. (a) Intracerebroventricular administration of 200 nmol of the competitive NMDAR antagonist D-AP5 (triangle), 10 min before LFS-900 attenuated LFS induced LTD (200 nmol D-AP5: 73.2 ± 3.9%, n = 5; P < 0.05 compared with Veh: 51.1 ± 3.0%, n = 7; and P < 0.05 compared with 200 nmol of the NMDAR low-affinity stereoisomer of D-AP5, L-AP5: 51.4 ± 2.1%, n = 5; one-way ANOVA-Sidak). The magnitude of LTD in animals injected with the lower dose 100 nmol D-AP5 was not significantly different from vehicle or 200 nmol D-AP5 (100 nmol D-AP5; 61.3 ± 4.5%, n = 5). All groups were significantly different from their respective baselines (paired t). (b) Summary of the mean EPSP amplitude data in (a). (c) Furthermore, i.c.v. application of the non-competitive Ro 25-6981 (2 nmol, triangle) strongly inhibited LTD (2 nmol Ro: 88.2 ± 3.9%, n = 5, P > 0.05 compared with baseline; Veh: 53.8 ± 3.1%, n = 8, P < 0.05 compared with baseline and between groups; two-way ANOVA RM-Sidak). (d) Summary of the mean EPSP data in (c). (e) I.c.v. injection of the lower dose Ro 25-6981 (0.5 nmol, triangle) alone did not affect LTD induction but completely prevented LTD when given in combination with MTEP (3 mg/kg, i.p., 1 h pre-LFS) (0.5 nmol Ro: 57.7 ± 6.6%, n = 6, P < 0.05 compared with baseline; 0.5 nmol Ro + MTEP: 86.4 ± 3.0%, n = 5, P > 0.05 compared with baseline, P < 0.05 between groups; two-way ANOVA RM-Sidak). (f) Summary of the mean EPSP amplitude data in (e). *P < 0.05. Calibration bars: vertical, 1 mV; horizontal, 10 ms.
Given that, compared with LTP, NMDAR-dependent ‘paired-burst’ –induced LTD was reported to be preferentially inhibited by antagonists of GluN2B over GluN2A in vivo38,39 and the known importance of GluN2B subunit-containing NMDARs in mediating facilitatory effects of mGlu5R co-activation35, next we tested the effect of local injection of the very selective GluN2B antagonist Ro 25-6981 on its own. Indeed, treatment with a relatively high dose (2 nmol, i.c.v.) of Ro 25-6981 greatly diminished the magnitude of LTD induced by LFS, compared with vehicle-treated animals (Fig. 4c,d). Furthermore, Ro 25-6981, at the lower dose of 0.5 nmol that was without significant effect on LTD when given on its own, inhibited LTD when given in combination with MTEP (Fig. 4e,f).
GluN2A subunit-containing NMDARs may also be required for LTD40,41 and mGlu5Rs couple to Gαq subunit-containing G proteins that can enhance GluN2A NMDAR function41. Moreover, activation of mGlu5Rs has been reported to promote the tyrosine phosphorylation of both GluN2A and GluN2B subunits in hippocampal cultures42 and recombinant GluN2A NMDAR-mediated Ca2+ responses can be enhanced by stimulation of mGlu5R, in a tyrosine kinase-dependent manner43. Therefore we tested the effect of local i.c.v. injection of the GluN2A-preferring antagonist NVP-AAM077 (NVP). Treatment with NVP (0.5 or 1 nmol) completely blocked HFS-induced LTP (Supplementary Fig. S2a,b). However, NVP failed to affect the magnitude of LTD induced by LFS, when injected alone (1 nmol) or when combined (0.5 or 1 nmol) with the mGlu5R antagonist MTEP (Supplementary Fig. S2c,d).
We didn’t test the effect of high local dose of MTEP on LTD induction because systemic administration of the doses employed in the previous section (see above) achieve very high receptor occupancy23.
LFS-evoked endogenous glutamate release activates mGlu5Rs to facilitate the induction of LTD
The findings described above support the hypothesis that mGlu5R and GluN2B NMDAR co-activation during LFS enhances LTD induction. Therefore, we wondered if pharmacologically boosting endogenous glutamate activation of mGlu5Rs would facilitate the induction of LTD. We predicted that a positive allosteric modulator of these receptors should lower the threshold for LTD, enabling its induction by weak LFS. Therefore, we examined the effect of the mGlu5R positive allosteric modulator VU 0360172 (15 mg/kg, s.c.) on the ability of a peri-threshold induction protocol (300 high-intensity pulses at 1 Hz, LFS-300) to induce LTD. Consistent with the hypothesis, VU 0360172 facilitated the induction of robust LTD by the peri-threshold electrical LFS-300 in a manner that was inhibited by MTEP (Fig. 5a,b).

