
Abstract
Oligodendrocytes are the myelin-producing cells of the central nervous system (CNS). A variety of brain disorders from “classical” demyelinating diseases, such as multiple sclerosis, stroke, schizophrenia, depression, Down syndrome and autism, are shown myelination defects. Oligodendrocyte myelination is regulated by a complex interplay of intrinsic, epigenetic and extrinsic factors. Gpr17 (G protein-coupled receptor 17) is a G protein-coupled receptor, and has been identified to be a regulator for oligodendrocyte development. Here, we demonstrate that the absence of Gpr17 enhances remyelination in vivo with a toxin-induced model whereby focal demyelinated lesions are generated in spinal cord white matter of adult mice by localized injection of LPC(L-a-lysophosphatidylcholine). The increased expression of the activated form of Erk1/2 (phospho-Erk1/2) in lesion areas suggested the potential role of Erk1/2 activity on the Gpr17-dependent modulation of myelination. The absence of Gpr17 enhances remyelination is correlate with the activated Erk1/2 (phospho-Erk1/2).Being a membrane receptor, Gpr17 represents an ideal druggable target to be exploited for innovative regenerative approaches to acute and chronic CNS diseases.
Similar content being viewed by others


Introduction
Oligodendrocytes are the myelin-producing cells of the central nervous system (CNS), and as such, wrap layers of lipid-dense insulating myelin around axons1. Mature oligodendrocytes have also been shown to provide metabolic support to axons through transport systems within myelin, which may help prevent neurodegeneration2.
Oligodendrocytes are generated from oligodendrocyte precursor cells (OPCs), which migrate to and colonize the brain’s white matter (and sometimes gray matter as well) and spinal cord3,4,5. This process is tightly controlled not only by a complex intrinsic oligodendrocyte differentiation program6, but also by external reciprocal signaling processes such as the degree of neuronal differentiation7. Previous studies have demonstrated that adult SVZ progenitors can generate new OPCs/oligodendrocytes after demyelinating lesions of the corpus callosum8,9, seizures10, or stroke11,12. These OPCs may participate in myelin repair after injury13,14.
OPCs respond to demyelinating injury by first undergoing activation, colonization of the demyelinated area by proliferation and migration, and eventually differentiation into new myelin-forming oligodendrocytes15,16. Critical to this process is the switch from a proliferative/migratory state to the exiting from the cell cycle and differentiation into a nondividing, nonmigratory mature oligodendrocyte.
Identifying pathways and transcription factors involved in the regulation of OPC differentiation in myelination and especially remyelination that can potentially be manipulated pharmacologically represents a critical task in the development of new therapies for enhancing endogenous remyelination and thus axonal protection in MS and other myelin disorders17. At present, the factors that promote the initiation of OPC differentiation and overcome the block for successful remyelination in demyelinating diseases are poorly defined18.
Gpr17 is an orphan G-protein-coupled receptor that responds to both uracil nucleotides and cysteinyl leukotrienes (cysLTs)19,20. Endogenous ligands of Gpr17, such as UDP glucose and cysLTs, have been identified, and synthetic ligands, such as MDL29951 and pranlukast, have been developed to activate or antagonize Gpr17 activity, respectively19,21,22.
Activation of Gpr17 signaling upregulates the expression of a differentiation inhibitor, ID2, and promotes the nuclear translocation of ID2 and ID423. Overexpression of Gpr17 in the oligodendrocyte lineage causes defects in myelinogenesis in transgenic mice, and Gpr17 knock-out mice exhibit precocious myelination in the spinal cord at the neonatal stage23. The hypothesis that activation of Gpr17 delays oligodendrocyte maturation is supported by recent findings that Gpr17 desensitization by G-protein receptor kinase phosphorylation and subsequent internalization are necessary for terminal differentiation of OPCs24. Furthermore, Gpr17 has been shown to negatively regulate oligodendrocyte differentiation via the inactivation of intracellular protein kinase A (PKA) and cAMP-activated GTP exchange factor Epac125. In addition to the regulation of normal oligodendrocyte development, Gpr17 also functions as a sensor for extracellular damage signals under pathological conditions such as ischemia and brain trauma21,26,27,28. Remyelination is more rapid in Gpr17 knockout mice than in wild-type mice after a lysolecithin injection in the corpus callosum29. Gpr17 antagonism results in structural and functional rejuvenation of aged brains, suggesting a promising clinical application for a Gpr17-based intervention30.
In the present study, we chose the L-a-lysophosphatidylcholine (LPC) lesion as a model to examine the temporal response and transcription factor expression of endogenous OPCs following demyelination. We elucidated the role of Gpr17 in the survival and differentiation of oligodendrocytes in response to spinal cord demyelinating injury. The absence of Gpr17 enhances remyelination is correlate with the activated Erk1/2 (phospho-Erk1/2).
