
Abstract
This paper describes the wet-chemistry synthesis of highly crystalline hexagonal flakes of Ni-Fe layered double hydroxide (LDH) produced at temperature as low as 100 °C. The flakes with diameter in the range of 0.5–1.5 μm and the thickness between 15 and 20 nm were obtained by homogeneous precipitation method with the use of triethanolamine (TEA) and urea. By analyzing the intermediate products, it is suggested that, differently from previous reports, a thermodynamically metastable iron oxyhydroxide and Ni-TEA complex are firstly formed at room temperature. Subsequently, when the mixture is heated to 100 °C and the pH increases due to the thermal decomposition of urea, Ni2+ and Fe3+ are slowly released and then recombine, thus leading to formation of pure, highly-crystalline Ni-Fe LDH flakes. This material showed promising results as an electrocatalyst in oxygen evolution reaction (OER) providing an overpotential value of 0.36 V.
Similar content being viewed by others

Introduction
Ni-Fe LDH layered double hydroxide (LDH) have become the focus of an extensive scientific research, mainly due to the high electrocatalytic activity of this material in oxygen evolution reaction (OER), oxygen reduction reaction (ORR) and electrode material for supercapacitor1,2,3,4,5. LDH have a structure similar to brucite where single layers are made of edge sharing Mg(OH)6 octahedrons6. Contrary to brucite, the layers of LDH present a net positive charge due to the partial substitution of divalent cations with trivalent ones7. In order to ensure overall electrical charge neutrality, anions are intercalated within the interlayer space, which results in electrostatic interactions between the layers. These forces together with van der Waals interactions and hydrogen bonds keep the sheets together to form a three-dimensional layered framework7,8.
Recently it was suggested that Ni-Fe LDH catalytic activity is related not only to the surface area, but also to the number of open coordination sites at the edges9, so the design of simple synthesis routes for the preparation of crystalline, thin platelets with homogeneously distributed Ni and Fe is crucial in order to achieve reliable and highly active catalysts3. However, the synthesis of Fe3+-containing LDH with high crystallinity and a well-defined shape is challenging because usually gel-like, water-insoluble Fe(OH)3 precipitates at pH above 2, which impedes the further incorporation of Ni2+ within its structure10,11,12. Several techniques, such a as reversed co-precipitation13, ball milling14, topochemical routes10,15 or co-precipitation with the use of long-chain organic acid16, were implemented in order to synthesize high-quality Ni-Fe LDH. However, these synthesis routes do not necessarily assure phase purity and the desired morphology of the obtained material. For instance, samples prepared by co-precipitation methods17, are poorly crystalline and usually contaminated with metal hydroxides. Similarly, the ball milling approach is not suitable to produce the material with well-defined morphology14 and the obtained nanoparticles exhibit a broad size distribution. Furthermore, in this process the only source of iron ions are carbon steel balls, which corrode during the milling process and release Fe2+ and/or Fe3+. The presence of Fe2+ ions in the reaction mixture, together with other elements (such as Mn, C, S) used to make steel balls, might contaminate the LDH phase. Finally, the topochemical route relies on an initial precipitation of Fe2+ and its subsequent oxidation to Fe3+ using iodine10 or anthraquinone-2-sulfonate10,15. This additional synthesis step led to the formation of regular hexagonal platelets15, however Fe2+ ions and oxidizing agents are possible sources of contamination. Recently, in order to overcome these issues and produce regular platelets of Ni-Fe LDH, special attention has been placed on the use of capping agents, such as trisodium citrate or triethanolamine (TEA), which coordinate Fe3+, hence preventing the formation of its hydroxide while simultaneously facilitating the combination with Ni2+ 11,12.
The synthesis of Ni-Fe LDH with the use of TEA follows the so-called “atrane route”9. In a simple way, the formation of Fe-TEA complexes, which are inert to hydrolysis, prevents the precipitation of insoluble iron hydroxides. This inertness is controlled by pH and temperature, so when both parameters are increased4, Fe-TEA complex decomposes and Fe3+ ions recombine with other metal ions present in the solution to form mixed-metal LDH9. However, the hydrolysis of this atrane complex takes place at temperature exceeding the boiling point of the solvent (typically 150 °C)3,11, which, in case of wet-chemistry synthesis, involves the use of autoclaves. In general, the scalability of the hydrothermal methods is limited by the size of a pressure vessel and the total synthesis costs are significantly higher, which results from special equipment and high temperature requirements11.
