
Abstract
In marine environments, aerobic anoxygenic phototrophic (AAP) bacterial assemblages vary in space and along environmental gradients but the factors shaping their diversity and distribution at different taxonomic levels remain poorly identified. Using sets of sequences encoding the M sub-unit of the photosynthetic apparatus from different oceanic regions, we prioritized the processes underlying AAP bacterial biogeographical patterns. The present analysis offers novel insights into the ecological distribution of marine AAP bacteria and highlights that physiological constraints play a key role in structuring AAP bacterial assemblages at a global scale. Salinity especially seems to favor lineage-specific adaptations. Moreover, by inferring the evolutionary history of habitat transitions, a substantial congruence between habitat and evolutionary relatedness was highlighted. The identification of ecological cohesive clades for AAP bacteria suggests that prediction of AAP bacterial assemblages is possible from marine habitat properties.
Similar content being viewed by others

Introduction
Anoxygenic phototrophic bacteria were long considered to be ecological oddities from specialized habitats1 limiting their relevance for the biosphere. This view has been challenged when bacteriochlorophyll a (Bchl a), the primary pigment of anoxygenic photosynthesis, was found to be distributed in surface waters of the open ocean2. This finding attracted a substantial scientific interest because the light-based metabolic strategy of aerobic anoxygenic phototrophic (AAP) bacteria implied a possible significant revision of oceanic energy budget2. However, the ecology of AAP bacteria is still far from being understood3. Unlike classical photosynthesis, the light energy harvested by AAP bacteria does not fuel the CO2 fixation as in autotrophic cells4. Genomic and physiological evidences showed that they are photoheterotrophic bacteria, using both organic substrates and light for their carbon and energy requirements5. This puzzling life style challenged the classical view of bacteria being dependent on recycling dissolved organic matter and raised questions about the selective advantage of phototrophy for heterotrophic bacterial communities. From an ecological view, the parsimonious explanation was that photoheterotrophy enables microbes to survive adverse conditions and/or to outgrow competitors6. It was therefore postulated that the ability to use light may be especially beneficial in nutrient-poor marine environments2. But the hypothesis was repeatedly disproved as AAP bacteria are more abundant in productive marine areas7,8,9,10,11 illustrating that the link between trophic conditions and ecology of AAP bacteria is not trivial. Indeed, besides the unifying property to perform light-dependent energy transduction, AAP bacteria are very diverse in terms of physiology and metabolism2,12.
To gain a more accurate picture of the factors governing the ecology of AAP bacteria, their diversity was investigated in different oceanic regions (e.g. refs10,13,14,15,16), and several studies have attempted to connect the observed patterns with environmental variables (e.g. refs10,15). Although some trends began to emerge, the link between ecological niches, phylogeny and habitat of AAP bacteria remains patchy. Most studies hypothesized that multifactorial parameters, such as geographic distance (e.g. refs16,17) and environmental gradients (e.g. refs15,18,19,20) act in structuring AAP populations, but the respective importance of these factors has not really been explored. Moreover, recent reports suggest that the expected diversity of AAP bacteria was overestimated and that their biogeographical patterns are not so obvious15,20,21.
Determining how environmental conditions control the ecology of AAP bacteria, particularly at a fine taxonomic resolution (i.e., sufficient to identify lineages with distinct traits), is critical for understanding how these organisms populate the oceans and contribute to global carbon cycling. Following this idea, the objectives of the present study were (i) to provide elements establishing the role of the environmental context in structuring AAP bacterial diversity, (ii) to prioritize the processes (i.e, deterministic vs stochastic) responsible for generating AAP bacterial patterns in marine environments, (iii) to determine if a link between phylogeny and habitat preferendum exists. To this aim, we analyzed the sequence polymorphism of the gene encoding for the M subunit of the core photosynthetic apparatus (pufM gene) of AAP bacteria in contrasted oceanic provinces encompassing different marine regions, distinct nutrient status and oceanic regimes, various temperatures, salinities and depths.
Results
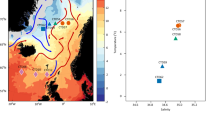
The pufM dataset used in this study was assembled from sequences previously generated from 27 samples collected during 4 cruises (PROSOPE, BOUM, ARCTIC and MALINA) which took place between 1999 and 2009 in different oceanic regions (Mediterranean, North Pacific to Western Beaufort Sea, Barents and Norwegian seas; Fig. 1a). The cruise transects encompass three major oceanic biomes (polar, westerly winds and coastal boundary zone) and six oceanic provinces [Atlantic Subarctic, Atlantic Arctic, Boreal Polar, Canary coastal, Pacific Subarctic gyres and Mediterranean Sea-Black Sea, Table S1] encompassing large ranges of salinities (Fig. 1b) and temperatures (Fig. 1c), distinct nutrient status (Fig. 1d–h) and oceanic regimes (Table S1) and various depths (Table S1).