Results
Gpr17 expression gradually increases during LPC induced demyelination
Gpr17−/− mice was deleted the entire Gpr17 coding region, replaced with histone 2b–fused GFP (h2b-GFP) to trace individual endogenous Gpr17-expressing cells23. To assess the function of Gpr17 in remyelination in vivo, we employed the LPC-induced demyelination in white matter of spinal cord. Stereotaxic injection of LPC into the adult spinal cord results in selective and focal myelin loss with minimal axonal damage in adjacent cells and axons, induces subsequent remyelination within 4 weeks31,32,33. The relatively short duration of the experiments and the easy analysis of the demyelinated area make it a convenient model to study demyelination ⁄ remyelination processes34. Myelin regenerates through an OPC recruitment phase at 7 days post lesion (dpl) and an oligodendrocyte regeneration and remyelination phase at 14 dpl35. LPC was injected into the ventrolateral column of 8-week-old male wild type (control), Gpr17+/−(control)23, and Gpr17−/− mice from the same litter to trigger demyelination(Fig. 1A). Increased cell density (as for instance shown by DAPI staining) represents the LPC lesions.Gpr17 expression, indicated by the expression of the reporter GFP, is almost absent at 3 dpl, but gradually increases at 7 dpl, 14 dpl and reaches its peak at around14 dpl. At 7 dpl, Gpr17 is observed in the perimeter of the lesion and outside the demyelination area, excluding the pia border. At 14 dpl, the expression of Gpr17 appeared to be more densely distributed than in adjacent normal white matter, reaches its peak around14 dpl (Fig. 1B,C), which is consistent with previous studies demonstrating increased oligodendrocyte densities in remyelinating regions36.

Gpr17 expression gradually increases during LPC induced demyelination. (A) The location of LPC-induced lesion (DAPI counterstaining, dashed lines in white matter) in the spinal cord. (B) Representative expression of Gpr17, visualized by the expression of the reporter protein GFP, in the LPC-induced demyelinating lesions (demarcated with dashed lines in white matter) in spinal cords of 8-week-old Gpr17–/–mice at 3 dpl,7 dpl, 14 dpl and 28 dpl. (C) Quantification of the numbers of GFP at 3 dpl,7 dpl, 14 dpl and 28 dpl; We compared GFP of the Gpr17−/− as follows,3 dpl vs. 7 dpl,7 dpl vs. 14 dpl, 14 dpl vs. 28 dpl. Image J was used to measure area of the lesion, count the cells. Sections were taken from the center of each lesion to control for lesion variability. White dashed line demonstrates lesion borders. Student’s t-test, Data are presented as Mean ± SEM, Error bars indicate SEM, **P < 0.01, ***P < 0.001; n = 3 animals for each genotype, Scale bar: (A) 25 μm; (B) 50 μm. LPC, L-a-lysophosphatidylcholine, Dpl: days post lesion; Ctrl, control; WM:white matter; GM:gray matter.
Loss of Gpr17 promotes remyelination after LPC -induced demyelination in the CNS
Previously, the analysis of Gpr17−/−mice showed that Gpr17 functioned as a cell-intrinsic factor that blocked oligodendrocyte terminal differentiation23. To determine whether the loss of Gpr17 facilitates remyelination after injury and accelerates the recovery of injured myelin sheaths, we analyzed the expressions of two myelin genes, Myelin basic protein (MBP) and proteolipid protein (PLP), in the lesions at 3dpl, 7dpl and 14 dpl. MBP, a maturing oligodendrocyte marker, which labels both premyelinating and myelinating oligodendrocytes. PLP exhibits transcriptional upregulation during differentiation from the immature progenitor stage to the mature oligodendrocyte stage37. At 3 dpl, both Gpr17 null and control mice exhibited comparable lesions with very little MBP mRNA expression as detected by in situ hybridizations within the lesion, indicating a similar loss of preexisting myelin and no remyelination occurred at this stage (Fig. 2A). However, at 7dpl and 14 dpl, the expression of PLP was comparable between Gpr17 null mice and control littermates (Fig. 2A,C). At 14 dpl and 28 dpl, Gpr17−/− mouse showed more profound expression of MBP in lesion region in comparison to control group (Fig. 2D). The average lesion size of Gpr17−/− was significantly smaller compared with control group (Fig. 2A,B). Importantly, as indicated by electron micrographs, a great number of hypermyelination of axons were detected in the lesions of Gpr17−/− mice than in controls (Fig. 3A). The thickness of newly generated myelin sheaths around axons was significantly increased in the gpr17 null mice (Fig. 3A). Myelinogenesis was comparable between control and Gpr17−/− mice as indicated by the integrity of the myelin sheath ultrastructure in the lesion (Fig. 3B,C). There were more remyelinated axons in Gpr17−/− mice both at 14 dpl and 28 dpl (Fig. 3C). These observations indicate that loss of Gpr17 promotes remyelination after demyelinating injury. Despite the discrepancy in the onset of remyelination, both the mutant and the control mice do not exhibited complete remyelination at 28 dpl (Fig. 2D, Supplematary data S1).

Gpr17 ablation promotes remyelination in LPC-induced demyelinating animal model. (A) In situ hybridization analysis of MBP and PLP in the lesion regions (demarcated with dashed lines) at 3 dpl,7 dpl and 14 dpl in spinal cords of 8-week-old control and Gpr17−/− mice; n = 3 animals for each genotype. (B) Quantification of the areas of lesion regions (demarcated with dashed lines) at 3 dpl,7 dpl and 14 dpl in spinal cords of 8-week-old control and Gpr17−/− mice. (C) Quantification of the numbers of PLP in the lesion regions (demarcated with dashed lines) at7 dpl and at 14 dpl. (D) Immunostaining of MBP in lesion regions at 14 dpl and 28 dpl in spinal cords of 8-week-old control and Gpr17−/− mice; n = 3 animals for each genotype. Data are presented as Mean ± SEM. *P < 0.05, ***P < 0.001; Student’s t-test. Scale bars, (A,D)100 μm.