Endogenous glutamate release activates mGlu5Rs to facilitate the induction of LTD in vivo. (a) The mGlu5R positive allosteric modulator VU 0360172 (15 mg/kg, s.c., asterisk) facilitated the induction of LTD by a peri-threshold electrical LFS-300 conditioning protocol (300 high-intensity pulses at 1 Hz) relative to vehicle controls (control: 90.1 ± 2.6%, n = 11; VU 0360172: 58.7 ± 4.0%, n = 8, P < 0.05 compared with baselines and between groups; one-way ANOVA-Sidak followed by paired t). The facilitation of LTD by VU 0360172 was prevented when the animals were co-treated with MTEP (3 mg/kg, i.p., 1 h pre-LFS, hash) (VU + MTEP: 81.4 ± 2.7%, n = 5; P < 0.05 compared with VU group). (b) Summary of the mean EPSP amplitude data in (a). (c) Local injection of a relatively high dose (2 nmol, i.c.v., triangle) of the GluN2B antagonist Ro 25-6981 prevented LTD induction by LFS-300 in the presence of VU 0360172 (asterisk), whereas systemic treatment (12 mg/kg, i.p., hash) did not (i.c.v. Ro 25-6981: 97.8 ± 3.0%, n = 5, P > 0.05 compared with baseline; i.p. Ro 25-6981: 51.4 ± 3.3%, n = 5, P < 0.05 compared with baseline and between groups; two-way ANOVA RM-Sidak). (d) Summary of the mean EPSP amplitude data in (c). *P < 0.05. Calibration bars: vertical, 1 mV; horizontal, 10 ms.
The data presented so far indicate that the relatively low potency of competitive and non-competitive NMDAR antagonists, when given on their own, in blocking LTD induction by LFS might be caused by co-activation of mGlu5Rs. To further test this hypothesis we compared the ability of systemic and local injection with the non-competitive GluN2B antagonist Ro 25-6981 to prevent LTD induced by weak LFS in the presence of the mGlu5R positive allosteric modulator. Whereas the induction of LTD by LFS-300 in the presence of VU 0360172 was not blocked by a standard systemic dose (12 mg/kg, i.p.) of Ro 25-6981, local high-dose (2 nmol, i.c.v.) injection of this antagonist completely blocked this form of LTD (Fig. 5c,d).
The observed positive allosteric mGlu5R modulator-mediated facilitation of LTD induction, and the requirement for high-dose local GluN2B antagonist to block this facilitated LTD provide complimentary support for the involvement of mGlu5-NMDA receptor co-activation in LFS-induced LTD.
NMDAR ion channel function is required for the induction of LTD in vivo
In the light of the controversy over the requirement for ion flux through the NMDAR channel in LFS induction of synaptic LTD in vitro4,5,6, next we examined the effect of the use-dependent NMDAR channel blocker MK-801 in vivo. In order to reduce the likelihood of significant systemic toxicity, we tested the effects of local injection of MK-801 in the brain. We found that MK-801 (60 nmol, i.c.v.) prevented LTD induction by electrical stimulation in a stereoselective manner. Consistent with a need for the ion channel function of the NMDARs in the induction of LTD, only the NMDAR-selective (+)-enantiomer (MK-801) reduced the magnitude of LTD whereas the control (−)-enantiomer (60 nmol) appeared inactive (Fig. 6a,b).

Necessity for NMDAR ion channel function in the induction of LTD in vivo. (a) A relatively high dose of locally-injected use-dependent NMDAR ion channel blocker MK-801 (60 nmol, i.c.v., triangle), in comparison with the control (−) stereoisomer, strongly inhibited LTD induction in vivo (i.c.v. MK-801: 89.5 ± 1.3%, n = 5; i.c.v.(−)-MK-801: 52.8 ± 3.7%, n = 5, P < 0.05 compared with baselines and between groups; two-way ANOVA RM-Sidak). (b) Summary of the mean EPSP amplitude data in (c). *P < 0.05. Calibration bars: vertical, 1 mV; horizontal, 10 ms.
These data provide strong evidence that the induction of synaptic LTD in vivo requires ion flux via NMDARs.
Discussion
In the present study, we have re-examined glutamate receptor mechanisms underlying the induction of LTD by LFS at CA3-to-CA1 synapses in the hippocampus in vivo. We discovered that local application of relatively high doses of competitive and non-competitive receptor antagonists, when given alone, was required to reveal the NMDAR-dependence of synaptic LTD induction. Remarkably, although mGlu5R antagonism alone failed to affect LTD, standard systemic doses of NMDAR antagonists were sufficient to prevent LFS-induced LTD when mGlu5Rs were simultaneously blocked. Complementing these findings, a positive allosteric modulator of mGlu5Rs lowered the threshold for electrically-induced LTD. Moreover, local application of a relatively high dose of NMDAR antagonist was again required to inhibit this mGlu5R positive allosteric modulator-facilitated LTD. We also found strong evidence of the obligatory requirement for the GluN2B subunit and the ion channel function of NMDARs in the induction of LTD by LFS in vivo. Taken together these findings attest that endogenously released glutamate activation of mGlu5Rs enhances LTD induction that requires the GluN2B subunit and ion channel function of NMDARs.