(a) Schematic representation of the location of stations sampled during cruise tracks depicted on Ocean Data View (http://odv.awi.de)45, map. (b–h) Box-plots displaying for each dataset: (b) Salinity (in g.L−1), (c) Temperature (in °C) and concentrations (in µmol.L−1) in (d) Nitrate, (e) Nitrite, (f) Phosphate, (g) Chlorophyll a (Chl a) and (h) Silicate. (i) Variables factor map and (j) Individuals factor map of the principal component analysis (PCA) performed using standardized quantitative values of the following quantitative ancillary variables: temperature (in °C), salinity (g.L−1), chlorophyll a (in µM), silicate (in µM), nitrite (in µM), nitrate (in µM) and phosphate (in µM); qualitative data (depth of Deep Chlorophyll Maximum (DCM) and distance to shore) were used as illustrative factors.
All of the ~300 pufM sequences were generated by classical cloning-sequencing approaches after amplification by a same primer pair, to give 245 bp PCR products, avoiding biases in the comparison of the relative abundance of OTUs. The sequences were grouped into OTUs, at 94% nucleic acid sequence similarity, using the furthest neighbor clustering method. The number of OTUs in each set of pufM sequences was variable (20 to 48 OTUs), presumably due to a different number of sequences in the primary datasets (Table S2). Despite those differences, coverage values indicated that most diversity in most sequence sets has been retrieved (Table S2). A total of 107 OTUs were identified for the overall dataset with a coverage value of 96% (Table S2, Supplemental material SI1). Only 6 OTUs exhibited a degree of paraphyly (paraphyly index (PI) comprised between 0.01 and 0.08), the other 101 OTUs were monophyletic (PI = 0, data not shown).
Comparisons of pufM datasets
We used community ecology methods to analyze the structure of AAP bacterial communities using two metrics that each emphasized different community characteristics. Bray-Curtis provides a measure of community composition differences between samples based on OTU counts, regardless of taxonomic assignment22. UniFrac quantifies community similarity based on the phylogenetic relatedness23. Both methods showed that AAP bacterial communities exhibited divergent structure of their diversity (Fig. 2a) and phylogenetic composition (Fig. S1) over stations. Nevertheless, at a broader scale, each oceanic region consistently clustered together (Fig. 2a, Fig. S1) illustrating that AAP bacterial signatures were unique to each oceanic region (Mediterranean Sea, Beaufort, Barents and Norwegian seas) (Fig. 2a).

(a) Hierarchical clustering (Bray-Curtis distance) of stations according to OTU relative abundance and heat map showing OTU abundance and distribution. Subcosmopolitan OTUs (OTUs found in at least 2 oceanic regions) and indicator OTUs [according to the concept of indicative species (Auguet et al. 2010)] are indicated at the right. (b–c) Venn diagrams showing the numbers (a) of unique and shared OTUs and (b) of unique and shared pufM sequences between PROSOPE, BOUM, ARCTIC and MALINA datasets, respectively.
To obtain deeper insights into the differences in AAP bacterial community composition, we also assessed the number of shared OTUs and sequences between marine regions. No OTU was common to all oceanic areas investigated here and many were only detected in one sample (Fig. 2a,b). A few dominant OTUs were shared across oceanic regions (Fig. 2b,c). For example, the 7 OTUs common to the Mediterranean (PROSOPE and BOUM datasets) and the Barents and the Norwegian Seas (ARCTIC dataset) covered more than 56% of the pufM sequences retrieved from these oceanic biomes (Fig. 2b,c). Similarly, the 3 OTUs shared by the ARCTIC and MALINA datasets grouped 104 pufM sequences (~20% of sequences; Fig. 2b,c). OTUs common to at least two oceanic regions, hereafter identified as subcosmopolitan OTUs, were affiliated to α- and γ-Proteobacteria within similar proportions (25% and 28%, respectively, Fig. 2a). In contrast, all OTUs affiliated to β-Proteobacteria were detected in only one oceanic region (Fig. 2a). Applying the ecological concept of indicator species according to Auguet et al.24 (i.e. specialist lineages the most frequently represented in most sites of an oceanic region), we identified only 8 indicator lineages (significant IndVal index (P < 0.05)) for the four marine environment analyzed (Fig. 2a). Half of them (OTUs 007, 008, 011, 020) were characteristic of the Barents and Norwegian seas, 2 (OTUs 001, 017) of the western Beaufort Sea, and the OTUs 006, 012 were specific of the Mediterranean Sea (Fig. 2a).
Spatial species turnover
The AAP community similarity between each pairwise set of samples decayed significantly (p < 0.000001) with the geographic distance (Fig. 3). To deeply understand the link between AAP bacterial communities and geographical distance, we compared them using similarity matrices and Mantel tests. Distance matrices for environmental variables and geographic distance were measured by the Euclidean distance between values at two stations. We used Mantel tests to determine the correlation between species similarity matrices and environmental and geographic distance (Table 1). The Mantel correlation (Mc) between species similarity and geographic distance (0.75, p = 0.000001) was higher than with environmental factors (0.45, p = 0.000001). Simple Mantel tests also revealed that environmental factors and geographic distance were significantly correlated (0.38, p = 0.000001). However, the Mc between species similarity and geographic distance, partialling out environmental factors, was not significant (0.08, p = 0.1). In contrast, Mc between environmental factors and species similarity, independent of distance, was significant (0.25, p = 0.00006) (Table 1).