In this work, pure, highly crystalline Ni-Fe LDH hexagonal platelets were produced by homogeneous precipitation at a relatively low temperature of 100 °C. In contrast to previous reports3,9, we used a large excess of the capping agent in relation to Fe3+ ions which caused the precipitation of a metastable solid iron(III) oxyhydroxide at room temperature, while nickel(II) remained in the solution coordinated by TEA molecules. Then, upon heating and pH increase, the reaction intermediates decompose and recombine forming Ni-Fe LDH hexagons. To the best of our knowledge, the transformation of iron(III) oxyhydroxide to LDH phase at the temperature as low as 100 °C has not been previously reported in literature. Moreover, this process was carried out in a conventional round-bottom flask, suggesting upscaling possibilities of the procedure. Preliminary electrochemical measurements suggest that this material might be potentially used as a catalyst for OER.
Results and Discussion
The morphological and structural characterization of Ni-Fe LDH hexagonal platelets
The morphological and structural characterization of the as-prepared LDH was carried by SEM, TEM, AFM, FT-IR and TGA analysis. SEM micrograph (Fig. 1a) revealed that the as synthesized sample is composed of flakes with a well-defined hexagonal shape and on the order of 1 µm in lateral dimension. The height profiles of the flakes, measured by AFM (Fig. 2), are in the range of 15 to 20 nm, which roughly corresponds to 18–25 layers.

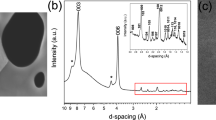
SEM and TEM micrograph (a,b); SAED pattern (c); XRD pattern (d); SEM- EDX elements mapping (e–h) of the Ni-Fe LDH hexagonal platelets.

AFM image and height profiles of Ni-Fe LDH hexagonal platelets.
The homogenous contrast observed in the TEM micrographs (Fig. 1b) suggests that the flakes have a uniform thickness and the SAED pattern (Fig. 1c) shows a hexagonal symmetry (a = 3.08 Å, c = 23.55 Å), which is confirmed by XRD patterns (Fig. 1d). The absence of additional reflections in the SAED pattern indicates that the sample is solely composed by Ni-Fe LDH. This is evidenced by the (\(101\)), (\(01\bar{1}\)), (\(1\bar{1}2\) and \((110)\) lattice planes (Fig. 1c).
The homogeneous distribution of the metal cations within the nanoflakes15 was confirmed via SEM-EDX mapping (Fig. 1e–h). Nickel, iron and oxygen are evenly distributed in the flakes surface, thus indicating a consistent substitution of Ni2+ by Fe3+ in the brucite-like layers of Ni(OH)2.
FT-IR analysis of the flakes (Fig. 3a) revealed the typical spectrum of LDH. Adsorbed water in the samples leads to the broad signal at 3450 cm−1, which is characteristic of the O-H stretching mode in brucite-like layers. The O-H bending vibrations in water molecules is also present at 1645 cm−1 18,19. A strong band at 690 cm−1 is usually ascribed to the υM-O lattice vibrations17. The absorption band observed at 1350 cm−1 is related to the stretching mode of CO32−17, while the weak peaks at 1155 cm−1 and 1040 cm−1 (green labels) are characteristic of the C-N stretching mode in tertiary amines20, suggesting a small contamination with TEA.

FT-IR spectrum (a) and TGA curve (b) of Ni-Fe LDH flakes.
LDH are known to thermally transform into mix-metal oxide and spinel phases21. The TGA curve (Fig. 3b) indicates that this process occurs in two steps11. Firstly, in the temperature range of 30 °C to 200 °C, the adsorbed and interlamellar water is removed, resulting in a 10% mass loss. Secondly, at temperatures of 200 to 800 °C, the weight loss is promoted by the dehydroxylation of the brucite-like layers and the decomposition of the counter anions17. The total mass loss of roughly 30% is consistent with other works reported on the thermal decomposition of LDH materials11,14,15. Finally, Ni2+/Fe3+ ratio was calculated as 3.54 by atomic absorption spectroscopy which, in combination with TGA results, led to the formula Ni0.78Fe0.22(OH)2(CO3)0.11∙0.6H2O.
