
Abstract
Despite the importance of precisely regulating stem cell division, the molecular basis for this control is still elusive. Here, we show that surface glia in the developing Drosophila brain play essential roles in regulating the proliferation of neural stem cells, neuroblasts (NBs). We found that two classes of extracellular factors, Dally-like (Dlp), a heparan sulfate proteoglycan, and Glass bottom boat (Gbb), a BMP homologue, are required for proper NB proliferation. Interestingly, Dlp expressed in perineural glia (PG), the most outer layer of the surface glia, is responsible for NB proliferation. Consistent with this finding, functional ablation of PG using a dominant-negative form of dynamin showed that PG has an instructive role in regulating NB proliferation. Gbb acts not only as an autocrine proliferation factor in NBs but also as a paracrine survival signal in the PG. We propose that bidirectional communication between NBs and glia through TGF-β signaling influences mutual development of these two cell types. We also discuss the possibility that PG and NBs communicate via direct membrane contact or transcytotic transport of membrane components. Thus, our study shows that the surface glia acts not only as a simple structural insulator but also a dynamic regulator of brain development.
Similar content being viewed by others


Introduction
The Drosophila CNS develops from neural stem cells called neuroblasts (NBs)1,2,3. The self-renewal and differentiation of NBs are thought to be controlled by both intrinsic programs in the NBs as well as extrinsic cues. Despite a wealth of knowledge about intrinsic factors that regulate NB development4, very little is known regarding non-autonomous factors that affect this process. In the embryonic CNS, it has been shown that NBs receive an extrinsic signal from the overlying epithelium to properly orient the mitotic spindle axis5. Several lines of evidence suggest that post-embryonic NBs are also controlled by extrinsic factors. One previous study showed that the activin receptor, Baboon (Babo), and its transcriptional mediator, Smad2 (Smox), regulate NB proliferation in the larval CNS, indicating that activin-like ligands may play a role in this process6. Furthermore, expression of a dominant-negative form of E-cadherin in glia reduced the number of proliferating NBs7. This observation indicated that physical contact of larval NBs with glia is critical to NB proliferation. Larval glia are also known to secrete Anachronism, which maintains NBs in a mitotically inactive state (the quiescent state) during early larval stages8. Thus, although there is no direct evidence that Drosophila larval glia have a niche function, it is assumed that division and differentiation of larval brain NBs may be controlled by surrounding glial cells.
Recent studies demonstrated vital roles of heparan sulfate proteoglycans (HSPGs) as important regulators of stem cell behaviors9,10,11,12,13,14. HSPGs are a class of carbohydrate-modified proteins involved in a variety of biological processes such as growth factor signaling, cell adhesion, and enzymatic catalysis15. Importantly, these molecules serve as co-receptors for growth factor signaling, regulating reception and tissue distribution of secreted signaling proteins such as FGFs, BMPs, Wnts, and Hedgehog on the cell surface16,17,18,19,20,21. Although HSPGs are generally thought to regulate growth factor signaling on the surface of signal-receiving cells, they can also activate signaling in trans from adjacent cells to play non-autonomous roles9,11,22,23.
The TGF-β pathway is an evolutionarily conserved signal transduction module that mediates diverse biological processes in animals, including stem cell control. Like in mammals, both the BMP and Activin branches are required for Drosophila development and homeostasis24. BMPs are involved in numerous processes throughout all developmental stages, from patterning of the embryo25,26 and imaginal discs27,28 to stem cell maintenance29. Activin branch ligands also play diverse roles in growth and patterning in Drosophila, including control of imaginal cell proliferation30,31,32, neuroblast proliferation6, neuron remodeling33,34, axon guidance35,36, and sugar homeostasis37.