Distance-decay curves for AAP bacterial communities. The solid black line denotes the least-squares linear regression across all spatial scales. Linear regression: P < 0.01.
Environmental gradients driving AAP bacteria structure and identification of AAP bacteria ecoclades
Multivariate regression trees (MRT) were performed to explore and predict relationships between the relative abundance of phylogroups (Fig. 4a) and of OTUs (Fig. 4b) to environmental determinants. The MRT analysis for phylogroups showed a six-leaf tree ordination (explaining 89% of the standardized variance) primarily based on salinity, and followed by Chla, nitrate and depth (Fig. 4a). Pie charts show how the relative abundance of each phylogroup contributed to the separation and composition of the leaves (Fig. 4a). The MRT analysis carried out for OTUs explained 61% of the standardized variance, with salinity, temperature and nitrate mainly responsible of branch splits (Fig. 4b). Whether for phylogroups or OTUs, salinity explained a significant part of the distribution patterns (71% for phylogroups, 32% for OTUs, Fig. 4). The AAP bacteria belonging to γ- and β-Proteobacteria roughly shared more similar distribution patterns within the lineage than between lineages whereas members of α-AAP were widespread (Fig. 4). The indicator OTUs (Fig. 2a) were mainly responsible for the regression tree topology observed and enabled us to identify 5 habitat categories (C1 to C5, Fig. 4b, Supplemental material SI1).

Multivariate regression tree (MRT) analyses of the interaction between environmental parameters and (a) AAP bacterial phylogroups and (b) OTUs abundance (in terms of sequence number). The model explained 89% and 61% of the variance in the whole data set for phylogroups and OTUs, respectively. Pies (a) and multiple value bar chars (b) under each leaf represent the mean of normalized phylogroups (a) and OTUs (b) abundance for each lineage significantly correlated with environmental parameters. Number on the bar chars (b) indicated the number of the corresponding OTUs. Asterisks show indicator OTUs at a significance threshold of P = 0.05. C1 to C5 indicated the 5 habitats identified for AAP from the MRT analysis (e.g. habitat C1: salinity ≥35 g.L−1 and temperature ≥17 °C). [Note that stations Z65 and Z59 from ARCTIC and stations ARC2 and PAC2 from MALINA were not used in MRT analyses because of the lack of environmental data (see Table S1)].
In MRT analyses, most of the variance of the AAP bacterial composition was explained, at different taxonomic level, by environmental parameters (Fig. 4). This suggests that it should be possible to demarcate pufM sequences into ecologically cohesive clades, sharing a common projected habitat which reflected their relative abundance in the environmental categories defined previously (C1 to C5, Fig. 4b, Supplemental material SI1). We used AdaptML to demarcate ecoclades by inferring the evolutionary history of habitat transitions. The resulting observations suggest that AAP bacteria resolved into a striking number of ecologically distinct ecoclades with clearly identifiable preferences (Fig. 5a). The analysis identified 5 distinct inferred habitats (H0 to H5), for the 48 ecoclades, with strong signals from salinity and other environmental settings (Fig. 5b). The OTUs affiliated to α- and β-like AAP bacteria did not contain mixed environmental signal and ecoclades represented coherent phylogenetic clusters. However, numerous dominant γ-like AAP bacterial populations exhibited distinct habitat preferences within a same OTU (Figs 5a, S2).

(a) Maximum likelihood phylogenetic tree showing environmental characteristics and habitat predictions. Characteristics of the sampling locations are plotted on the inner and outer rings: the inner ring indicates the phylogroup, the intermediate ring indicates high (≥35 ppt), medium (32≤ salt ≤35) or low (<32 ppt) salinity and the outer ring indicates the microenvironment (according to the identified habitat C1 to C5 in the MRT analysis). Ecological habitats predicted by the model are identified by colored circles at the parent nodes on the tree. Ecoclades which contained at least 10 pufM sequences and passed a post hoc empirical significance threshold (P-value < 0.01) are indicated by alternating blue and gray shading of clusters. (b) The distribution of each habitat among microenvironments and salinity. The habitat and environment colors match the legend in (a).
Discussion
Recent results gathered from sampling expeditions (e.g. refs17,18,20) and from high-throughput sequencing approaches to achieve pufM gene polymorphism (e.g. refs17,19,25) question the actual extent of the diversity but also the existence of biogeographical patterns for AAP bacteria in marine systems. Indeed, (i) most pufM sequences identified from newly explored ecosystems, including some isolated and extreme environments, are similar to sequences retrieved elsewhere (e.g. refs17,18,20), (ii) next generation sequencing approaches such as 454 pyrosequencing19,25 or Illumina MiSeq amplicon17 did not yield a significant higher number of OTUs than classical clone library methods (e.g. refs15,16,26). We previously addressed these issues and highlighted the need to rely on comprehensive analyses integrating comparable datasets of pufM sequences from different oceanic regions20. This study is in this line and although the sampling was not exhaustive, it offers a comparison of the diversity of AAP bacterial communities in different oceanic provinces and it allows us to identify factors shaping their structure across different marine biomes in the northern hemisphere.