Electrocatalytic activity in OER
The performance of the Ni0.78Fe0.22(OH)2(CO3)0.11∙0.6H2O as an electrocatalyst for OER was tested by linear sweep voltammetry polarization (Fig. 4).

Linear sweep voltammetry curves for the Ni-Fe LDH hexagons on Ni foam and a bare Ni foam at a scan rate of 5 mV∙s−1 in 1 M KOH. Inset: Cyclic voltammetry of Ni-Fe LDH hexagons on Ni foam in 1 M KOH.
As expected the synthesized Ni-Fe LDH exhibits electrocatalytic activity towards OER, presenting an overpotential of 0.36 V, which is similar to other nanostructured Ni-Fe LDH materials2,22,23,24,25,26. The overpotential of the support (Ni-foam) was also measured resulting in a value of 0.43 V. The cyclic voltammograms shown in the inset of Fig. 4 reveal clear anodic and cathodic peaks which are associated with the redox couple NiII/NiIII.2 This is a typical behaviour of Ni-Fe LDH and it has origin in the insertion/desertion of OH− ions from the LDH structure. A similar response was observed for the bare Ni foam (Fig. S1 in ESI†).
These preliminaries results indicate that the two-dimensional Ni-Fe LDH hexagons described in this paper might represent a viable option for OER applications. Therefore, further characterization and optimization of the LDH hexagons will be considered for future work.
Possible formation mechanism of Ni-Fe LDH platelets
Results presented so far indicate that homogeneous precipitation at low temperature is a very interesting technique for the synthesis of crystalline LDH hexagons with promising electrocatalytic properties. However, the processes behind this approach are not fully understood. Further experimental work allowed us to come forth with a hypothetical model for the LDH formation by homogeneous precipitation at low temperature. First, the addition of TEA to the reaction mixture drastically changes the pH of the starting solution (from 2.5 to 6.6). At these conditions, a gel-like brown precipitate was formed at room temperature (Fig. 5a), which upon heating to 100 °C and pH increase transformed to Ni-Fe LDH hexagonal platelets. In order to try to clarify how the final product is produced we split the synthesis process between two steps: at room temperature and at 100 °C.

Reaction mixture after overnight stirring at room temperature (a); FT-IR spectrum (b); XRD pattern (c); TEM micrograph (inset is associated SAED pattern) (d) of the dried precipitate.
Step 1- Room Temperature
The formation of the iron oxyhydroxide precipitate at room temperature was investigated by UV-Vis measurements performed at 350 nm, which is linearly related to the Fe3+ concentration (Ni2+ has a negligible absorbance at that wavelength, Fig. S2 in ESI†). Before recording the UV-Vis spectra the reaction mixture was centrifuged (3000 rpm/10 min.) in order to remove the precipitate and monitor the Fe3+ concentration in the solution. As presented in the Fig. S3 in ESI†, the formation of the precipitate has an induction period of 2 hours, at which the Fe3+ concentration decreases slightly, followed by an exponential decay, which results in negligible concentration of Fe3+ in solution after 9 hours, whilst maintaining a constant pH of 6.6. This experiment led us to find out the minimum time required to precipitate all Fe3+ from the starting solution, which is approximately 9 hours.
The precipitate formed after 24 hours of stirring was isolated, washed several times with water, dried at room temperature and analyzed by FT-IR, XRD and TEM (Fig. 5b–d).
In the FT-IR spectrum (Fig. 5b) a broad adsorption band with a maximum at 3270 cm−1 is attributed to υO-H stretching mode, which is typical for hydroxides and also to structural/adsorbed water. The peak at 1640 cm−1 additionally confirms the presence of water in the precipitate. Furthermore, the peaks at 1470 cm−1 and 1360 cm−1, assigned to the Fe-O and Fe-OH vibrations, respectively, suggest that the precipitate is most probably one of the forms of ferric oxyhydroxide27,28. The very strong absorption at wavenumber <1000 cm−1 can be explained by the deformation vibrations of surface O-H groups in ferric hydroxides28. The weak absorption bands at the region of 1200–1000 cm−1 might be related to the partial complexation of Fe3+ by TEA molecules29.