Glial cells in the Drosophila larval brain have been classified into a few different types based on their positions, morphology, and gene expression profiles, including perineural glia (PG), subperineural glia (SPG), cortex glia, and astrocyte-like glia, which send processes into the neuropil38,39,40. Two surface glia cell types, PG and SPG, provide a structural basis for an insulation barrier between the CNS and hemolymph known as the blood-brain barrier (BBB)41,42,43. The most outer layer is formed by PG, which is characterized by the formation of extensive, thin cell protrusions. The SPG are located immediately below the PG. The SPG forms septate junctions (SJs) and are central for the Drosophila BBB function44,45. The BBB restricts molecular movement at the interface between blood and nervous tissues, and it plays critical roles in maintaining a regulated microenvironment for reliable neural signaling46,47. On the other hand, the BBB is a major obstacle for pharmacologic treatments to the brain. Past studies have focused on the functions of surface glia as an effective shield, which separates the nervous system from the open circulatory system, to maintain brain homeostasis42. However, the biological role of surface glia in regulating neural development remains to be elucidated.
In this study, we identified bidirectional interactions of NBs and surface glial cells in the developing larval brain. We found that Dally-like (Dlp), a HSPG of the glypican family, is expressed in PG and is critical for proper NB proliferation. Selective ablation of glial cells supported the idea that PG play an instructive role in regulating NB proliferation. We also found that Glass bottom boat (Gbb), a BMP homologue, is expressed in NBs and functions with Dlp to regulate normal proliferation of NBs. In addition to serving as an autocrine signal in NBs, Gbb also acts as a paracrine survival signal in the PG. Thus, the development of NBs and glia is controlled by reciprocal communication between these cells through TGF-β signaling pathways in the NSC niche. Finally, we discuss the possibility that PG make physical membrane contact with NBs.
Results
Dlp is required for normal NB proliferation
HS-deficient mutants, tout velu (ttv) and sulfateless, show a small brain phenotype mainly because the sizes of brain lobes and ventral ganglion do not properly increase during third larval instar stage. A previous study showed that mutations in activin signaling components cause a similar small brain phenotype, which is associated with altered innervation of photoreceptor (PR) axons (Fig. 1B)6. The PR axon innervation abnormality is due to a reduction in the size of PR target field in the CNS, the lamina and medulla, rather than defects of PR cells. Thus, PR axon projection is a sensitive readout of brain growth. Similar to a mutant for baboon (babo), the Drosophila activin type I receptor, ttv mutant brains show PR axon targeting defect revealed by staining with anti-Chaoptin antibody (22B10) (Fig. 1C). This observation suggests that one or more HSPG molecules are required for normal NB proliferation. No obvious small brain phenotype is caused by mutations in dally, one of two integral-membrane HSPGs of the glypican type (Fig. 1D), or dSyndecan, the only Drosophila Syndecan homologue (data not shown). We found that brains mutant for dlp, the second glypican homologue, exhibit a very severe small brain phenotype with abnormalities of PR axon projection (Fig. 1E), suggesting that dlp is required for normal brain growth.

dlp regulates NB proliferation in Drosophila larval brain. (A–E) Morphology of brains from control (yw) (A), and baboFd4 (B), ttv524 (C), dallygem (D) and dlp1 (E) mutants. Photoreceptor (PR) axon projections are assessed by 24B10 staining (green). Propidium iodide (red) staining shows brain lobe (BL), eye disc (ED) and ventral nerve cord (VNC). In control and dally brains, the PR axons form a neural plexus at the lamina (lm) and project to the medulla (md). babo, ttv and dlp brains show reduced brain size and abnormally bundled growth cones. Although most of dlp1 homozygous mutants die during embryogenesis, we obtained rare survivors (E). Anterior to the top. Scale bar: 100 µm. (F) A schematic illustration of Drosophila larval brain and ventral nerve cord (VNC). A and P shows anterior-posterior axis. Most images in this paper are horizontal confocal optic sections (black plate) unless otherwise stated. (G–I) BrdU incorporation into NBs in control (G) and dlp1/dlpA187 (H), and PG-Gal4 dlpA187/UAS-dlp dlp1 (I). NB proliferation was impaired in dlp mutants. dlp expression in PG fully rescued the NB proliferation defect of dlp mutants. (J and K) Expression of Dlp. Anti-Dlp antibody staining of yw (J) and dlp1/dlpA187 (K) brains. Images are shown for surface glial cell focal plane. Dlp is detected on the surface of the brain lobe largely overlapping with PG. Loss of the signal confirmed the specificity of anti-Dlp staining. (L and M) Cross-sections of brains from PG > mCD8ChRFP (L) and SPG > mCD8ChRFP (M) larvae stained with anti-Dlp antibody. Signals for anti-Dlp and mCD8ChRFP are shown red and green, respectively. (N) Summary of PI for each genotype. Numerical figures depict the mean ± SE. In this, and the following figures, data were analyzed by one-way ANOVA with Bonferroni post hoc test. n.s.: not significant; ***p < 0.001.