Gpr17 ablation promotes remyelination in LPC-induced demyelinating animal model. (A) Representative electron micrographs of spinal cords of 8-week-old control and Gpr17−/− mice at 14 dpl and 28 dpl, n = 3 animals for each genotype. Blue arrow indicates the newly formed thin myelin sheath. Red arrow indicates hypermyelination of axons. (B) Quantification of g-ratio of newly myelinated axons with diameter of 1 μm in spinal lesions of 8-week-old control and Gpr17−/− mice at 14 dpl and 28 dpl; P < 0.001, Student’s t-test. (C) Results of morphometry to quantify remyelination at 14 dpl and 28 dpl (see Materials and Methods). Data are presented as Mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; Student’s t-test. Scale bar, (A) 1 μm.
Enhanced oligodendrocyte differentiation was recruited toward demyelinated lesions
During the remyelination process, OPCs are recruited toward demyelinated lesions38. Olig2, which marks a subset of glioblasts and maturing OPCs, is known to be critical for oligodendrocyte specification and differentiation39,40. We performed immunohistochemistry for Olig2 at 7dpl and 14dpl. To investigate whether there were changes in the proliferation of Olig2 + cells in response to injury in the mutant mice, we administered the thymidine analog BrdU (5-bromo-2′-deoxyuridine, Sigma-Aldrich,) to mice 2 hours before sacrifice, thereby labeling cycling cells across this time period via its incorporation into DNA during replication. We found that the number of BrdU+/Olig2 + cells in the lesion ventrolateral column increased at 7dpl and 14 dpl (Fig. 4A–D), indicating significant increase in Olig2 + cell proliferation.

The expression of Olig2 proliferation was increased in lesion regions in Gpr17−/− mice. (A,B) and (C) Immunostaining and quantification of for expression of OL lineage cells marker Olig2 and BrdU in lesion regions at 7 dpl and 14 dpl in spinal cords of 8-week-old control and Gpr17−/− mice; n = 3 animals for each genotype. White dashed line demonstrates lesion borders. (D) Quantitative real-time PCR (qRT-PCR) analysis of OPCs marker PDGFRa expression in lesion regions. n = 3 animals for each genotype. Student’s t-test. Data are presented as Mean ± SEM. **P < 0.01; Scale bar: 50 μm.
To investigate whether there were changes in OPCs in response to injury, we performed quantitative real-time PCR (qRT-PCR) for OPC marker PDGF receptor α. (PDGFRα). The expression was not significantly different between the two genotypes at 7dpl (Fig. 4E), suggesting that the generation of OPCs was not affected by the deletion of Gpr17.
Sox10 is an oligodendrocyte differentiation-promoting factor, required for terminal differentiation of the oligodendrocyte lineage41,42,43. In Sox10-deficient mice progenitors develop, but terminal differentiation is disrupted43. We performed immunohistochemistry for Sox10. In contrast, Gpr17−/− mice showed a dramatic increase in Sox10 + immunoreactivity in the demyelinated area, particularly around the edges of the lesion (Fig. 5A,B). Small amounts of Sox10 + immunoreactivity were occasionally seen scattered throughout the lesion in control mice (Fig. 5A,B). Taken together, the enhanced oligodendrocyte differentiation was recruited toward demyelinated lesions, we hypothesized that the recruitment is correlates with Sox10.

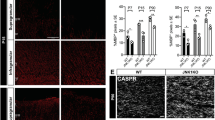
Gpr17-dependent inhibition of oligodendrocyte differentiation is mediated by Erk phosphorylation. (A and B) Immunostaining and quantification of expression of oligodendrocyte differentiation-promoting factor Sox10 and GFP in lesion regions at 7 dpl and 14 dpl in spinal cords of 8-week-old control and Gpr17−/− mice; n = 3 animals for each genotype. Arrow indicates double labeled with sox10 and GFP. White dashed line demonstrates lesion borders. (C and D) Western blot analysis of p-Erk1/2 expression in lesion regions at 14 dpl in spinal cords of 8-week-old control and Gpr17−/− mice; n = 4 animals for each genotype. Student’s t-test. Data are presented as Mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; Scale bar: 50 μm.
Gpr17-dependent inhibition of oligodendrocyte differentiation is mediated by Erk phosphorylation
The extracellular signal-regulated kinases, Erk1 and Erk2, are prototypic members of the mitogen activated protein kinase (MAPK) family44. Erk1/2 activation has been studied extensively in oligodendrocyte development, where it promotes differentiation and increased myelin thickness45. In mice, genetic loss of Erk1/2 in the oligodendrocyte lineage results in normal numbers of OPCs and oligodendrocytes but widespread hypomyelination, while constitutive activation of Erk1/2 results in a profound increase in the extent of remyelination after toxin-induced demyelinating injury46,47. To explore whether the effect of GPR17 on oligodendrocyte differentiation is influenced by Erk1/2, we performed Western Blotting of phospho-Erk1/2 at 14 dpl. We detected increased expression of phospho-Erk1/2 in Gpr17−/− mice (Figs 5C,D, S3). Taken together, our results indicate that the inhibitory action of Gpr17 in oligodendrocyte maturation (that is, late-stage differentiation) and myelination are correlated with Erk1/2 pathway.