The XRD pattern of the precipitate (Fig. 5c) shows that the material presents a low degree of crystallinity, as only two broad peaks at about 2.6 Å and 1.5 Å are detected, which are typical for iron oxyhydroxides previously reported30,31,32. Additional characterization by TEM and SAED (Fig. 5d) demonstrate that the material presents an irregular morphology and the particles are strongly agglomerated. The SAED (inset in Fig. 5d) highlights only two bright, diffused rings which positions fit well to the pattern obtained by XRD. TEM results are in a good agreement with those found in the literature suggesting that the precipitate formed at room temperature is most probably one of the forms of ferrihydrite33. This thermodynamically metastable Fe3+ oxyhydroxides finally transform to more crystalline forms depending on the temperature and pH34,35. Considering that those parameters are both changing during the further thermal decomposition of urea, we hypothesize that, in presence of Ni2+ and CO32−, the ferrihydrite transforms into Ni-Fe LDH.
Since most of the Fe3+ precipitates as ferrihydrite, the remaining solution is expected to be composed of Ni2+, NO3−, TEA and urea. In order to understand the coordination of Ni2+, the reaction mixture was analyzed by NMR and UV-Vis spectroscopy and compared to the spectra of pure Ni(NO3)2 and TEA (Fig. 6).

1H NMR and 13C NMR spectra of the reaction mixture and TEA (a,b); UV-Vis spectra of the reaction mixture and Ni(NO3)2 (c); absorbance at 350 nm related to Fe3+concentration and pH value during 48 hours of heating the reaction mixture at 100 °C (d).
Firstly, the reaction mixture was characterized by 1H and 13C NMR spectroscopy and compared to the spectrum of pure TEA (Fig. 6a,b). The changes in both 1H and 13C environment are clearly visible: 1H NMR spectrum of pure TEA shows two triplets for the methylene groups bonded to N and O, respectively, which are observed to be broadened and shifted downfield in the reaction mixture due to the presence of Ni2+. This is in good agreement with the theoretical predictions, suggesting that the loss of electron-density upon coordination to the metal results in a shift to higher frequency of the protons adjacent to the ligand36. In contrast, in the 13C NMR spectrum the signals are shifted upfield, which was also previously observed in metal-organic compounds37, which is an additional proof of the formation Ni2+-TEA complex. Several coordination compounds of Ni-TEA-NO3− have been described in the form of blue crystals38,39, which differs from the clear, pale green/blue solution obtained in the present study. This difference is most probably the result of an insufficient amount of TEA in relation to Ni2+, since the TEA:Ni2+ ratio is 4:3 instead of 2:1 to form [Ni(TEA)2](NO3)2 in a solid form.
The UV-Vis absorption spectra of both Ni(NO3)2 and the reaction mixture (Fig. 6c) show two weak bands assigned to d-d transitions corresponding to an octahedral coordination geometry of Ni2+ 38,40,41. A sharp peak centred at 390 nm can be assigned to 3A2g → 3T1g(P) transition in both cases38,40, while the absorbance is higher in case of the reaction mixture, which probably is a result of metal coordination by TEA ligands. Moreover, the doublet with maxima at 656 nm and 726 nm ascribed to 3A2g → 3T1g(F) transition typical of nickel(II) aqua complex ([Ni(H2O)6]2+)41,42 is slightly shifted towards lower wavelengths in the reaction mixture. Higher absorbance of the peak at 390 nm and a blue shift at 656–720 nm were previously observed by Agarwala et al.42. for various Ni-TEA complexes in solution including [Ni(TEA)2]2+, [Ni(TEA)2(OH)2]2+ and [Ni(TEA)]2+.
Therefore, on the basis of UV-Vis and NMR spectroscopy, nickel(II) is most probably present in the reaction mixture as an octahedral Ni-TEA metal complex, however the chemical formula cannot be accurately defined, due to many possibilities of ligand coordination42,43, and most probably it consists as a mixture of different Ni-TEA derivatives in quick kinetic equilibrium.