To determine if the small brain phenotype of dlp mutants is caused by reduced proliferation of NBs, we analyzed NB proliferation in control and mutant brains by BrdU labeling (Fig. 1G–I). In control (yw) third instar larval brains, where NBs are actively dividing, BrdU incorporation showed a unique and robust pattern in NBs and their progeny (Fig. 1G). Although dlp null mutants have been described as embryonic lethal, trans allelic heterozygotes of certain dlp loss-of-function mutations (dlp1/dlpA187) survive to late third larval instar stage. We found that dlp1/dlpA187 mutant brain shows significantly impaired NB proliferation (Fig. 1H and N; Proliferation Index (PI) = 0.74).
Interestingly, Dlp is predominantly, although not exclusively, expressed at the surface of larval brains (Fig. 1J and K). To further define the localization of Dlp on the brain surface, we used well-established surface glia Gal4 drivers, NP6293-Gal4 and NP2276-Gal4. We carefully assessed the expression patterns of these Gal4 drivers and confirmed that NP6293-Gal4 and NP2276-Gal4 are specifically expressed in PG and SPG, respectively (Fig. S1, data not shown), consistent with previous studies. Therefore, hereafter, we will call these drivers PG-Gal4 and SPG-Gal4, respectively, in this paper. To define Dlp-expressing cells, fluorescent UAS transgene reporters were induced to mark glia membranes using PG-Gal4 and SPG-Gal4. Anti-Dlp antibody staining of brains from PG > mCD8ChRFP (animals bearing PG-Gal4 and UAS-mCD8ChRFP) and SPG > mCD8ChRFP showed that the area of Dlp expression is largely overlapping with PG (Fig. 1L and M).
To determine if Dlp on the surface glia non-autonomously regulates NB development, we specifically expressed dlp in PG of dlp mutants using PG-Gal4. We found that the reduced NB proliferation of dlp mutants was completely restored by PG-specific expression of dlp (Fig. 1I and N; PI = 1.07). Since no leaky expression of PG-Gal4 is detectable in NBs or any parts of the brain inside the surface glia (Fig. S1), this rescue experiment demonstrated that Dlp expressed in PG can sufficiently function to maintain normal NB proliferation.
Perineural glia are required for normal development of NBs
To examine the role of PG in NB development, we tested the effects of functionally ablating glial cells in vivo using a temperature-sensitive dominant-negative form of shibire (shi), the Drosophila dynamin48. Since dynamin is required for the formation of membrane vesicles49, expression of this dominant negative form of shi (shits) can block various types of membrane trafficking events, including endocytosis and protein secretion, and has been effectively used to disrupt glial cell function50,51,52. We expressed UAS-shits specifically in PG using PG-Gal4 (PG > shits) and assessed its effect on NB proliferation. We found that the functional ablation of PG by expression of shits severely disrupted NB cell proliferation (Fig. 2D and K; PI = 1.01 and 0.43 at permissive (18 °C) and restrictive (32 °C) temperatures, respectively), which resulted in a small brain phenotype (data not shown). Normal NB proliferation was observed in control (yw) brains for both temperature conditions (Fig. 2A and B). PG > shits reared at 32 °C also showed a decreased number of M-phase cells labeled with anti-phosphorylated histone H3 (pH3) (Fig. 2E and F). Brains from various control animals (PG > shits reared at the permissive temperature, UAS > shits without Gal4 driver) were indistinguishable from wild-type (Fig. 2C and E, data not shown). These observations show that functional ablation of PG disrupts normal proliferation of neighboring NBs.