We observed that AAP bacteria exhibited a significant turnover of taxa (beta-diversity) across the oceanic regions investigated here providing strong evidence of biogeographical patterns for these microbes, the distance-decay relationship being one example (Fig. 3). Selection and dispersal of microbes are commonly accepted as main drivers of biogeographical patterns27. To further investigate the underlying mechanisms of AAP bacterial patterns, we tested whether samples were more similar within than across ocean regions. If the dispersal limitation rather than the environmental selection dominated, a higher similarity within than across ocean regions was expected. By contrast, if the environmental selection fully explained biogeographic patterns, we would expect environmental factors to correlate with community similarity.
Our analyses revealed that the environmental selection was the largest main-effect factor contributing to AAP bacteria taxonomic variation between oceanic regions (Table 1). However, the interaction of environment and distance better explained the variation than either of the main-effect factors, indicating a role for as yet unexplained covariance between environment and separation distance (Table 1). We also identified subcosmopolite OTUs (i.e., OTUs shared by at least two oceanic regions) which were mainly representative of dominant AAP bacterial populations (Fig. 2). In line with Bibiloni-Isaksson et al.17, this result could reinforce the idea that key AAP bacterial groups are widely distributed across similar environments.
Overall, our results suggest that geographic distance plays a subordinate role on the composition of AAP marine microbial communities and rather selective processes shape their community composition, a hypothesis summed up by microbiologists as, “everything is everywhere-the environment selects”28,29. Therefore, a distance-decay curve is observed because environmental variables tend to be spatially auto correlated and AAP bacteria with differing niche preferences are selected from the available pool of taxa as the environment changes with distance.
Accordingly, deterministic processes (i.e., selection by environmental variables) are important for non-random spatial distributions of marine AAP bacteria, suggesting that these microbes are specialized on particular habitats. This is consistent with former meta-analyses on natural bacterial assemblages (e.g., refs30,31,32). We showed well-defined community patterns along broad environmental conditions and habitat types (Fig. 4). Taken together, our data revealed salinity as the major environmental factor shaping taxonomic AAP bacterial community composition in the ocean (Fig. 4) while trophic status (i.e., Chl a and nitrate concentrations) and temperature were subsequent explaining factors (Fig. 4). Therefore, our analysis highlighted that physiological constraints play a key role in the AAP bacterial assemblages, and beyond, salinity especially seems to encourage such lineage-specific adaptations. Previous studies on prokaryotic assemblages have shown that salinity is the major determinant structuring bacterial30 and archaeal communities24. Its influence exceeded that of temperature and/or light, recognized as selective parameters for some of other marine microbial taxa33,34. Such commonality in the types of determinant factors suggests that, at global scale, adaptative strategies (physiological constraints) determine the occurrence of AAP in accordance to their heterotrophic status. This does not exclude that at a local scale, other determinants (e.g., light) act in structuring AAP bacterial populations (e.g., refs15,18,20,21).
The AAP bacteria belonging to γ- and β-proteobacteria roughly shared more similar distribution patterns (and probably more similar physiological traits) within the lineage than between lineages whereas members of α-AAP bacteria were widespread. For example, γ- and β-AAP bacteria favor high and low level of salinity, respectively. The preference of Betaproteobacteria for low salt levels is consistent with previous studies that reported their dominance in brackish and freshwater environments [3] and references therein). But by highlighting the predilection of gammaproteobacterial AAP bacteria for high salinity marine systems, this analysis sheds a new light on their ecology. This finding is well supported by their overwhelming dominance in the Mediterranean Sea15,35 where salinity ranged from 36.2 to >39‰36 and provides an interesting framework for designing future culture efforts to expand the diversity of cultivable γ-AAP bacteria.
A global dispersal potential for microorganisms37 and subsequent environmental selection may represent a mechanism for driving patterns of microbial biogeography34. At the same time, local adaptations by natural selection will lead to differences in spatially distant populations of phylogenetically similar organisms34. We found that AAP bacteria resolve into a striking number of ecologically distinct ecoclades with clearly identifiable preferences (Fig. 5a). We identified five distinct inferred habitats (H0 to H5) with strong signals from salinity and other environmental settings (Fig. 5b). OTUs affiliated to α- and β-like AAP bacteria did not contain mixed environmental signal and ecoclades are coherent in terms of phylogeny indicating that ecological niches for AAP bacteria are expressed at the species (i.e., OTU) level. However, numerous dominant γ-like AAP bacterial populations exhibited distinct habitat preferences within a same OTU, reinforcing the previously suggested idea15 that ecotypes exist for γ- AAP bacteria.