Step 2-Heating to 100 °C
The heating of the reaction mixture (gel-like ferrihydrite and Ni2+-TEA complex) to 100 °C was monitored by UV-Vis measurements (Fig. 6d). Within the first 4 hours of heating the mixture, a dramatic raise of Fe3+ concentration was observed, suggesting the decomposition of ferrihydrite. The Fe3+ concentration augmented for a maximum of 8 hours and started decreasing until the end of the measurements. Nevertheless, after 48 hours of heating the reaction mixture, some unbounded Fe3+ ions remained in the solution, as observed in Fig. 6d. This is consistent with the Ni2+/Fe3+ ratio in the final product (3.54), which is slightly higher than initially used (3.00) suggesting that not all Fe3+ was incorporated to the Ni-Fe LDH.
The evolution of the pH is also plotted in Fig. 6d. During the first 12 hours, a linear increase of the pH is observed due to the urea hydrolysis at 100 °C. After this time, the consumption of OH− by metal cations leads to a slower pH increase. Carbonate anions produced from urea decomposition intercalate between the layers to neutralize a positive charge generated by Fe3+ substitution within Ni(OH)2.
The precipitates formed after 4, 12 and 24 hours of heating were analysed by SEM (Fig. S4 in ESI†), in order to investigate the platelets formation process. After 4 and 12 hours, hexagonal flakes with slightly jagged edges and diameters around 0.5 μm mixed with an amorphous precipitate (most probably, remaining ferrihydrite) were observed. Further heating for an additional 12 hours lead to the complete consumption of the amorphous precipitate and only hexagonal platelets remained. Nevertheless, the heating was continued for additional 24 hours, in order to maximize the incorporation of Fe3+ to the LDH.
Conclusion
In summary, we obtained Ni-Fe LDH hexagonal platelets by a homogeneous precipitation method at 100 °C under atmospheric pressure. The flakes’ diameter is in the range of 0.5–1.5 μm and thicknesses between 15 and 20 nm. This material is highly crystalline and the metal cations are homogeneously distributed within a single flake. We believe that those platelets are formed in a two step process, where firstly reaction intermediates (iron oxyhydroxide and Ni-TEA complex mixture) are formed at room temperature, and then, upon heating to 100 °C, Ni2+ and Fe3+ are released from their precursors and react with OH− and CO32− (from urea hydrolysis) forming regular hexagons of Ni-Fe LDH. The preliminary results suggested that the obtained material showed favourable electrocatalytic activity towards OER, exhibiting an overpotential of 0.36 V vs RHE, which is consistent with literature reports.
We have demonstrated in this paper that high quality Fe3+-containing LDH platelets can be produced by a simple method at 100 °C in a conventional synthesis setup.
Methods
Chemicals
Nickel(II) nitrate hexahydrate (Ni(NO3)2∙6H2O, extra pure, Fisher Scientific), Iron(III) nitrate nonahydrate (Fe(NO3)3∙9H2O, purity <98%, Sigma Aldrich), urea (CH4N2O2, purity ≥99.3%, Alfa Aesar) and TEA (C6H15NO3, purity ≥99%, Sigma Aldrich) were used as received without further purification.
Synthesis of Ni-Fe LDH hexagonal platelets
Ni(NO3)2∙6H2O, Fe(NO3)3∙9H2O and urea were dissolved in 80 ml of deionized water to a final concentration of 7.5, 2.5 and 17.5 mM, respectively. Then, 0.8 mmol of TEA were added and the solution was stirred at room temperature for 24 hours, which led to the formation of gel-like brown precipitate. After that, the reaction mixture was transferred to 100 ml round-bottom flask and immersed in an oil bath previously heated to 100 °C under reflux conditions and heated for 48 hours. The flask was naturally cooled down to room temperature and the obtained precipitate was collected by centrifugation (3000 rpm/ 10 min) and washed with water several times.