Functional ablation of PG caused a NB proliferation defect. (A–F) Blocking shibire disrupts normal proliferation of NBs. BrdU incorporation in control (A and B) and PG > shits (C and D) brains at permissive (18 °C; A and C) and restrictive (32 °C; B and D) temperatures. (E and F) shits expression reduces pH3-positive M phase cells. Anti-pH3 staining of PG > shits at permissive (E) and restrictive (F) temperatures. (G–J) shits expression affects membrane distribution. Membrane distribution was visualized in PG-Gal4/+; UAS-mCD8GFP/UAS-shits at permissive (G) and restrictive (H) temperatures. The localization of ManII-eGFP was examined in PG-Gal4/+; UAS-ManII-eGFP/UAS-shits at permissive (I) and restrictive (J) temperatures. (K) Summary of PI for each genotype. (L) Dlp localization in PG > shits brains at restrictive temperature. shits expression disrupts cell surface localization of Dlp. This image includes tissue debris with unknown origin on the top of the brain. n.s.: not significant; ***p < 0.001.
To further explore the cellular and molecular basis for the role of PG in NB proliferation, we examined the effect of shits on membrane structures in PG. We observed a gross abnormality in patterns of a membrane-bound form of GFP (mCD8GFP) in PG > shits at the restrictive temperature (Fig. 2G and H). This cell membrane marker accumulated in restricted areas, forming patches, and failed to uniformly distribute on the PG plasma membrane. Expression of shits in PG also affected a marker for the Golgi (ManII-eGFP) (Fig. 2I and J). Altered patterns of these membrane markers suggested that normal membrane trafficking is required for proper control of NB development.
We next asked if Dlp localization is affected by shits expression. In a section near the brain surface, the PG focal plane, anti-Dlp staining showed abnormal aggregations of Dlp-containing membrane in PG > shits brain at 32 °C (Fig. 2L, compare to Fig. 1J). This observation suggests that the mislocalization of Dlp in the PG cell membrane may contribute to the NB proliferation defect of shits brains. Altogether, these results support the idea that Dlp on the PG surface regulates NB proliferation.
Gbb signaling regulates NB proliferation
The requirement for Dlp in NB proliferation suggests that a signaling molecule controls NB development in a HSPG-dependent manner. Previous studies have shown that most HS-dependent ligands are dispensable for NB development at the third instar larval stage53,54, or they are not expressed in the corresponding regions54,55,56. An exception is Gbb, a BMP-like ligand, which has been found to be expressed in the developing larval CNS by RNA in situ hybridization57. We monitored gbb expression at higher resolution, using gbb-lacZ and gbb-LexA > LexAop-rCD2GFP, in which gbb enhancers drive expression of a membrane-bound form of GFP, to determine the specific cell types that express gbb. As shown in Fig. 3A, a reporter assay revealed that gbb expression overlaps with a NB-marker, Mira. We also asked if gbb is expressed in PG. In this and subsequent experiments, we used an antibody against a nuclear receptor Seven-up (Svp) as a specific marker for PG since we have found Svp expression extensively overlaps with PG-Gal4-positive cells (Fig. S1B-B”). Thus, anti-Svp is a useful tool to label PG. We observed no overlap in expression between gbb-lacZ and Svp (Fig. S2). Thus, gbb is specifically expressed in the NBs but not in PG.

gbb controls NB proliferation. (A–A”) gbb is expressed in NBs. Expression of gbb-LexAVP16 was monitored by LexAop-rCD2GFP (A) and NBs are labeled with anti-Mira antibody (A’). (A”) Merged image of A and A’. IOA: inner optic anlage. (B–G) BrdU incorporation of control (yw reared at 32 °C; B), gbb1/gbb4 (reared at 32 °C; C), gbb1/gbb2 (D), gbb1 gbb-Gal4/gbb2 UAS-gbb (E), gbb1 pros-Gal4/gbb2 UAS-gbb (F), and gbb1/+; dlp1/dlpA187 (G) brains. NB proliferation was severely disrupted in gbb mutants. This phenotype is rescued by gbb expression in NBs. Heterozygosity for gbb enhanced the NB proliferation defect of dlp mutants, suggesting that these two genes function in the same pathway. (H and I) Summary of PI for each genotype. Numerical figures depict the mean ± SE. n.s.: not significant; **p < 0.01; ***p < 0.001.