The induction of LTD by LFS was inhibited only by local application of relatively high doses of a variety of NMDAR antagonists that act via different mechanisms: (1) bind competitively to the orthosteric glutamate binding site (D-AP5), (2) non-competitively block the GluN2B subunit (negative allosteric modulator Ro 25-6981) or (3) non-competitively block the open ion channel (MK-801). The lack of significant inhibitory effect of standard doses of these and similar agents previously, led us to wrongly assume that high-intensity LFS induction of synaptic LTD in vivo is NMDAR-independent19. These results reinforce the need to reassess LTD induced by other electrical stimulation protocols that are currently considered by many not to require NMDAR activation, on a case by case basis9,44,45, see also46. This requirement for local application of relatively high doses of NMDAR antagonists is unlikely to be solely because LFS increases glutamate release, since the standard doses of D-AP5 and CPP used here, that failed to inhibit LTD, completely block the induction of LTP by electrical high-frequency conditioning stimulation that greatly increases glutamate release36,47. Moreover, by definition, non-competitive blockade of NMDARs will be relatively independent of ambient glutamate concentration especially at synapses with low receptor reserve. The requirement for a relatively high concentration of antagonist to achieve significant block of LTD induction therefore could be caused by a recruitment of additional functional NMDAR numbers, perhaps as a consequence of glutamate spillover to extrasynaptic NMDARs which may be preferentially blocked by GluN2B selective antagonists21,48, but see49. Our finding that blocking mGlu5Rs lowered the dose of NMDAR antagonist required to inhibit LTD by LFS is consistent with, but does not prove, the interpretation that mGlu5R co-activation is critically involved. Previously NMDAR-dependent LTD induction in rats was reported to be blocked by i.c.v. injection of either D-AP5 or the mGlu5R antagonist MPEP alone50. The apparent differences from our findings may be caused by different recording (freely behaving versus anesthetized) or stimulation (high-intensity LFS used here) conditions. Moreover MPEP, unlike MTEP, can also block NMDARs if the local concentration reaches above ~10 µM23,51,52. Interestingly, mGlu5 and NMDA receptors are associated as part of an interactome16 and co-activation of these receptors enhances NMDAR-mediated synaptic function24,25,26. Moreover, a PKC-dependent activation by mGlu1Rs leading to increased numbers of functional NMDA receptors and increased mean channel open time has been proposed as a basis for modulating synaptic plasticity53,54,55. Although we did not find evidence for a role of mGlu1R, since activation of mGlu5R also increases PKC activation, similar modulatory mechanisms may apply for this receptor subtype. Previous research on hippocampal slices from young rats found that although LFS induced LTD of the NMDAR-mediated component of synaptic transmission required mGlu1R activation, LTD of the AMPAR-mediated component was not34 (see also refs7,45,56,57). Further, complementary, confirmation of the role of mGlu5Rs in the direct regulation of NMDAR function in vivo was our finding that a positive allosteric modulator at mGlu5Rs lowers the threshold for the induction of LTD by LFS. Importantly, similar to LTD induced by standard LFS, relatively high-dose NMDAR antagonist also was required to inhibit this pharmacologically potentiated LTD. Although it is possible that mGlu5R-mediated depolarization58 or dis-inhibition59 may be involved in the facilitation of LTD, it is unclear how such an action would increase the dose requirement for non-competitive NMDA receptor antagonist to block LTD. Future research, including high-resolution confocal microscopy in vivo, will be necessary to directly assess if mGlu5R activation boosts functional NMDAR numbers or possibly interacts at the level of downstream mediators such as P38 MAP kinase which has been implicated in mediating both mGluR- and NMDAR-dependent LTD1,60.
Recent research35 emphasizes a key role for GluN2B subunits in mediating mGlu5R enhancement of NMDAR excitability, where co-activation of mGlu5R/NMDAR activates Src kinases, synergistically leading to GluN2B(Tyr1472) phosphorylation. Similar phosphorylation prevents GluN2B internalization, stabilizing GluN2B-containing NMDARs in the plasma membrane61,62. In striatal neurons activation of mGlu5R dissociates CaMKIIα from the receptor thereby promoting its binding to any adjacent GluN2B subunit, which enables CaMKIIα to phosphorylate GluN2B63. Such phosphorylation stabilizes GluN2B in the membrane and enhances NMDAR function63,64. Indeed our data demonstrate that GluN2B subunits are critical for LTD induction by LFS, consistent with previous in vivo38,65, and many in vitro studies, e.g.66,67, reviewed in40 but see68,69,70. Similarly, although an apparent resistance of LTD to inhibition by NMDAR antagonists, including D-AP5 and CPP, in vitro has been attributed to a preferential involvement of GluN2C/D subunit-containing NMDARs in juvenile rat hippocampus71,72 the present data strongly indicate a requirement for GluN2B in LTD in the adult rat in vivo. GluN2 subunit expression and localization changes markedly across development, with a higher ratio of GluN2A:GluN2B, and a shift of GluN2B from synaptic to a predominantly perisynaptic and extrasynaptic location, in adulthood73. Small changes in age during development affect the magnitude or even the presence of LTD74, consistent with a critical role for GluN2B in the induction of LTD. Based on our findings with Ro 25-6981, GluN1/GluN2B diheteromers are required for synaptic LTD induced by high-intensity LFS in the adult rat in vivo. The finding that a dose of NVP that completely inhibited LTP, given alone did not affect LFS-induced LTD, is consistent with previous in vivo findings that LTP is more sensitive to NVP than paired burst-induced LTD38,39. However, it does not rule out roles for GluN1/GluN2A diheteromers or GluN1/GluN2A/GluN2B triheteromeric NMDARs40,41,75,76,77,78. The inability of a relatively low dose of NVP to inhibit LTD when given in combination with MTEP contrasts with our finding of a lower dose requirement for Ro 25-6981 in the presence of MTEP. Future research should determine if GluN2A and GluN2B differentially couple to mGlu5Rs in the adult rat hippocampus.