Equipment and characterization techniques
Scanning electron microscopy (SEM) images were acquired using a Zeiss Utra Plus (Carl Zeiss AG, Germany) operated at 2–3 kV, while energy dispersive X-ray spectroscopy (EDX) mapping was performed at 15 kV. Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) were conducted using an FEI Titan (FEI, Oregon, USA) microscope operated at 80 keV. Atomic force microscopy (AFM) images were taken on an Asylum Research MFP 3D microscope working in a tapping mode. Fourier-transform infrared (FT-IR) spectra were recorded using a Perkin Elmer Spectrum 100 FT-IR spectrometer via attenuated total reflectance (ATR) method. Powder X-ray diffraction (XRD) was measured in a Bruker Advance Powder X-ray diffractometer equipped with a Mo-Kα emission source (λ = 0.7107 Å) in the Bragg-Brentano configuration. The metals content was measured with a Varian 55 Atomic Absorption spectrometer. Thermogravimetric analysis (TGA) was carried out in a Perkin Elmer Pyris 1 TGA (the samples were heated up to 850 °C at a rate of 10 °C min−1) under air atmosphere. Ultraviolet-visible (UV-Vis) spectroscopy measurements were conducted using a Biochrom Libra S22 UV/Vis Spectrophotometer. 1H and 13C nuclear magnetic resonance (NMR) analyses were performed using a Bruker Avance HD 400 NMR equipped with a BBFO probe. An ultrasonic spray deposition (USI Prism Ultracoat 300) was used to deposit the sample dispersion onto nickel foam substrates (0.25 cm2) and prepare electrodes with an average mass load of 0.3 mg∙cm−2. Electrochemical measurements were conducted in a BioLogic VMP 300. A platinum wire and Ag/AgCl were used as counter and reference electrodes in 1 M KOH electrolyte solution, respectively. Both cyclic and linear sweep voltammetry curves were acquired in the range of 0 V to 0.6 V at a scan rate of 5 mV∙s−1 against Ag/AgCl. All the potentials were calibrated with respect to a reversible hydrogen electrode (RHE) reference as follows:2 VRHE = VAg/AgCl + VAg/AgCl(vs RHE) + (0.059 × pH). All voltammetry curves shown are compensated for solution resistance.
References
Gong, M. & Dai, H. A mini review of NiFe-based materials as highly active oxygen evolution reaction electrocatalysts. Nano Res. 8, 23–39 (2015).
Oliver-Tolentino, M. A. et al. An Approach to Understanding the Electrocatalytic Activity Enhancement by Superexchange Interaction toward OER in Alkaline Media of Ni–Fe LDH. J. Phys. Chem. C 118, 22432–22438 (2014).
Song, F. & Hu, X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 5, 1–9 (2014).
Long, X., Wang, Z., Xiao, S., An, Y. & Yang, S. Transition metal based layered double hydroxides tailored for energy conversion and storage. Mater. Today 19, 213–226 (2016).
Gao, X. et al. Low-cost and high-performance of a vertically grown 3D Ni–Fe layered double hydroxide/graphene aerogel supercapacitor electrode material. RSC Adv. 6, 107278–107285 (2016).
Mariko Adachi-Pagano, C. F. a. J.-P. B. J. Mater. Chem. 13, 1988–1993 (2003).
Ma, R., Liu, Z., Li, L., Iyi, N. & Sasaki, T. Exfoliating layered double hydroxides in formamide: a method to obtain positively charged nanosheets. J. Mater. Chem. 16, 3809–3813 (2006).
Ma, R. & Sasaki, T. Nanosheets of Oxides and Hydroxides: Ultimate 2D Charge‐Bearing Functional Crystallites. Adv. Mater. 22, 5082–5104 (2010).
Ni, B. & Wang, X. Edge overgrowth of spiral bimetallic hydroxides ultrathin-nanosheets for water oxidation. Chem. Sci. 6, 3572–3576 (2015).
Ma, R. et al. Synthesis and exfoliation of Co2+−Fe3+ layered double hydroxides: An innovative topochemical approach. J. Am. Chem. Soc. 129, 5257–5263 (2007).
Abellán, G., Coronado, E., Martí-Gastaldo, C., Pinilla-Cienfuegos, E. & Ribera, A. Hexagonal nanosheets from the exfoliation of Ni2+− Fe3+ LDHs: a route towards layered multifunctional materials. J. Mater. Chem. 20, 7451–7455 (2010).
Han, Y. et al. Preparation of Ni2+− Fe3+ layered double hydroxide material with high crystallinity and well-defined hexagonal shapes. Chem. Mater. 20, 360–363 (2007).
Li, F., Liu, J., Evans, D. G. & Duan, X. Stoichiometric synthesis of pure MFe2O4 (M = Mg, Co, and Ni) spinel ferrites from tailored layered double hydroxide (hydrotalcite-like) precursors. Chem. Mater. 16, 1597–1602 (2004).