It is quite surprising to find both phylogenetic and ecological coherence within AAP bacterial populations, since those affiliated to α- and β-Proteobacteria, expressing a photosynthetic gene cluster supposed to be submitted to lateral gene transfers (LGT). The LGT theory was actually pointed out to be a reasonable and likely hypothesis to explain the patchy distribution of photosynthesis among different bacterial lineages38. But, our ecological interpretation favors the hypothesis of recurrent losses of photosynthetic capacity in different lineages, descendant from a photosynthetic common ancestor39. Indeed, the congruence between habitat and evolutionary relatedness suggest that AAP bacteria acquired their phototrophic abilities a long time ago. That scheme also enables us to better understand why AAP are phototrophs, as the ecological benefit of their phototrophy is not clear yet. This hypothesis is consistent with recent analyses suggesting that the capacity to synthesize Bchl a originated only once in a phototrophic bacterium that pre-dated -at the very least- the radiation event that gave rise to the phylum Chloroflexi, Chlorobi, Acidobacteria, and Proteobacteria40. This places Bchl a synthesis at an early stage during the evolution of bacteria and implies that phototrophy might have been a common trait in ancestral populations of bacteria during the Archean40. However, γ-like AAP challenge that hypothesis (Fig. 5). This result suggests that gammaproteobacterial AAPs acquired phototrophic capacity via a different evolutionary scenario and may evolve under different constraints illustrating again that the evolution of phototrophy in Proteobacteria is a very complex process3,41,42,43. To explore this question, comparing whole genomes of gammaproteobacterial AAPs will be important to identify specific changes leading to adaptative evolution.
The identification of ecoclades is a major advance in the understanding of the ecology of AAP bacteria. Since our analysis was based on a dataset of only 1,306 sequences and four marine regions, we acknowledge that ecoclade identification should be controversial. To determine if these ecoclades have an ecological consistency beyond the oceanic regions investigated here, a set of pufM sequences amplified from surface waters of the Pacific Ocean16 was added to the primary dataset. Although these pufM sequences were not obtained using the primer set that we used, 76% of them fell in 9 predicted ecoclades (ecoclade No. 1, 11, 12, 14, 17, 27, 35, 38, and 47). These ecoclades are mainly affiliated to Gammaproteobacteria with projected habitats H0 (15%), H3 (46%) and H4 (15%). The moderate to high salinity levels of these projected habitats are consistent with the environmental settings reported by the authors16.
In conclusion, our results clearly indicate a dominant role of deterministic processes in influencing the continental-scale structuring of AAP bacteria at different taxonomic levels and reveal that AAP bacteria show strong habitat associations that have likely emerged through evolutionary adaptation. Moreover, we showed that the distribution and structure of AAP bacterial communities can largely be understood in terms of habitat properties solely allowing identifying cohesive ecological clades with a surprising ecological and phylogenetic coherence. This result suggests that it would be possible to predict AAP bacterial assemblages from habitat properties in the marine environment.
Methods
Brief description of the pufM dataset
The dataset was assembled from studies15,18,20,35 examining AAP bacterial communities using pufM sequences amplified using the PufMF forward13 coupled with the PufM_WAW reverse44 primers to give a ~245 bp PCR product. For more details on PCR amplification conditions and clone library construction, see Lehours et al.15. We analyzed a total of 1,306 pufM sequences (see supplemental informations) for which at least one representative sequences of each operational taxonomic unit (OTU) are available in Genbank under accession n° HQ871842-HQ871863, JF421730-JF421749, GQ468944-GQ468986, JN248465-JN248539, and KM654564-KM654598.
Sampling locations and ancillary data
The pufM sequences were recovered from samples collected during four oceanographic cruises, namely PROSOPE15 and BOUM35 in the Mediterranean Sea, MALINA18 from the North Pacific Ocean to the Western Beaufort Sea, and ARCTIC20 at the boundary between the Norwegian, Greenland, and Barents Seas. The location of the stations sampled during the four cruise tracks are depicted on the Ocean Data View (http://odv.awi.de)45, map (Fig. 1a). Stations were affiliated to oceanic biomes and to oceanic provinces defined by Longhurst46,47 using the Longhurst Biogeographical map of arcgis (http://www.arcgis.com). The geographical distances between stations were calculated using the Geographic Distance Matrix Generator (http://biodiversityinformatics.amnh.org/open_source/gdmg/download.php). More details on the area sampled, the sampling procedures and the ancillary parameters characterizing each sampling area were described previously15,18,20,35.
The main ancillary parameters characterizing each sampling area, summarized in Table S1, were plotted in box plots generated using R software48 and used to perform a Principal Component Analysis (PCA) generated using FactoMineR package (http://cran.r-project.org/web/packages/FactoMineR/index.html).
Sequence clustering into OTU
A conservative value of 94% nucleic acid sequence similarity49 was chosen for clustering the 1,306 pufM sequences into Operational Taxonomic Units (OTUs) using the furthest neighbor clustering method. Sequence clustering was performed using MOTHUR (http://www.mothur.org/)50. MOTHUR was also used to generate a heatmap displaying the relative abundance of each OTU for each station. To compensate for the sequencing depth bias per sample in the heatmap generation, sequence abundance values within each OTU were normalized for comparison of OTU abundance between samples. A clustering, based on OTU composition and abundance (Bray-Curtis distance) at the different stations, was also performed and plotted on top of the heatmap. The Venn diagram was generated using MOTHUR and an “in house” developed Perl script.