The ability of the open-channel, use-dependent, blocker of NMDARs MK-80149, to inhibit LTD strongly indicates that the LTD induction is dependent on ion flux through NMDARs. The widely accepted view that the faster and greater Ca2+ influx through NMDARs during high-frequency stimulation protocols leads to LTP at hippocampal synapses while slower and smaller Ca2+ influx through NMDARs results in LTD, has been challenged recently6,79. Significantly, although we did not examine the requirement for a metabotropic function of NMDARs, the present study showing that LTD was blocked by MK-801strongly supports the view that Ca2+ influx through NMDARs is required for LTD induction in vivo. Thus, although our data are consistent with a physical interaction between mGlu5Rs and GluN2B facilitating LTD induction, the obligatory involvement of NMDAR ion channel function in triggering LTD in vivo emerges as paramount.
In view of the predominant localization of mGlu5Rs at peri- and extra-synaptic sites12,13,14,15, our finding of a facilitatory role of mGlu5Rs in NMDAR-dependent LTD induction is consistent with the interpretation that significant co-activation of mGlu5Rs and NMDARs at non-synaptic sites is involved. The likelihood of this occurring will be dependent on the spatial pattern of synaptic activation, with electrical field stimulation, as used in the present experiments, providing highly favorable conditions for glutamate spillover-mediated co-activation of mGlu5 and NMDA receptors, whereas NMDAR-dependent LTD induced by relatively sparse optogenetic stimulation appears to be independent of mGlu5Rs80. Nearby synapses are especially likely to be co-activated under pathological conditions such as epilepsy and spillover to neighboring synapses will be increased when glutamate homeostasis is compromised, such as occurs in psychiatric and neurological illnesses. Undoubtedly, hippocampal mGlu5Rs act as sentinels of glutamate spillover and, when activated in concert with GluN2B, trigger a persistent down-regulation of potentially redundant or inappropriate synaptic transmission, thus placing mGlu5R facilitation of LTD in the centre of the synaptic physiology-pathology continuum.
Materials and Methods
Animal surgery, electrode and cannula implantation
Animal care and experimental protocols were carried out in accordance with the approval of the Health Products Regulatory Authority, Ireland. Adult male Wistar and Lister hooded rats, supplied by Trinity College Comparative Medicine, were housed in a 12-h light-dark cycle at room temperature (19–22 °C). Electrode implantation/recording was performed on 4.5–6 month-old (400–500 g) animals, anaesthetized with urethane (1.5–1.6 g/kg, i.p.) and core body temperature was maintained at 37 °C while placed in a stereotaxic apparatus for the duration of the experiment. A twisted bipolar Teflon-coated tungsten wire (inner core diameter 50 μm, external diameter 75 µm) stimulating electrode was lowered into the ipsilateral stratum radiatum of area CA3 at coordinates −4.2 mm posterior to the coronal suture, and −3.8 mm lateral to the sagittal suture, measured from bregma. In some animals, which had also previously undergone surgery to transduce channelrhodopsin in CA3 cells, a 200 μm optical fiber cable was also attached to the stimulating electrode, but the light responses were unsuitable for recording. A monopolar Teflon-coated tungsten wire (inner core diameter 75 μm, external diameter 112 μm) was lowered into the stratum radiatum of area CA1 at coordinates −3.5 mm posterior to the coronal suture, and −2.5 mm lateral to the sagittal suture, measured from bregma. Screw electrodes located over the contralateral cortex were used for reference and earth. The final placement of electrodes was optimized by using electrophysiological criteria and confirmed via post-mortem analysis.
Where necessary, a stainless-steel cannula (22 gauge, 0.7 mm outer diameter) was implanted above the ipsilateral-to-recording ventricle (0.5 mm posterior to the coronal suture, and 1 mm lateral to the sagittal suture, lowered 4.2 mm from the surface of the skull) and secured in place with dental cement. The solutions were injected through an internal cannula (28 gauge, 0.36 mm outer diameter) no quicker than 1 μl per min. Verification of the placement of the cannula was performed post-mortem.
In vivo recording
Single square-wave electric pulse (0.2 ms duration) test stimulation was delivered to the Schaffer-collateral/commissural pathway every 30 s, at a frequency of 0.033 Hz, to evoke fEPSPs at a low intensity until stabilisation of a baseline response. Input/output (i/o) curves were generated for each rat separately to ascertain half maximum stimulation intensity that was used for test pulse stimulation. Baseline transmission had to be stable for a minimum of 30 min before injection of vehicle/drug. LTD was induced using an electrical LFS protocol consisting of 900 pulses run at 1 Hz, with the stimulation intensity increased to 95% maximum amplitude. A relatively weak LFS protocol (LFS-300), used to study the facilitation of LTD, consisted of 300 pulses at 1 Hz, with an intensity that evoked 95% maximum amplitude. fEPSPs were recorded for at least 1 h after LFS before a final end-run i/o curve was measured. Background EEG in the hippocampus was monitored (LabChart, AD instruments) throughout the experiment. None of the treatments had any discernable effect on baseline synaptic excitability or general EEG parameters. Upon completion of the recording session rats were perfused with 4% paraformaldehyde (Sigma) and the brain was stored at 4 °C.