Discussion
Rushton (1951) proposed that for a given axon diameter, there is a specific myelin thickness that maximizes conduction velocity48. Alterations in this ratio affect the speed of nerve impulse transmission and thus the timing of neuronal signals, potentially leading to aberrant circuit connectivity, and/or rendering axons vulnerable to damage47. A better understanding of the molecular mechanisms and signaling pathways that drive the process of myelin sheath formation is therefore important for the development of novel therapeutics designed to target remyelination45. Here, we identify Gpr17 as a negative regulator of oligodendrocyte maturation (late-stage differentiation) in the adult mouse spinal cord after demyelinating injury.
To test if the absence of Gpr17 enhances remyelination in vivo, we used a toxin-induced model whereby focal demyelinated lesions are generated in spinal cord’s white matter of adult mice by localized injection of LPC. In animal lesions, demyelination is completed within four days, after which OPCs are recruited into the lesion. Widespread remyelination does not normally commence until 14–21 days post lesion (dpl), which provides a defined window from days 4–14 to test the efficacy of drugs to enhance the extent and rate of remyelination31. Gpr17 expression, which is almost absent at 3 dpl, gradually increases at 7 dpl and 14 dpl, reaches its peak around14 dpl, this result is consistent with previous studies indicated Gpr17 almost absent in early OPCs, gradually increases in more mature precursors, reaches a plateau in immature/pre-oligodendrocytes, and then gradually decreases during terminal differentiation23,27,49. In various in vivo neurodegenerative models characterized by myelin loss (stroke, trauma, demyelination, and experimental autoimmune encephalomyelitis), Gpr17 was abnormally up-regulated in OPCs around lesion sites21,27,50. In rat brain, Gpr17 is expressed in OPCs and not in mature oligodendrocytes49. The deletion of Gpr17 resulted in an earlier onset of remyelination in mice, which is confirmed by the expression of myelin gene expression MBP and Plp at 7dpl, 14 dpl, 28 dpl, and myelin sheath ultrastructure at 14 dpl and 28 dpl (Figs 2A,D, 3A). Which is consistence with previous research showing that remyelination is more rapid in Gpr17 knockout mice than in wild-type mice in the corpus callosum29.
Myelination is mediated by oligodendrocyte precursor cells (OPCs) that are widely distributed throughout both the gray and white matters of the CNS throughout life51,52. Adult-born oligodendrocyte can continue to proliferate and produce compact myelin51. Although a significant fraction of OPCs remains undifferentiated, particularly in gray matter, many will eventually differentiate to become myelinating oligodendrocytes53.Development of the oligodendrocytes lineage is controlled by an intricate regulatory transcriptional network that includes multiple inhibitory and stimulatory factors54,55. We found that the absence of Gpr17 leads to increase expression of Olig2 and Sox10, However, the OPCs marker, PDGFRα, was not significantly different between the two genotypes. Which is consistent with previous analysis that loss of Olig1 function has no obvious effect on the recruitment of progenitor cells but progenitors are impaired their ability to differentiate after demyelination56. Olig2, an oligodendrocyte lineage specific marker, expressed continuously throughout the lineage57. Sox10 is an oligodendrocyte differentiation-promoting factor, required for terminal differentiation of the oligodendrocyte lineage41,42,43. The absence of Gpr17 the enhanced oligodendrocyte differentiation was recruited toward demyelinated lesions, we hypothesized that this recruitment is correlate with Sox10.
It is critical to clearly understand the cellular and molecular mechanisms that regulate myelination in order to develop novel therapies to target remyelination15,35,45,58,59. We found the activated form of Erk1/2 (p-Erk1/2) was increased in lesion areas at 14dpl, suggests that Gpr17 negatively regulates the remyelination is correlate with Erk activation. In support of previous work showing that Erk1/2 function as late-stage regulators of CNS myelination and that the control of myelin thickness is independent of oligodendrocyte development and initiation of myelin wrapping46, enhanced activation of Erk1/2 in oligodendrocytes does not alter oligodendrocyte survival after LPC-induced demyelination47.
Activate Erk1/2 in oligodendrocytes to directly or indirectly target a cohort of genes in myelinating oligodendrocytes that work together to upregulate the major myelin/cytoskeletal proteins above a basal level to promote the assembly and continued wrapping of the myelin sheath, thus increasing myelin thickness46. In the future, it will be important to define the upstream and downstream effectors of Erk1/2 and to understand the interplay between Gpr17 and Erk1/2 in the integration of biosynthetic and cytoskeletal pathways that are pivotal for proper CNS myelination46. Endogenous and synthetic ligands of Gpr17 have been developed to activate or antagonize Gpr17 activity, respectively19,22. Being a membrane receptor, Gpr17 represents an ideal ‘druggable’ target to be exploited for innovative regenerative approaches to acute and chronic CNS diseases. Therapies targeted to Gpr17 through Erk1/2 pathways may prove useful not only to drive accelerated remyelination, but also to generate thicker myelin sheaths potentially rendering CNS axons less vulnerable to future episodes of demyelination.