Phylogenetic analyses of pufM sequences
All pufM sequences were aligned using ARB (http://www.arb-home.de/)51, and added using ADD-BY-PARSIMONY algorithm to a robust pufM tree constructed as described previously15. Phylogenetic tree display and annotation were performed with iTOL software (http://itol.embl.de/)52. AAP bacterial communities retrieved from the 4 studies (PROSOPE, BOUM, ARCTIC and MALINA) were compared using phylogenetic information with Unifrac distance metric (http://bmf2.colorado.edu/unifrac/index.psp)23. The following phylogenetic analyses were performed with Unifrac1: “Unifrac significance”, comparing each pair of environments, which gives a P-value dissimilarity matrix2, “Cluster environments” determining using UPGMA method which environments have similar microbial communities3, “Jackknife environment clusters” performing Jackknife analysis of environment clusters (100 resamplings) produced with the Cluster environments analysis option to determine the robustness of the analysis. We calculated the paraphyly index (PI) as described by Koeppel & Wu53.
Resemblance matrices for biogeographic analyses
Three square resemblance matrices were performed using R software48: the biotic similarity matrix (derived from OTU abundance using Morista-Horn index), the environmental-similarity matrix (derived from environmental data matrix after standard normalization and using Euclidean distance), the geographic distance matrix (derived from the site-location matrix including latitude and longitude values for each station). The environmental parameters integrated were the following: depth, salinity, concentrations in nitrate, phosphate, nitrite, silicate and chlorophyll a, distance to shore, and the position according to DCM (Table S1). To investigate the relationship between AAP bacterial community similarity, geographic distance, and environmental distance across spatial scales, we performed simple and partial Mantel tests with 1,000,000 iterations, using zt software (http://bioinformatics.psb.ugent.be/software/details/ZT)54.
Taxon-based approach and demarcation of “ecoclades”
To identify AAP bacterial taxa which may be analogous to the concept of ‘indicator species’24, tables of relative abundances for phylogroups and for OTUs were constructed. We used the indicator value (IndVal) index, which combines the relative abundance and relative frequency of occurrence of OTUs55. Multivariate regression trees were computed with the R package mvpart56 in order to represent the relationship between phylogroup or OTU relative abundances and the environmental matrix. The quantitative environmental parameters used in the MRT were the following: depth, salinity, concentrations in nitrate, phosphate, nitrite, silicate and chlorophyll a, latitude and longitude. The qualitative parameters used in the MRT were: the trophic status of the sampling location (defined according to phosphate concentrations as eutrophic (>20 µg.L−1), mesotrophic (10–20 µg.L−1) or oligotrophic (<10 µg.L−1)); the distance to shore (coastal or offshore), the oceanic region and the sampling depth relative to the DCM (Table S1). We used AdaptML (available at http://almlab.mit.edu/adaptml/)57, to demarcate ecoclades in our marine AAP bacteria dataset. AdaptML is a maximum likelihood method that employs a hidden Markov model to learn ‘projected habitats’ (distribution patterns among environmental categories) and ecologically cohesive ‘populations’ (groups of related strains sharing the same projected habitat). Our AdaptML analysis used the 5 habitats (C1 to C5) predicted from the MRT analysis. The habitat learning and clustering steps of AdaptML were performed using the default settings.
Data availability
Sequence representatives of each OTU are available in Genbank under accession n° HQ871842-HQ871863, JF421730-JF421749, GQ468944-GQ468986, JN248465-JN248539, and KM654564-KM654598. All sequences, ADAPTML results and OTU cluster file are provided as supplemental material (Supplemental material SI1, Fig. S2)
References
Moran, M. A. & Miller, W. L. Resourceful heterotrophs make the most of light in the coastal ocean. Nat. Rev. Microbiol. 5, 792–800 (2007).
Kolber, Z. S., Van Dover, C. L., Niederman, R. A. & Falkowski, P. G. Bacterial photosynthesis in surface waters of the open Ocean. Nature 407, 177–179 (2000).
Koblížek, M. Ecology of aerobic anoxygenic phototrophs in aquatic environments. FEMS Microbiol. Rev. 39, 854–870 (2015).
Koblížek, M. et al. Isolation and characterization of Erythrobacter sp. strains from the upper ocean. Arch. Microbiol. 180, 327–338 (2003).
Yurkov, V. & Csotonyi, J. T. New light on aerobic anoxygenic phototrophs in The purple phototrophic bacteria (Eds Hunter, N., Daldal, F., Thurnauer, M.C., Beatty, J.T.) 31–55 (Springer Science + Business Media BV, 2008).
Cottrell, M. T. & Kirchman, D. L. Photoheterotrophic microbes in the Arctic Ocean in summer and winter. Appl. Environ. Microbiol. 75, 4958–4966 (2009).