Agents
CPP ((R,S)-3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid) (Abcam), MTEP (3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride) (Abcam), D-AP5 (D-(−)-2-amino-5-phosphonopentanoic acid) and L-AP5 (L-(+)-2-Amino-5-phosphonopentanoic acid) (both Tocris), MK-801 ((5 S,10 R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate) (dizocilpine) and its less active (−) enantiomer (both Tocris), and NVP-AAM077, also known as PEAQX, (tetrasodium; 5-[[[(1S)-1-(4-bromophenyl)ethyl]amino]-phosphonatomethyl]quinoxaline-2,3-diolate;hydrate) (Alamone), were prepared in ultra-clean water as stock solutions. The stock solutions were further diluted into 1 ml saline for i.p. injection; 1 ml saline was used as vehicle control. Ro 25-6981 ([R-(R,S)]-α-(4-Hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol hydrochloride hydrate) (Sigma) and VU 0360172 (N-Cyclobutyl-6-[2-(3-fluorophenyl)ethynyl]-3-pyridinecarboxamide hydrochloride) (Tocris) were dissolved in DMSO and diluted in saline; a 1 ml 10% v/v solution of DMSO in saline was used as vehicle control. LY341495 ((2S)-2-Amino-2-[(1 S,2 S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl)propanoic acid) (Abcam), was dissolved in 1.2 molar equivalent sodium hydroxide (NaOH, Sigma) and diluted in saline; saline was used as vehicle control. JNJ16259685 ((3,4-dihydro-2H-pyrano[2,3]b quinolin-7-yl) (cis-4-methoxycyclohexyl) methanone) (R&D Systems), was initially dissolved in 70% ethanol and subsequently diluted by a factor of 40 in saline.
The dose of CPP was chosen based on previous publications including our finding that 10 mg/kg, i.p., completely blocked high-frequency stimulation induced LTP22. The pre-injection interval was based on pilot studies and the literature81. In the case of MTEP, we started with a dose of 3 mg/kg, which we found completely blocked Aβ-facilitated LTD19. MTEP is known to achieve >90% receptor occupancy after i.p. injection with either 3 or 6 mg/kg in 10% Tween 80 in awake rats23. Ro 25-6981, at a dose of 6 mg/kg prevents Aβ-mediated LTP inhibition47. We chose doses for i.c.v. injection of Ro 25-6981 based on pilot studies and prior research82. The MK-801 dose of 60 nmol, i.c.v., for local injection is based on previous research83. (−)-MK-801 is approximately one-seventh as potent as its active enantiomer MK-801 at NMDARs84. The initial dose of D-AP5 was chosen on the basis of previous reports including our finding that LTP is blocked by 100 nmol after i.c.v. injection36. D-AP5′s stereoisomer L-AP5 is known to be at least 30-fold less potent as an NMDAR antagonist than D-AP537. A dose of 3 mg/kg, i.p., LY341495 has been reported to impair recognition memory in rats85. This relatively low dose is used to strongly block group II mGluRs but may also at least partly block group III mGluRs86,87,88. Based on previous research89 and pilot studies we chose a dose of 15 mg/kg s.c. for VU 0360172. JNJ16259685 has been reported to achieve >90% receptor occupancy after s.c. injection with 0.5 mg/kg90. The dose of NVP was chosen based on its known ability to inhibit LTP in the range of 0.25–1 nmol after i.c.v. injection in vivo47. NVP is a competitive antagonist with an ~11-fold preference for GluN1/2 A over GluN1/2B receptors, which will likely only maintain its selectivity to block only GluN2A diheteromeric or GluN2A/B triheteromeric NMDAR-mediated synaptic responses when used at relatively low, non-saturating concentrations77.
Data analysis
Values are expressed as the mean ± s.e.m. % of the baseline fEPSP amplitude measured 30 or 60 min before an injection. The magnitude of LTD was measured over the last 10 min at 1 h after LFS. Similar results were obtained when fEPSP slope was analysed. No data were excluded, and control experiments were interleaved randomly throughout. For two groups with two time points, two-way ANOVA with repeated measures with Sidak’s multiple comparison test (two-way ANOVA RM-Sidak) was used. To compare between groups or time points of three or more, one-way ANOVA with Sidak’s multiple comparisons (one-way ANOVA-Sidak) was used. Two-tailed paired Student’s t-tests (paired t) were used to compare pre- and post-LFS data within one group. A value of P < 0.05 was considered statistically significant.
References
Collingridge, G. L., Peineau, S., Howland, J. G. & Wang, Y. T. Long-term depression in the CNS. Nat Rev Neurosci 11, 459–473 (2010).
Nabavi, S. et al. Engineering a memory with LTD and LTP. Nature 511, 348–352 (2014).
Holtmaat, A. & Caroni, P. Functional and structural underpinnings of neuronal assembly formation in learning. Nat Neurosci 19, 1553–1562 (2016).
Babiec, W. E. et al. Ionotropic NMDA receptor signaling is required for the induction of long-term depression in the mouse hippocampal CA1 region. J Neurosci 34, 5285–5290 (2014).
Gray, J. A., Zito, K. & Hell, J. W. Non-ionotropic signaling by the NMDA receptor: controversy and opportunity. F1000Res 5, 1010 (2016).
Nabavi, S. et al. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc Natl Acad Sci USA 110, 4027–4032 (2013).
Volk, L. J., Daly, C. A. & Huber, K. M. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol 95, 2427–2438 (2006).