Methods
Animals
C57BL/6 mice were purchased and maintained in the Sichuan University Laboratory Animal Center. Gpr17−/− mice were generated as described previously23 (Chen et al., 2009). All mice were maintained in the Sichuan University Laboratory Animal Center. The mice were housed in specific pathogen free (SPF) cages under standard laboratory conditions on a 12 h light/dark cycle with constant access to food and water. Animals of both sexes were used in the study (for spinal cord injury experiments, males only), and littermates were used as controls. There were no blinding assessors of experimental group at any stage of the experiments. All animal use and studies were approved by ethical committees of Sichuan University and by the Institutional Animal Care and Use Committee of Sichuan University. All experiments were performed in accordance with relevant guidelines and regulations.
LPC-induced demyelinating injury in the spinal cord
LPC-induced demyelination was carried out in the ventrolateral spinal white matter of 8-week-old male mice. Anesthesia was induced and maintained by peritoneal injection of a mixture of ketamine (90 mg per kilogram body weight) and xylazine (10 mg per kilogram body weight). We make a vertical incision (about 1.5 cm) over the laminectomy site spanning from about thoracic vertebrae T8 to T13.Lift the skin, we can see the ribs. By tracing the ribs backward, we identify T10-T12. After exposing the spinal vertebrae at the level of T10–T12, meningeal tissue in the intervertebral space was cleared and the dura mater was pierced with a dental needle. One percent LPC (L-a-lysophosphatidylcholine, Sigma; 0.5 μl) via a Hamilton syringe attached to a glass micropipette was injected into the ventrolateral white matter via a stereotactic apparatus. Inject LPC at a rate of 1 μl/5 seconds. After injecting, wait 10 seconds and retract the needle. Importantly, charcoal was used to mark the site of LPC injection so that the area of tissue at the lesion center could be unambiguously identified even after remyelination was complete.Spinal cord tissues carrying the lesions were collected at time points as follows: 7 dpl, representing peak OPC recruitment60,61, 14 dpl, representing OL differentiation and new myelin sheath formation62, and 28 dpl, representing new myelin sheath formation (at least 9 mice per control and mutant groups were used for each time point analysis).
Tissue processing and histochemistry
Animals were anesthetized and perfused transcardially with PBS briefly, followed by 4% (w/v) paraformaldehyde (PFA,Sigma-Alorich,441244) in sodium phosphate buffer (pH 7.4), for perfusion and immersion fixation is Formaldehyde which is dissolved PFA. Tissues were postfixed for 2 hours before cryoprotection with 30% sucrose in PBS overnight. Unfixed tissues were used for Quantitative real-time PCR and Western Blot. The tissue surrounding the injection site was dissected. The tissue processing and immunohistochemical staining procedures were performed as described previously63. Briefly, for tissue immunostaining, 18 μm cryosections were incubated overnight in primary antibodies diluted in block solution (PBS with 5% v/v normal goat serum (Sigma-Aldrich, St Louis) and 0.3% v/v Triton X-100). After washing with PBS, sections were then incubated overnight at 4 °C with corresponding Cy2 or Cy3 fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch). Secondary antibodies were used at 1:1000. For BrdU staining, tissue sections were denatured with 0.1 N HCl for 1 hour in a 37 °C water bath. After denaturation, sections were neutralized with 0.1 M Borax pH 8.5 (Sigma-Aldrich, St Louis) for 10 min. Sections were washed with 0.3% Triton X-100/1-PBS (wash buffer) for three times and blocked with 5% normal donkey serum (Sigma-Aldrich) containing wash buffer for 1 hour at room temperature. Mouse anti-BrdU (BD Bioscience, 550891, 1:500) antibody was used to label BrdU overnight at 4 °C. Samples were mounted in Fluoromount G (SouthernBiotech) for fluorescent microscopy. For BrdU incorporation analysis, control and Gpr17−/− littermates were injected with BrdU (Sigma-Aldrich) (100 mg per kilogram body weight) 2 hours before anaesthesia. Primary antibodies used were as follows: Olig2 (Millipore,AB9610, 1:1,000), BrdU (BD Bioscience, 550891, 1:500), Sox10(Santa Cruz, sc-17343, 1:300),MBP(Abcam,ab40390,1:400). We define the lesion border by the increased cell density, shown by DAPI staining. Cell counted using ImageJ (National Institutes of Health).
RNA in situ hybridization
RNA in situ hybridization was performed using digoxigenin-labelled riboprobes as described previously64,65. Briefly, following pretreatments (Proteinase K, postfixation, acetic anhydride), 18 μm cryosections were then prehybridized for 3–4 hours at 65 °C. Hybridization was carried out for 16 hours using 1–2 mg/ml of probe, in plastic slide mailers containing sufficient probe solution to immerse the part of the slides containing the sections. Probes used in this way could be re-used up to six or seven times without appreciable loss of signal. The alkaline phosphatase reaction product was developed using the NBT/BCIP reagents. Sections were prewashed with levamisole, and development was carried out for 2–20 hours depending upon the abundance of the target mRNA. Wash buffers containing CHAPS detergent were re-used to economize. Probes used for in situ hybridization were: Mbp and Plp, the Plp probe also recognizes DM-20.