We next examined whether gbb is required for NB proliferation. Blocking gbb activity at the third instar using a temperature-sensitive allelic combination (gbb1/gbb4) severely disrupted NB proliferation (Fig. 3C and H; PI = 0.72 at permissive temperature, 0.26 at restrictive temperature). An even more severe NB division defect was observed in the early third instar survivors of gbb null (gbb1/gbb2) mutants (Fig. 3D and I; PI = 0.07). This gbb1/gbb2 phenotype was fully rescued by induction of UAS-gbb using either gbb-Gal4 or pros-Gal4, another NB-specific driver (Fig. 3E,F and I; PI = 1.21 and 1.05, respectively). These results confirmed that Gbb produced in NBs is responsible for normal cell proliferation of NBs (Fig. 3E and F).
To determine if gbb and dlp function in the same pathway to regulate NB development, we examined genetic interactions between the two genes. The NB proliferation defect of dlp mutants was significantly aggravated by heterozygosity for gbb in gbb1/+; dlp1/dlpA187 brains (Fig. 3G and I, compare to Fig. 1B; PI = 0.29), while gbb heterozygotes (gbb1/+) show normal NB proliferation (data not shown). This synergistic effect is consistent with the notion that the gbb and dlp genes function in the same pathway to regulate NB proliferation.
Role of Gbb autocrine signaling in NB proliferation
We next asked which cells in the developing brain respond to the Gbb signal using dad-lacZ58, a widely-used reporter of BMP signaling activity. Interestingly, we detected specific dad-lacZ signals in both Dpn-positive NBs and Svp-positive PG (Fig. 4A–B”), showing that Gbb signaling is received by both NBs and PG. Most dad-lacZ expression was lost in gbb mutant brains (Fig. 4C–C”). This result indicated that gbb is responsible for most, if not all, BMP signaling in this region of the brain at third instar larval stage. When we specifically blocked Gbb signaling in NBs by using gbb-Gal4 to express UAS-dad, an inhibitor of BMP signaling, NB proliferation was strongly inhibited (Fig. 4D and K; PI = 0.17). Since surrounding glia are wild-type, this result demonstrated an autonomous role for Gbb signaling in NB proliferation.

Gbb autocrine signaling in NB proliferation. (A–B”) dad-lacZ/+ brains were stained with anti-β-galactosidase (A and B) and anti-Dpn (a NB marker; A’) or anti-Svp (a PG marker; B’) antibodies. (A” and B”) are merged images of A and A’, B and B’, respectively. Arrows show overlapped expression of dad-lacZ with Dpn (A”) and Svp (B”). dad-lacZ expression was detected in both NBs and PG. (C–C”) dad-lacZ expression (C) and anti-Dpn (C’) staining in gbb1/gbb4 (reared at 32 °C) brain. C” is merged image of C and C’. dad-lacZ signal was significantly diminished in gbb mutants. (D) BrdU incorporation of gbb-Gal4/UAS-dad brain. (E-G) dad-lacZ expression (green) and anti-Dpn staining (magenta) in control (E), PG> shibirets (F and G) brains at permissive (F) and restrictive (E and G) temperatures. Gbb signaling is impaired by PG functional ablation. (H–H”) dad-lacZ expression (H) and anti-Dpn staining (H’) of dlp1/dlpA187 brain. H” is a merged image of H and H’. The number of dad-lacZ-positive NBs was significantly decreased in dlp mutants. (I and J) BrdU incorporation in control (yw; I) and PG-Gal4/UAS-dlp (J) brains reared at 29 °C. (K) Summary of PI for each genotype. n.s.: not significant; ***p < 0.001.