Oliet, S. H., Malenka, R. C. & Nicoll, R. A. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18, 969–982 (1997).
Luscher, C. & Huber, K. M. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459 (2010).
Izumi, Y. & Zorumski, C. F. NMDA receptors, mGluR5, and endocannabinoids are involved in a cascade leading to hippocampal long-term depression. Neuropsychopharmacology 37, 609–617 (2012).
Palmer, M. J., Irving, A. J., Seabrook, G. R., Jane, D. E. & Collingridge, G. L. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology 36, 1517–1532 (1997).
Lujan, R., Nusser, Z., Roberts, J. D., Shigemoto, R. & Somogyi, P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci 8, 1488–1500 (1996).
Lujan, R., Roberts, J. D., Shigemoto, R., Ohishi, H. & Somogyi, P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat 13, 219–241 (1997).
Ferraguti, F. & Shigemoto, R. Metabotropic glutamate receptors. Cell Tissue Res 326, 483–504 (2006).
Hainmuller, T., Krieglstein, K., Kulik, A. & Bartos, M. Joint CP-AMPA and group I mGlu receptor activation is required for synaptic plasticity in dentate gyrus fast-spiking interneurons. Proc Natl Acad Sci USA 111, 13211–13216 (2014).
Darnell, J. C. & Klann, E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16, 1530–1536 (2013).
Xu, L., Anwyl, R. & Rowan, M. J. Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387, 497–500 (1997).
Staubli, U. & Scafidi, J. Studies on long-term depression in area CA1 of the anesthetized and freely moving rat. J Neurosci 17, 4820–4828 (1997).
Hu, N. W. et al. mGlu5 receptors and cellular prion protein mediate amyloid-beta-facilitated synaptic long-term depression in vivo. Nat Commun 5, 3374 (2014).
Arnth-Jensen, N., Jabaudon, D. & Scanziani, M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci 5, 325–331 (2002).
Scimemi, A., Fine, A., Kullmann, D. M. & Rusakov, D. A. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci 24, 4767–4777 (2004).
Doyle, C., Holscher, C., Rowan, M. J. & Anwyl, R. The selective neuronal NO synthase inhibitor 7-nitro-indazole blocks both long-term potentiation and depotentiation of field EPSPs in rat hippocampal CA1 in vivo. J Neurosci 16, 418–424 (1996).
Busse, C. S. et al. The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 29, 1971–1979 (2004).
Doherty, A. J., Palmer, M. J., Henley, J. M., Collingridge, G. L. & Jane, D. E. RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but not mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology 36, 265–267 (1997).
Kotecha, S. A. et al. Co-stimulation of mGluR5 and N-methyl-D-aspartate receptors is required for potentiation of excitatory synaptic transmission in hippocampal neurons. J Biol Chem 278, 27742–27749 (2003).
Mannaioni, G., Marino, M. J., Valenti, O., Traynelis, S. F. & Conn, P. J. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21, 5925–5934 (2001).
Tamaru, Y., Nomura, S., Mizuno, N. & Shigemoto, R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience 106, 481–503 (2001).
Trepanier, C., Lei, G., Xie, Y. F. & MacDonald, J. F. Group II metabotropic glutamate receptors modify N-methyl-D-aspartate receptors via Src kinase. Sci Rep 3, 926 (2013).
Rosenberg, N., Gerber, U. & Ster, J. Activation of Group II Metabotropic Glutamate Receptors Promotes LTP Induction at Schaffer Collateral-CA1 Pyramidal Cell Synapses by Priming NMDA Receptors. J Neurosci 36, 11521–11531 (2016).
Manahan-Vaughan, D. Group 1 and 2 metabotropic glutamate receptors play differential roles in hippocampal long-term depression and long-term potentiation in freely moving rats. J Neurosci 17, 3303–3311 (1997).
Li, S. T. et al. Calcineurin plays different roles in group II metabotropic glutamate receptor- and NMDA receptor-dependent long-term depression. J Neurosci 22, 5034–5041 (2002).
Santschi, L. A., Zhang, X. L. & Stanton, P. K. Activation of receptors negatively coupled to adenylate cyclase is required for induction of long-term synaptic depression at Schaffer collateral-CA1 synapses. J Neurobiol 66, 205–219 (2006).
Baude, A. et al. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron 11, 771–787 (1993).
Bhouri, M. et al. mGlu1 receptor-induced LTD of NMDA receptor transmission selectively at Schaffer collateral-CA1 synapses mediates metaplasticity. J Neurosci 34, 12223–12229 (2014).
Sarantis, K., Tsiamaki, E., Kouvaros, S., Papatheodoropoulos, C. & Angelatou, F. Adenosine A(2)A receptors permit mGluR5-evoked tyrosine phosphorylation of NR2B (Tyr1472) in rat hippocampus: a possible key mechanism in NMDA receptor modulation. J Neurochem 135, 714–726 (2015).
Hu, N. W., Smith, I. M., Walsh, D. M. & Rowan, M. J. Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain 131, 2414–2424 (2008).
Watkins, J. C. & Evans, R. H. Excitatory amino acid transmitters. Annu Rev Pharmacol Toxicol 21, 165–204 (1981).
Fox, C. J., Russell, K. I., Wang, Y. T. & Christie, B. R. Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus 16, 907–915 (2006).