Western Blot Analysis
The tissues or cells were homogenized in lysis buffer (50 mM Tris, pH 7.4, 150 mMNaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS) containing protease inhibitors (Roche Applied Science). The lysates were clarified by centrifugation at 13,000 × g at 4 °C for 20 min, and the supernatants were collected and normalized for protein concentration. Proteins were separated by 8% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Immobilon-P, Millipore). After blocking with PBS containing 5% skim milk and 0.05% Tween 20, the membranes were incubated with primary antibodies.For detection, a fluorescence-conjugated secondary antibody and an electrogenerated chemiluminescence system (GE Healthcare) were used. The membrane was exposed to an imaging system (LAS-3000, Fujifilm) according to the manufacturer’s specifications. The protein bands were quantified using ImageJ 1.44p software. The following antibodies were used: rabbit anti-p-ERK1/2 (1:1000,Santa Cruz, sc-7383); rabbit anti-ERK1/2 (1:1000,Santa Cruz,sc-292838), rabbit anti-GAPDH (1:5000, Abcam, ab128915). Horseradish peroxidase-conjugated rabbit IgG-specific (1:5000, Cell Signaling Technology, 7074 S) were used for secondary antibodies. Full-length gel is available at Supplementary data.
Electron microscopy
The spinal cord regions from 8-week-old Gpr17−/− or control mice were dissected and fixed in a solution of 2% paraformaldehyde, 2% glutaraldehyde (v/v), and 0.1 m cacodylic acid, pH 7.2, and processed for electron microscopy as described previously23 (Chen et al., 2009). Sections of 1 μm were cut, stained with toluidine blue, and examined by light microscopy, from which remyelination was identified using standard morphological criteria. G-ratio calculations of axons in the area of interest were calculated by dividing the diameter of an axon by the diameter of the axon plus the associated myelin sheath. A total of 120 axons of 1 μm diameter for each group of 3–4 animals were used. Images of transverse ultra-thin sections through the lesion were observed at a magnification of x10000 and analyzed using Image J. For remyelination analysis, the number of remyelinated axons was counted, excluding non-myelinated and non-demyelinated axons (G-ratio ≤ 0.55). Digitized and calibrated images were analyzed using ImageJ (National Institutes of Health). Overall G ratio was compared by unpaired t-test. Statistical significance was set at P < 0.05.
Quantitative real-time PCR analysis
RNAs were isolated with Trizol (Invitrogen Inc.) from snap-frozen tissues. Reverse transcription was performed with the cDNA Reverse Transcription Kit (Bio-Rad) with iQ SYBR Green Supermix (170–8880). qRT–PCR was carried out using the Bio-Rad CFX96 Real-Time System using GAPDH as an internal control. Each analysis was performed in triplicates and the results were normalized to GAPDH for each sample. The qRT–PCR primer sequences are listed in Supplementary Table 1. Relative expression was calculated using Comparative Ct method66.
Statistical analysis
The data for two-group comparisons were analyzed for statistical significance using two-tailed Student’s t tests. Error bars represent SEM. Values of p < 0.05 were considered significant.
Data availability statement format guidelines
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Boulanger, J. J. & Messier, C. From precursors to myelinating oligodendrocytes: contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 269, 343–366, https://doi.org/10.1016/j.neuroscience.2014.03.063 (2014).
Lee, Y. et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448, https://doi.org/10.1038/nature11314 (2012).
Thomas, J. L. et al. Spatiotemporal development of oligodendrocytes in the embryonic brain. J Neurosci Res 59, 471–476, https://doi.org/10.1002/(SICI)1097-4547(20000215)59:4<471::AID-JNR1>3.0.CO;2-3 (2000).
Qi, Y., Stapp, D. & Qiu, M. Origin and molecular specification of oligodendrocytes in the telencephalon. Trends Neurosci 25, 223–225 (2002).
Kessaris, N. et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci 9, 173–179, https://doi.org/10.1038/nn1620 (2006).
Brinkmann, B. G. et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 59, 581–595, https://doi.org/10.1016/j.neuron.2008.06.028 (2008).
Bradl, M. & Lassmann, H. Oligodendrocytes: biology and pathology. Acta Neuropathol 119, 37–53, https://doi.org/10.1007/s00401-009-0601-5 (2010).
Nait-Oumesmar, B. et al. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci 11, 4357–4366 (1999).
Picard-Riera, N. et al. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci USA 99, 13211–13216, https://doi.org/10.1073/pnas.192314199 (2002).
Parent, J. M. von dem Bussche, N. & Lowenstein, D. H. Prolonged seizures recruit caudal subventricular zone glial progenitors into the injured hippocampus. Hippocampus 16, 321–328, https://doi.org/10.1002/hipo.20166 (2006).
Li, L. et al. Focal cerebral ischemia induces a multilineage cytogenic response from adult subventricular zone that is predominantly gliogenic. Glia 58, 1610–1619, https://doi.org/10.1002/glia.21033 (2010).
Zhang, R. L. et al. Ascl1 lineage cells contribute to ischemia-induced neurogenesis and oligodendrogenesis. J Cereb Blood Flow Metab 31, 614–625, https://doi.org/10.1038/jcbfm.2010.134 (2011).
Gensert, J. M. & Goldman, J. E. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 19, 197–203 (1997).
Chari, D. M. & Blakemore, W. F. Efficient recolonisation of progenitor-depleted areas of the CNS by adult oligodendrocyte progenitor cells. Glia 37, 307–313 (2002).
Franklin, R. J. & Ffrench-Constant, C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci 9, 839–855, https://doi.org/10.1038/nrn2480 (2008).
Moyon, S. et al. Demyelination causes adult CNS progenitors to revert to an immature state and express immune cues that support their migration. J Neurosci 35, 4–20, https://doi.org/10.1523/jneurosci.0849-14.2015 (2015).