Iwasaki, T., Yoshii, H., Nakamura, H. & Watano, S. Simple and rapid synthesis of Ni–Fe layered double hydroxide by a new mechanochemical method. Appl. Clay Sci. 58, 120–124 (2012).
Ma, W. et al. A superlattice of alternately stacked Ni–Fe hydroxide nanosheets and graphene for efficient splitting of water. ACS nano 9, 1977–1984 (2015).
Hou, W., Kang, L., Sun, R. & Liu, Z.-H. Exfoliation of layered double hydroxides by an electrostatic repulsion in aqueous solution. Colloids Surf., A 312, 92–98 (2008).
Auerbach, S. M., Carrado, K. A. & Dutta, P. K. Handbook of layered materials. 373-474, 509-540 (CRC Press, 2004).
Kuśtrowski, P. et al. Influence of thermal treatment conditions on the activity of hydrotalcite-derived Mg–Al oxides in the aldol condensation of acetone. Microporous Mesoporous Mater. 78, 11–22 (2005).
Ureña-Amate, M. D. N. D. B. & María del Mar Socias-Viciana, E. G.-P. Controlled release of nitrate from hydrotalcite modified formulations. Appl. Clay Sci. 52, 368–373 (2011).
Coates, J. Interpretation of infrared spectra, a practical approach. Encyclopedia of analytical chemistry (2000).
Stanimirova, T., Piperov, N., Petrova, N. & Kirov, G. Thermal evolution of Mg-Al-CO3 hydrotalcites. Clay Miner 39, 177–191 (2004).
Tang, C. et al. Spatially Confined Hybridization of Nanometer‐Sized NiFe Hydroxides into Nitrogen‐Doped Graphene Frameworks Leading to Superior Oxygen Evolution Reactivity. Adv. Mater. 27, 4516–4522 (2015).
Liu, Q. et al. Bifunctional Ni 1− x Fe x layered double hydroxides/Ni foam electrodes for high-efficient overall water splitting: A study on compositional tuning and valence state evolution. Int. J. Hydrogen Energy 42, 5560–5568 (2017).
Hunter, B. M., Hieringer, W., Winkler, J., Gray, H. & Müller, A. Effect of interlayer anions on [NiFe]-LDH nanosheet water oxidation activity. Energy Environ. Sci. 9, 1734–1743 (2016).
Li, X. et al. Atomically thin layered NiFe double hydroxides assembled 3D microspheres with promoted electrochemical performances. J. Power Sources 325, 675–681 (2016).
Luo, Q., Peng, M., Sun, X., Luo, Y. & Asiri, A. M. Efficient electrochemical water splitting catalyzed by electrodeposited NiFe nanosheets film. Int. J. Hydrogen Energy 41, 8785–8792 (2016).
Xu, Z., Yu, J. & Xiao, W. Microemulsion‐Assisted Preparation of a Mesoporous Ferrihydrite/SiO2 Composite for the Efficient Removal of Formaldehyde from Air. Chem. Eur. J. 19, 9592–9598 (2013).
Russell, J. Infrared spectroscopy of ferrihydrite: evidence for the presence of structural hydroxyl groups. Clay Miner 14, 109–114 (1979).
Naiini, A. A., Young, V. & Verkade, J. G. New complexes of thriethanolamine (Tea): Novel structural features of [Y (TEA) 2](ClO 4) 3· 3C 5 H 5 N and [Cd (TEA) 2](NO 3) 2. Polyhedron 14, 393–400 (1995).
Vodyanitskii, Y. N. Iron hydroxides in soils: a review of publications. Eurasian Soil Sci. 43, 1244–1254 (2010).
Rout, K., Mohapatra, M. & Anand, S. 2-Line ferrihydrite: synthesis, characterization and its adsorption behaviour for removal of Pb (II), Cd (II), Cu (II) and Zn (II) from aqueous solutions. Dalton Trans. 41, 3302–3312 (2012).
Michel, F. et al. Similarities in 2-and 6-line ferrihydrite based on pair distribution function analysis of X-ray total scattering. Chem. Mater. 19, 1489–1496 (2007).