If PG support NB proliferation through activation and/or maintenance of Gbb signaling, one could expect that shits expression in PG would impair Gbb signaling in NBs. We found that this was the case. Gbb signaling in NBs can be monitored by co-expression of dad-lacZ and Dpn (arrows in Fig. 4E and F). Expression of dad-lacZ in Dpn-positive cells was eliminated by PG-specific expression of shits (Fig. 4G). In addition, we also observed that dad-lacZ signal was diminished in dlp mutants (Fig. 4H–H”). Together with the genetic interactions between gbb and dlp shown above, these results indicated that Dlp in the PG potentiates autocrine Gbb signaling in NB to promote its proliferation. We also evaluated the effects of gbb or dlp overexpression on the NB phenotype, but neither caused any significant change in NB proliferation (Fig. 4K; PI = 0.91 and 1.16, respectively).
We have previously shown that Dlp acts as a co-receptor for Gbb in vitro using a trans signaling assay11. This assay measures the activity of a co-receptor expressed in a contacting neighboring cell to regulate signaling in trans in the signal-receiving cell. This in vitro data supports our model that Dlp (on PG surface) can act as a trans co-receptor for Gbb (received by NB) in the brain. Since no previous work has shown a direct binding between Dlp and Gbb, we attempted to show Dlp-Gbb complex formation by co-immunoprecipitation of the proteins expressed in S2 cells. However, the Gbb-Dlp complex was not detectable, probably because the level of HS modification onto the Dlp core-protein in S2 cells is too low. As shown in Fig. S3A and a previous work59, Gbb binds to heparin-sepharose beads. Thus, similar to many other BMP members, Gbb is a heparin-binding protein59, characteristic of HS-dependent factors.
We also analyzed the cellular localization of Gbb and Dlp by expressing Gbb-HA and Dlp-Myc in S2 cells. Immunotaining with anti-HA and anti-Myc antibodies using an extracellular staining protocol revealed that Gbb and Dlp extensively co-localize on the S2 cell surface (Fig. S3B). Together, our results and previous reports suggest that Dlp serves as a co-receptor for Gbb signaling to regulate NB proliferation in the larval brain.
Role of Gbb signaling in perineural glia
The observation that both NBs and PG receive Gbb signaling (monitored by dad-lacZ, Fig. 4A–B”) raised the question of what role this pathway plays in PG. Remarkably, we found that the number of PG cells, monitored by anti-Svp antibody staining, was substantially reduced in gbb null mutant brains (Figs 5B,G and 6B; PG Index (PGI) = 0.44). This phenotype is unique to the gbb null condition since gbb hypomorphic mutations did not affect PG cell numbers (Fig. 5C and G; PGI = 0.94). The loss of PG cells in gbb null mutants was rescued by UAS-gbb expression in NBs using gbb-Gal4 (Fig. 5D and G; PGI = 0.89). Thus, in addition to NB proliferation, Gbb signaling regulates PG maintenance. The lack of this phenotype in gbb1/gbb4 and dlp mutants (Fig. 5G; PGI = 0.95) suggests that the critical threshold required for PG maintenance is lower than that for NB proliferation. Consistent with the phenotype of gbb null mutants, blocking BMP signaling in PG by UAS-dad expression using PG-Gal4 also reduced the PG cell number (Fig. 5E and G; PGI = 0.35). This treatment also resulted in impaired NB proliferation (Figs 5F and 4K; PI = 0.32), consistent with the essential role of PG in the normal proliferation of NBs.

Function of Gbb paracrine signaling in PG maintenance. (A–E) Anti-Svp antibody staining of control (yw reared at 32 °C; A), gbb1/gbb2 (B), gbb1/gbb4 (reared at 32 °C; C), gbb1 gbb-Gal4/gbb2 UAS-gbb (D), and PG-Gal4/UAS-dad (E) brains. (F) BrdU incorporation in a PG-Gal4/UAS-dad brain. The number of Svp-positive PG was significantly reduced in gbb1/gbb2 but not in gbb1/gbb4 animals. PG number and NB proliferation were also diminished by induction of UAS-dad using PG-Gal4. (G and H) Summary of PG Index (PGI). To calculate PGI, the number of PG cells in brains of each genotype was divided by that of control brains (yw in G, PG-Gal4/UAS-nlsGFP in H). The number of PG cells was counted as Svp-positive cells (Svp, G) or GFP-positive cells in PG-Gal4/UAS-nlsGFP brains (GFP). The genotypes used in H are: PG-Gal4/UAS-nlsGFP, PG-Gal4 gbb1/gbb2; UAS-nlsGFP/+, and PG-Gal4 gbb1/UAS-p35 gbb2; UAS-nlsGFP/+. Numerical figures depict the mean ± SE. *p < 0.05; **p < 0.01.