Ge, Y. et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci USA 107, 16697–16702 (2010).
Shipton, O. A. & Paulsen, O. GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos Trans R Soc Lond B Biol Sci 369, 20130163 (2014).
Yang, K., Jackson, M. F. & MacDonald, J. F. Recent progress in understanding subtype specific regulation of NMDA receptors by G Protein Coupled Receptors (GPCRs). Int J Mol Sci 15, 3003–3024 (2014).
Takagi, N., Besshoh, S., Marunouchi, T., Takeo, S. & Tanonaka, K. Metabotropic glutamate receptor 5 activation enhances tyrosine phosphorylation of the N-methyl-D-aspartate (NMDA) receptor and NMDA-induced cell death in hippocampal cultured neurons. Biol Pharm Bull 35, 2224–2229 (2012).
Collett, V. J. & Collingridge, G. L. Interactions between NMDA receptors and mGlu5 receptors expressed in HEK293 cells. Br J Pharmacol 142, 991–1001 (2004).
Kemp, N. & Bashir, Z. I. NMDA receptor-dependent and -independent long-term depression in the CA1 region of the adult rat hippocampus in vitro. Neuropharmacology 36, 397–399 (1997).
Eng, A. G., Kelver, D. A., Hedrick, T. P. & Swanson, G. T. Transduction of group I mGluR-mediated synaptic plasticity by beta-arrestin2 signalling. Nat Commun 7, 13571 (2016).
Toft, A. K., Lundbye, C. J. & Banke, T. G. Dysregulated NMDA-Receptor Signaling Inhibits Long-Term Depression in a Mouse Model of Fragile X Syndrome. J Neurosci 36, 9817–9827 (2016).
Hu, N. W., Klyubin, I., Anwyl, R. & Rowan, M. J. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci USA 106, 20504–20509 (2009).
Tovar, K. R. & Westbrook, G. L. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19, 4180–4188 (1999).
Harris, A. Z. & Pettit, D. L. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol 584, 509–519 (2007).
Popkirov, S. G. & Manahan-Vaughan, D. Involvement of the metabotropic glutamate receptor mGluR5 in NMDA receptor-dependent, learning-facilitated long-term depression in CA1 synapses. Cereb Cortex 21, 501–509 (2011).
Harney, S. C., Rowan, M. & Anwyl, R. Long-term depression of NMDA receptor-mediated synaptic transmission is dependent on activation of metabotropic glutamate receptors and is altered to long-term potentiation by low intracellular calcium buffering. J Neurosci 26, 1128–1132 (2006).
O’Leary, D. M., Movsesyan, V., Vicini, S. & Faden, A. I. Selective mGluR5 antagonists MPEP and SIB-1893 decrease NMDA or glutamate-mediated neuronal toxicity through actions that reflect NMDA receptor antagonism. Br J Pharmacol 131, 1429–1437 (2000).
Lan, J. Y. et al. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4, 382–390 (2001).
Lan, J. Y. et al. Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J Neurosci 21, 6058–6068 (2001).
Skeberdis, V. A. et al. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology 40, 856–865 (2001).
Neyman, S. & Manahan-Vaughan, D. Metabotropic glutamate receptor 1 (mGluR1) and 5 (mGluR5) regulate late phases of LTP and LTD in the hippocampal CA1 region in vitro. Eur J Neurosci 27, 1345–1352 (2008).
Peng, Y. et al. Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor-mediated synaptic responses. Hippocampus 20, 646–658 (2010).
Pisani, A., Calabresi, P., Centonze, D. & Bernardi, G. Enhancement of NMDA responses by group I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol 120, 1007–1014 (1997).
Olmo, I. G., Ferreira-Vieira, T. H. & Ribeiro, F. M. Dissecting the Signaling Pathways Involved in the Crosstalk between Metabotropic Glutamate 5 and Cannabinoid Type 1 Receptors. Mol Pharmacol 90, 609–619 (2016).
Sanderson, T. M., Hogg, E. L., Collingridge, G. L. & Correa, S. A. Hippocampal metabotropic glutamate receptor long-term depression in health and disease: focus on mitogen-activated protein kinase pathways. J Neurochem 139(2), 200–214 (2016).
Prybylowski, K. et al. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47, 845–857 (2005).
Roche, K. W. et al. Molecular determinants of NMDA receptor internalization. Nat Neurosci 4, 794–802 (2001).
Jin, D. Z., Guo, M. L., Xue, B., Mao, L. M. & Wang, J. Q. Differential regulation of CaMKIIalpha interactions with mGluR5 and NMDA receptors by Ca(2+) in neurons. J Neurochem 127, 620–631 (2013).
Jin, D. Z., Xue, B., Mao, L. M. & Wang, J. Q. Metabotropic glutamate receptor 5 upregulates surface NMDA receptor expression in striatal neurons via CaMKII. Brain Res 1624, 414–423 (2015).
Wong, T. P. et al. Hippocampal long-term depression mediates acute stress-induced spatial memory retrieval impairment. Proc Natl Acad Sci USA 104, 11471–11476 (2007).
Liu, L. et al. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304, 1021–1024 (2004).
France, G. et al. Multiple roles of GluN2B-containing NMDA receptors in synaptic plasticity in juvenile hippocampus. Neuropharmacology 112, 76–83 (2017).