Fancy, S. P. et al. Overcoming remyelination failure in multiple sclerosis and other myelin disorders. Experimental neurology 225, 18–23, https://doi.org/10.1016/j.expneurol.2009.12.020 (2010).
He, D. et al. lncRNA Functional Networks in Oligodendrocytes Reveal Stage-Specific Myelination Control by an lncOL1/Suz12 Complex in the CNS. Neuron 93, 362–378, https://doi.org/10.1016/j.neuron.2016.11.044 (2017).
Ciana, P. et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. The EMBO journal 25, 4615–4627, https://doi.org/10.1038/sj.emboj.7601341 (2006).
Fratangeli, A. et al. The regulated expression, intracellular trafficking, and membrane recycling of the P2Y-like receptor GPR17 in Oli-neu oligodendroglial cells. The Journal of biological chemistry 288, 5241–5256, https://doi.org/10.1074/jbc.M112.404996 (2013).
Lecca, D. et al. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PloS one 3, e3579, https://doi.org/10.1371/journal.pone.0003579 (2008).
Hennen, S. et al. Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Science signaling 6, ra93, https://doi.org/10.1126/scisignal.2004350 (2013).
Chen, Y. et al. The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat Neurosci 12, 1398–1406, https://doi.org/10.1038/nn.2410 (2009).
Daniele, S. et al. Does GRK-beta arrestin machinery work as a “switch on” for GPR17-mediated activation of intracellular signaling pathways? Cellular signalling 26, 1310–1325, https://doi.org/10.1016/j.cellsig.2014.02.016 (2014).
Simon, K. et al. The Orphan G Protein-coupled Receptor GPR17 Negatively Regulates Oligodendrocyte Differentiation via Galphai/o and Its Downstream Effector Molecules. The Journal of biological chemistry 291, 705–718, https://doi.org/10.1074/jbc.M115.683953 (2016).
Ceruti, S. et al. The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain: a journal of neurology 132, 2206–2218, https://doi.org/10.1093/brain/awp147 (2009).
Boda, E. et al. The GPR17 receptor in NG2 expressing cells: focus on in vivo cell maturation and participation in acute trauma and chronic damage. Glia 59, 1958–1973, https://doi.org/10.1002/glia.21237 (2011).
Zhao, B. et al. The new P2Y-like receptor G protein-coupled receptor 17 mediates acute neuronal injury and late microgliosis after focal cerebral ischemia in rats. Neuroscience 202, 42–57, https://doi.org/10.1016/j.neuroscience.2011.11.066 (2012).
Ou, Z. et al. Olig2-Targeted G-Protein-Coupled Receptor Gpr17 Regulates Oligodendrocyte Survival in Response to Lysolecithin-Induced Demyelination. J Neurosci 36, 10560–10573, https://doi.org/10.1523/jneurosci.0898-16.2016 (2016).
Marschallinger, J. et al. Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nature communications 6, 8466, https://doi.org/10.1038/ncomms9466 (2015).
Jeffery, N. D. & Blakemore, W. F. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. Journal of neurocytology 24, 775–781 (1995).
Woodruff, R. H. & Franklin, R. J. Demyelination and remyelination of the caudal cerebellar peduncle of adult rats following stereotaxic injections of lysolecithin, ethidium bromide, and complement/anti-galactocerebroside: a comparative study. Glia 25, 216–228 (1999).
Hall, S. M. The effect of injections of lysophosphatidyl choline into white matter of the adult mouse spinal cord. Journal of cell science 10, 535–546 (1972).
Magalon, K., Cantarella, C., Monti, G., Cayre, M. & Durbec, P. Enriched environment promotes adult neural progenitor cell mobilization in mouse demyelination models. The European journal of neuroscience 25, 761–771, https://doi.org/10.1111/j.1460-9568.2007.05335.x (2007).
Franklin, R. J. Why does remyelination fail in multiple sclerosis? Nature reviews. Neuroscience 3, 705–714, https://doi.org/10.1038/nrn917 (2002).
Blakemore, W. F. & Keirstead, H. S. The origin of remyelinating cells in the central nervous system. Journal of neuroimmunology 98, 69–76 (1999).
Shigemoto-Mogami, Y., Hoshikawa, K., Goldman, J. E., Sekino, Y. & Sato, K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci 34, 2231–2243, https://doi.org/10.1523/jneurosci.1619-13.2014 (2014).
Takahashi, C., Muramatsu, R., Fujimura, H., Mochizuki, H. & Yamashita, T. Prostacyclin promotes oligodendrocyte precursor recruitment and remyelination after spinal cord demyelination. Cell death & disease 4, e795, https://doi.org/10.1038/cddis.2013.335 (2013).
Masahira, N. et al. Olig2-positive progenitors in the embryonic spinal cord give rise not only to motoneurons and oligodendrocytes, but also to a subset of astrocytes and ependymal cells. Developmental biology 293, 358–369, https://doi.org/10.1016/j.ydbio.2006.02.029 (2006).
Fu, H. et al. Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development (Cambridge, England) 129, 681–693 (2002).
Lopez-Anido, C. et al. Differential Sox10 genomic occupancy in myelinating glia. Glia. https://doi.org/10.1002/glia.22855 (2015).
Srinivasan, R. et al. Genome-wide analysis of EGR2/SOX10 binding in myelinating peripheral nerve. Nucleic acids research 40, 6449–6460, https://doi.org/10.1093/nar/gks313 (2012).