Schwalbach, M. S. & Fuhrman, J. A. Wide-ranging abundances of aerobic anoxygenic phototrophic bacteria in the world ocean revealed by epifluorescence microscopy and quantitative PCR. Limnol. Oceanogr. 50, 620–628 (2005).
Sieracki, M. E., Gilg, I. C., Their, E. C., Poulton, N. J. & Goericke, R. Distribution of planktonic aerobic anoxygenic photoheterotrophic bacteria in the northwest Atlantic. Limnol. Oceanogr. 51, 38–46 (2006).
Jiao, N., Zhang, F. & Hong, N. Significant roles of bacteriochlorophyll a supplemental to chlorophyll a in the ocean. ISME J. 4, 595–597 (2010).
Jiao, N. et al. Distinct distribution pattern of abundance and diversity of aerobic anoxygenic phototrophic bacteria in the global ocean. Environ. Microbiol. 9, 3091–3099 (2007).
Lamy, D. et al. Ecology of aerobic anoxygenic phototrophic bacteria along an oligotrophic gradient in the Mediterranean Sea. Biogeosciences 8, 973–985 (2011).
Rathgeber, C., Beatty, J. T. & Yurkov, V. Aerobic phototrophic bacteria: new evidence for the diversity, ecological importance and applied potential of this previously overlooked group. Photosyn. Res. 81, 113–128 (2004).
Béjà, O. et al. Unsuspected diversity among marine aerobic anoxygenic phototrophs. Nature 415, 630–633 (2002).
Yutin, N. et al. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific Oceans using the Global Ocean Sampling expedition metagenomes. Environ Microbiol 9, 1464–1475 (2007).
Lehours, A.-C., Cottrell, M. T., Dahan, O., Kirchman, D. L. & Jeanthon, C. Summer distribution and diversity of aerobic anoxygenic phototrophic bacteria in the Mediterranean Sea in relation to environmental variables. FEMS Microbiol. Ecol. 74, 39–409 (2010).
Ritchie, A. E. & Johnson, Z. I. Abundance and genetic diversity of aerobic anoxygenic phototrophic bacteria of coastal regions of the Pacific Ocean. Appl. Environ. Microbiol. 78, 2858–2866 (2012).
Bibiloni-Isaksson, J. et al. Spatial and temporal variability of aerobic anoxygenic photoheterotrophic bacteria along the east coast of Australia. Environ. Microbiol. 18, 4485–4500 (2016).
Bœuf, D., Cottrell, M. T., Kirchman, D. L., Lebaron, P. & Jeanthon, C. Summer community structure of aerobic anoxygenic phototrophic bacteria in the western Arctic Ocean. FEMS Microbiol. Ecol. 85, 417–32 (2013).
Ferrera, I., Borrego, C. M., Salazar, G. & Gasol, J. M. Marked seasonality of aerobic anoxygenic phototrophic bacteria in the coastal NW Mediterranean Sea as revealed by cell abundance, pigment concentration and pyrosequencing of pufM gene. Environ. Microbiol. 16, 2953–2965 (2014).
Lehours, A. C. & Jeanthon, C. The hydrological context determines the beta-diversity of aerobic anoxygenic phototrophic bacteria in European Arctic seas but does not favor endemism. Front. Microbiol. 6, 638 (2015).
Bœuf, D., Humily, F. & Jeanthon, C. Diversity of Arctic pelagic Bacteria with an emphasis on photoheterotrophs: a review. Biogeosciences 11, 3309–3322 (2014).
Bray, J. R. & Curtis, J. T. An ordination of upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Auguet, J. C., Barberan, A. & Casamayor, E. O. Global ecological patterns in uncultured Archaea. ISME J. 4, 182–190 (2010).
Zheng, Q., Liu, Y., Steindler, L. & Jiao, N. Pyrosequencing analysis of aerobic anoxygenic phototrophic bacterial community structure in the oligotrophic western Pacific Ocean. FEMS Microbiol. Lett. 362, fnv034 (2015).
Cottrell, M. T., Ras, J. & Kirchman, D. L. Bacteriochlorophyll and community structure of aerobic anoxygenic phototrophic bacteria in a particle-rich estuary. ISME J. 4, 945–954 (2010).
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C. & Martiny, J. B. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506 (2012).
Baas Becking, L. Geobiologie of Inleiding Tot De Milieukunde. (W.P. Van Stockum & Zoon, 1934).
De Wit, R. & Bouvier, T. ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ. Microbiol. 8, 755–758 (2006).
Lozupone, C. A. & Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. 104, 11436–11440 (2007).
Nemergut, D. R. et al. Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 13, 135–144 (2011).
Zinger, L. et al. Global patterns of bacterial Beta-diversity in seafloor and seawater ecosystems. PLoS One 6, e24570 (2011).
Johnson, Z. I. et al. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 31, 1737–1740 (2006).
Sunagawa, S. et al. Structure and function of the global ocean microbiome. Science 348, 1261359 (2015).