Bartlett, T. E. et al. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 52, 60–70 (2007).
Kollen, M., Dutar, P. & Jouvenceau, A. The magnitude of hippocampal long term depression depends on the synaptic location of activated NR2-containing N-methyl-D-aspartate receptors. Neuroscience 154, 1308–1317 (2008).
Morishita, W. et al. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology 52, 71–76 (2007).
Hrabetova, S. & Sacktor, T. C. Long-term potentiation and long-term depression are induced through pharmacologically distinct NMDA receptors. Neurosci Lett 226, 107–110 (1997).
Hrabetova, S. et al. Distinct NMDA receptor subpopulations contribute to long-term potentiation and long-term depression induction. J Neurosci 20, RC81 (2000).
Yashiro, K. & Philpot, B. D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094 (2008).
Ku, H. Y., Huang, Y. F., Chao, P. H., Huang, C. C. & Hsu, K. S. Neonatal isolation delays the developmental decline of long-term depression in the CA1 region of rat hippocampus. Neuropsychopharmacology 33, 2847–2859 (2008).
Izumi, Y. & Zorumski, C. F. Sensitivity of N-methyl-D-aspartate receptor-mediated excitatory postsynaptic potentials and synaptic plasticity to TCN 201 and TCN 213 in rat hippocampal slices. J Pharmacol Exp Ther 352, 267–273 (2015).
Hansen, K. B., Ogden, K. K., Yuan, H. & Traynelis, S. F. Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 81, 1084–1096 (2014).
Lind, G. E. et al. Structural basis of subunit selectivity for competitive NMDA receptor antagonists with preference for GluN2A over GluN2B subunits. Proc Natl Acad Sci USA 114, E6942–E6951 (2017).
Stroebel, D., Casado, M. & Paoletti, P. Triheteromeric NMDA receptors: from structure to synaptic physiology. Curr Opin Physiol 2, 1–12 (2018).
Tigaret, C. M., Olivo, V., Sadowski, J. H., Ashby, M. C. & Mellor, J. R. Coordinated activation of distinct Ca(2+) sources and metabotropic glutamate receptors encodes Hebbian synaptic plasticity. Nat Commun 7, 10289 (2016).
O’Riordan, K. J., Hu, N.-W. & Rowan, M. J. Aß facilitates LTD at Schaffer collateral synapses preferentially in the left hippocampus. Cell Reports 22, 2053–2065 (2018).
Yen, W., Williamson, J., Bertram, E. H. & Kapur, J. A comparison of three NMDA receptor antagonists in the treatment of prolonged status epilepticus. Epilepsy Res 59, 43–50 (2004).
Zhong, W. X. et al. N-methyl-D-aspartate receptor-dependent long-term potentiation in CA1 region affects synaptic expression of glutamate receptor subunits and associated proteins in the whole hippocampus. Neuroscience 141, 1399–1413 (2006).
Yoshiyama, M. & de Groat, W. C. Supraspinal and spinal alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid and N-methyl-D-aspartate glutamatergic control of the micturition reflex in the urethane-anesthetized rat. Neuroscience 132, 1017–1026 (2005).
Wong, E. H. et al. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc Natl Acad Sci USA 83, 7104–7108 (1986).
Pitsikas, N., Kaffe, E. & Markou, A. The metabotropic glutamate 2/3 receptor antagonist LY341495 differentially affects recognition memory in rats. Behav Brain Res 230, 374–379 (2012).
Bond, A. et al. Neuroprotective effects of LY379268, a selective mGlu2/3 receptor agonist: investigations into possible mechanism of action in vivo. J Pharmacol Exp Ther 294, 800–809 (2000).
Kingston, A. E. et al. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 37, 1–12 (1998).
Ornstein, P. L. et al. 2-substituted (2SR)-2-amino-2-((1SR,2SR)-2-carboxycycloprop-1-yl)glycines as potent and selective antagonists of group II metabotropic glutamate receptors. 2. Effects of aromatic substitution, pharmacological characterization, and bioavailability. J Med Chem 41, 358–378 (1998).
Noetzel, M. J. et al. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol 81, 120–133 (2012).
Lavreysen, H. et al. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology 47, 961–972 (2004).
Acknowledgements
Science Foundation Ireland (10/IN.1/B3001 and 14/IA/2571) the Irish Health Research Board (HRA-POR-2015-1102), and National Natural Science Foundation of China (No. 81471114).
Author information
Authors and Affiliations
Contributions
K.O’R. contributed to experimental design, performed experiments and data analysis. N.-W.H. contributed to experimental design, performed experiments and data analysis. M.J.R. conceived and supervised the project. All authors wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
O’Riordan, K.J., Hu, NW. & Rowan, M.J. Physiological activation of mGlu5 receptors supports the ion channel function of NMDA receptors in hippocampal LTD induction in vivo. Sci Rep 8, 4391 (2018). https://doi.org/10.1038/s41598-018-22768-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22768-x
This article is cited by
-
Bioactive human Alzheimer brain soluble Aβ: pathophysiology and therapeutic opportunities
Molecular Psychiatry (2022)
-
Dendritic autophagy degrades postsynaptic proteins and is required for long-term synaptic depression in mice
Nature Communications (2022)
-
Effects of blocking mGluR5 on primate dorsolateral prefrontal cortical neuronal firing and working memory performance
Psychopharmacology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