Stolt, C. C. et al. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes & development 16, 165–170, https://doi.org/10.1101/gad.215802 (2002).
Rubinfeld, H. & Seger, R. The ERK cascade: a prototype of MAPK signaling. Molecular biotechnology 31, 151–174, https://doi.org/10.1385/mb:31:2:151 (2005).
Gaesser, J. M. & Fyffe-Maricich, S. L. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Experimental neurology 283, 501–511, https://doi.org/10.1016/j.expneurol.2016.03.008 (2016).
Ishii, A., Fyffe-Maricich, S. L., Furusho, M., Miller, R. H. & Bansal, R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. The Journal of neuroscience: the official journal of the Society for Neuroscience 32, 8855–8864, https://doi.org/10.1523/jneurosci.0137-12.2012 (2012).
Fyffe-Maricich, S. L., Schott, A., Karl, M., Krasno, J. & Miller, R. H. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. The Journal of neuroscience: the official journal of the Society for Neuroscience 33, 18402–18408, https://doi.org/10.1523/jneurosci.2381-13.2013 (2013).
Rushton, W. A. A theory of the effects of fibre size in medullated nerve. The Journal of physiology 115, 101–122 (1951).
Fumagalli, M. et al. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. The Journal of biological chemistry 286, 10593–10604, https://doi.org/10.1074/jbc.M110.162867 (2011).
Fumagalli, M. et al. The ubiquitin ligase Mdm2 controls oligodendrocyte maturation by intertwining mTOR with G protein-coupled receptor kinase 2 in the regulation of GPR17 receptor desensitization. Glia 63, 2327–2339, https://doi.org/10.1002/glia.22896 (2015).
Young, K. M. et al. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885, https://doi.org/10.1016/j.neuron.2013.01.006 (2013).
Dawson, M. R., Polito, A., Levine, J. M. & Reynolds, R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Molecular and cellular neurosciences 24, 476–488 (2003).
Baumann, N. & Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 81, 871–927 (2001).
Emery, B. & Lu, Q. R. Transcriptional and Epigenetic Regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harbor perspectives in biology 7, a020461, https://doi.org/10.1101/cshperspect.a020461 (2015).
Hernandez, M. & Casaccia, P. Interplay between transcriptional control and chromatin regulation in the oligodendrocyte lineage. Glia 63, 1357–1375, https://doi.org/10.1002/glia.22818 (2015).
Arnett, H. A. et al. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science 306, 2111–2115, https://doi.org/10.1126/science.1103709 (2004).
He, D. et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nature neuroscience 19, 678–689, https://doi.org/10.1038/nn.4258 (2016).
Fancy, S. P., Chan, J. R., Baranzini, S. E., Franklin, R. J. & Rowitch, D. H. Myelin regeneration: a recapitulation of development? Annual review of neuroscience 34, 21–43, https://doi.org/10.1146/annurev-neuro-061010-113629 (2011).
Kotter, M. R., Stadelmann, C. & Hartung, H. P. Enhancing remyelination in disease–can we wrap it up? Brain: a journal of neurology 134, 1882–1900, https://doi.org/10.1093/brain/awr014 (2011).
Wegener, A. et al. Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain: a journal of neurology 138, 120–135, https://doi.org/10.1093/brain/awu375 (2015).
Sahel, A. et al. Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Frontiers in cellular neuroscience 9, 77, https://doi.org/10.3389/fncel.2015.00077 (2015).
Fancy, S. P. et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci 14, 1009–1016, https://doi.org/10.1038/nn.2855 (2011).
Yu, Y. et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261, https://doi.org/10.1016/j.cell.2012.12.006 (2013).
Lu, Q. R. et al. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109, 75–86 (2002).
Birren, S. J., Lo, L. & Anderson, D. J. Sympathetic neuroblasts undergo a developmental switch in trophic dependence. Development (Cambridge, England) 119, 597–610 (1993).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Author information
Authors and Affiliations
Contributions
C.L. contributed to the study design, analyzed data, and wrote the manuscript. L.D. and H.Z. performed the spinal cord injury analyzed the data, Q.L. and G.H. helped with the genotype of the mouse, S.B. performed the Quantitative real-time PCR (qRT-PCR) analysis of OPCs marker in lesion regions. L.L. conceived and supervised the project. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, C., Dong, L., Zhou, H. et al. G-Protein-Coupled Receptor Gpr17 Regulates Oligodendrocyte Differentiation in Response to Lysolecithin-Induced Demyelination. Sci Rep 8, 4502 (2018). https://doi.org/10.1038/s41598-018-22452-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22452-0
This article is cited by
-
The aminosteroid U73122 promotes oligodendrocytes generation and myelin formation
Acta Pharmacologica Sinica (2024)
-
The landscape of targets and lead molecules for remyelination
Nature Chemical Biology (2022)
-
Prenatal Stress Impairs Spinal Cord Oligodendrocyte Maturation via BDNF Signaling in the Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis
Cellular and Molecular Neurobiology (2022)
-
A review of non-prostanoid, eicosanoid receptors: expression, characterization, regulation, and mechanism of action
Journal of Cell Communication and Signaling (2022)
-
The Roles of Orphan G Protein-Coupled Receptors in Autoimmune Diseases
Clinical Reviews in Allergy & Immunology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