Janney, D. E., Cowley, J. M. & Buseck, P. R. Transmission electron microscopy of synthetic 2-and 6-line ferrihydrite. Clays Clay Miner. 48, 111–119 (2000).
Kukkadapu, R. K. et al. Transformation of 2-line ferrihydrite to 6-line ferrihydrite under oxic and anoxic conditions. Am. Mineral. 88, 1903–1914 (2003).
Das, S., Hendry, M. J. & Essilfie-Dughan, J. Transformation of two-line ferrihydrite to goethite and hematite as a function of pH and temperature. Environ. Sci. Technol. 45, 268–275 (2010).
Pastor, A. & Martínez-Viviente, E. NMR spectroscopy in coordination supramolecular chemistry: A unique and powerful methodology. Coord. Chem. Rev. 252, 2314–2345 (2008).
Fang, J., Wei, X., Sapp, J. B. & Deng, Y. Novel platinum (II) complexes containing diaminocyclohexane and thiourea derivative ligands: Synthesis and X-ray crystal structure of (trans-1, 2-diaminocyclohexane) dithioureaplatinum (II) nitrate monohydrate. Inorg. Chim. Acta 411, 5–10 (2014).
Hughes, M. & Rutt, K. Complexes of triethanolamine with cobalt (II), nickel (II) and copper (II) salts. J. Chem. Soc. A, 2788–2790 (1968).
Nielsen, K., Hazell, R. G. & Rasmussen, S. The Crystal Structure of Di-triethanolamine-Ni (II)-dinitrate. Acta Chem. Scand 26, 3 (1972).
Yeşilel, O. & Ölmez, H. Syntheses and spectrothermal studies of triethanolamine complexes of Co (II), Ni (II), Cu (II) and Cd (II) squarates. J. Therm. Anal. Calorim. 89, 261–265 (2006).
Hausinger, R. P. Biochemistry of nickel. Vol. 12 (Springer Science & Business Media, 2013).
Agarwala, G., Vishal, B. & Bjerrum, J. Metal Ammine Formation in Solution. XXII. The Nickel (II) Triethanolamine System. Acta Chem. Scand. 36, 459–464 (1982).
Cazacu, M., Bargan, A., Shova, S. & Turta, C. Cu (II) and Ni (II) complexes with a tri-, tetra-or hexadentate triethanolamine ligand: Structural characterization and properties. Polyhedron 50, 255–263 (2013).
Acknowledgements
This work was funded by the European Research Council (ERC) under the project 3D2DPrint (Grant Agreement 681544), ERC PoC TC2D, Science Foundation Ireland AMBER, SFI PIYRA and 7th Framework Programme ITN MoWSeS. The authors would like to thank Advanced Microscopy Laboratory, CRANN Trinity College Dublin.
Author information
Authors and Affiliations
Contributions
S.J. and V.N. designed and discussed the project. S.J. synthesised and characterized the materials. C.H. performed TEM analysis. A.S. provided the AFM and XRD measurements. A.S. and D.T. discussed FT-IR, XRD and TGA characterization. J.C. and M.B. performed and analysed OER measurements. T.S. discussed the possible formation mechanism. S.J. and J.C. wrote the paper with all authors contributing to preparation of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jaśkaniec, S., Hobbs, C., Seral-Ascaso, A. et al. Low-temperature synthesis and investigation into the formation mechanism of high quality Ni-Fe layered double hydroxides hexagonal platelets. Sci Rep 8, 4179 (2018). https://doi.org/10.1038/s41598-018-22630-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22630-0
This article is cited by
-
Photocatalytic transfer of aqueous nitrogen into ammonia using nickel-titanium-layered double hydroxide
Environmental Science and Pollution Research (2022)
-
Postsynthetic treatment of nickel–iron layered double hydroxides for the optimum catalysis of the oxygen evolution reaction
npj 2D Materials and Applications (2021)
-
TEM and EELS characterization of Ni–Fe layered double hydroxide decompositions caused by electron beam irradiation
npj 2D Materials and Applications (2021)
-
Removal of Cu2+ metal ions from water using Mg-Fe layered double hydroxide and Mg-Fe LDH/5-(3-nitrophenyllazo)-6-aminouracil nanocomposite for enhancing adsorption properties
Environmental Science and Pollution Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