Jeanthon, C. et al. Diversity of cultivated and metabolically active aerobic anoxygenic phototrophic bacteria along an oligotrophic gradient in the Mediterranean Sea. Biogeosciences 8, 4421–4457 (2011).
Bethoux, J. P. & Gentili, B. Functioning of the Mediterranean Sea: past and present changes related to freshwater input and climate changes. J. Mar. Syst. 20, 33–47 (1999).
Finlay, B. J. Global dispersal of free-living microbial eukaryote species. Science 296, 1061–1063 (2002).
Boucher, Y. et al. Lateral gene transfer and the origins of prokaryotic groups. Annu. Rev. Genet. 37, 283–328 (2003).
Woese, C. R. Bacterial evolution. Microbiol. Rev. 51, 221–271 (1987).
Cardona, T. Origin of bacteriochlorophyll a and the early diversification of photosynthesis. PlosOne, https://doi.org/10.1371/journal.pone.0151250 (2016).
Nagashima, K. V. P. et al. Horizontal transfer of genes coding for the photosynthetic reaction centers of purple bacteria. J. Mol. Evol. 45, 131–136 (1997).
Koblížek, M. et al. Regressive evolution of photosynthesis in the Roseobacter clade. Adv. Bot. Res. 66, 385–405 (2013).
Boldareva-Nuianzina, E. N. et al. Distribution and origin of oxygen-dependent and oxygen-independent forms of Mg-protoporphyrin monomethylester cyclase among phototrophic proteobacteria. Appl. Environ. Microb. 79, 2596–604 (2013).
Yutin, N., Suzuki, M. T. & Béjà, O. Novel primers reveal wider diversity among marine aerobic anoxygenic phototrophs. Appl. Environ. Microbiol. 71, 8958–8962 (2005).
Schlitzer, R. Ocean Data View, http://odv.awi.de (2017).
Longhurst, A. Biomes: The Primary Partition in Ecological Geography of the Sea (ed. Loghurst, A. R.) 89–102 (Academic Press, 2007).
Longhurst, A. Provinces: The Secondary Compartments (ed. Loghurst, A. R.) 103–114 (Academic Press, 2007).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org/ (2012).
Zeng, Y. H., Chen, X. H. & Jiao, N. Z. Genetic diversity assessment of anoxygenic photosynthetic bacteria by distance based grouping analysis of pufM sequences. Lett. Appl. Microbiol. 45, 639–645 (2007).
Schloss, P. D. et al. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Letunic, I. & Bork, P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2006).
Koeppel, A. F. & Wu, M. Surprisingly extensive mixed phylogenetic and ecological signals among bacterial Operational Taxonomic Units. Nucleic Acids Res. 41, 5175–5188 (2013).
Bonnet, E. & Van de Peer, Y. Zt: a software tool for simple and partial Mantel tests. J. Stat. Softw. 7, 1–12 (2002).
Dufrene, M. & Legendre, P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67, 345–366 (1997).
De’Ath, G. Multivariate regression trees: a new technique for modeling species-environment relationships. Ecology 83, 1105–1117 (2002).
Hunt, D. E. et al. Resource partitioning and sympatric differentiation among closely related bacterioplankton. Science 320, 1081–1085 (2008).
Acknowledgements
We wish to thank the crews of R/V La Thalassa, R/V L’Atalante, CCGS Amundsen, F/F Johan Hjort for outstanding shipboard operations during the PROSOPE, BOUM, MALINA, and ARCTIC 2002 cruises, respectively, and chief scientists H. Claustre, T. Moutin, M. Babin, and R. Ingvaldsen for coordinating these cruises and inviting us onboard. L. Garczarek, D. Marie, F. Partensky, and F. Not are acknowledged for collecting and providing samples. The research leading to these results was supported by the CNRS-INSU (LEFE-CYBER programme) and the MicroB3 project for C. Jeanthon. The Micro B3 project is funded from the European Union’s Seventh Framework Programme (Joint Call OCEAN.2011‐2: Marine microbial diversity–new insights into marine ecosystems functioning and its biotechnological potential) under the grant agreement no 287589. We are grateful to M. Perennou and G. Tanguy (Genomer sequencing platform-FR2424-Station Biologique de Roscoff) for help with sequencing. D. Boeuf was supported by a doctoral grant from the French Ministry of Higher Education and Research. ACL was supported by a postdoctoral fellowship from the CNRS during her postdoctoral research stay in Roscoff.
Author information
Authors and Affiliations
Contributions
A.C.L. and C.J. conceived the study; A.C.L., F.E. and C.J. designed research; A.C.L., F.E. and D.B. performed research; A.C.L., F.E., D.B. and C.J. analyzed data and A.C.L., F.E. and C.J. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lehours, AC., Enault, F., Boeuf, D. et al. Biogeographic patterns of aerobic anoxygenic phototrophic bacteria reveal an ecological consistency of phylogenetic clades in different oceanic biomes. Sci Rep 8, 4105 (2018). https://doi.org/10.1038/s41598-018-22413-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22413-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