Rescue of PG in gbb null mutants by UAS-p35 can restore NB proliferation. (A–C) PG marked by GFP expression in PG-Gal4/UAS-nlsGFP (A), PG-Gal4 gbb1/gbb2; UAS-nlsGFP (B), and PG-Gal4 gbb1/UAS-p35 gbb2; UAS-nlsGFP (C) brains. p35 expression partially restored PG number. (D–F) BrdU incorporation in yw (D), gbb1/gbb2 (E), and PG-Gal4 gbb1/gbb2; UAS-p35/+ (F) brains. p35 expression partially rescues the NB proliferation defect of gbb null mutants. (G–G”) daw is expressed in PG. A daw > nlsLacZ brain was stained with anti-β-galactosidase (G) and anti-Svp (G’) antibodies. (G”) Merged image of G and G’. daw is exclusively expressed in Svp-positive PG. (H) Summary of PI for each genotype. ***p < 0.001.
In order to determine if PG cells are lost by cell death in gbb null mutants, we expressed p35, an apoptosis inhibitor, in PG. In this experiment, we counted the PG cell number by marking PG with nlsGFP driven by PG-Gal4. We found that p35 expression significantly restored the PG in gbb null mutant brains (Figs 6A–C and 5H). Altogether, these results indicate that Gbb acts not only as an autocrine factor in NB but as a paracrine survival signal to inhibit PG apoptosis.
Interestingly, in addition to rescuing PG, p35 expression in gbb null mutants also partially restored NB proliferation (Fig. 6D–F and H; PI = 0.50). Given that this restoration occurs in the absence of Gbb function, this observation implies that additional factor(s) derived from PG promote NB division, independent of Gbb signaling. Possible candidates for such molecules are activin-like ligands since, as mentioned above, a previous study showed that mutations in baboon, the activin type I receptor, or dSmad2, the downstream activin signal transducer, strongly disrupt NB proliferation6. We examined the expression pattern of one of the Drosophila activin-like ligands, daw, in the CNS. Extensive co-localization of Svp and daw-Gal4 expression indicates that this ligand is exclusively expressed in PG (Fig. 6G–G”). This result is consistent with the idea that Daw, and possibly other activin-like ligands, serve as PG factors to control NB proliferation.
Since both dlp and activin signaling components show NB proliferation defect, we asked if they function in the same pathway (i.e. Dlp acts as a co-receptor for activins). We examined the genetic interactions between babo and dlp for brain growth using the PR projection assay. No synergistic effect was observed between babo and dlp mutations (data not shown), suggesting that activins regulate NB proliferation, independent of Gbb-Dlp signaling.
Possible mechanisms for communications between perineural glia and NBs
Our results showed that PG and NBs communicate to regulate each other’s development. Specifically, our findings support the idea that Dlp serves as a co-receptor for Gbb signaling during larval brain development. However, the mechanism by which these cells communicate across SPG, the major barrier component of the BBB, is unknown. One obvious possibility is that “transcytosis” in SPG may carry specific molecules, either alone or in complexes with lipid particles, to the other side of the BBB60. Alternatively, SPG may have unknown openings, which allow PG to directly contact NBs. Although we do not know the definite answer to this question, several observations described below are consistent with these possibilities.
When a membrane-bound form of GFP (mCD8GFP) was expressed in SPG by the SPG-Gal4 driver, GFP signal was detected non-uniformly, showing openings within the SPG layer (Fig. 7A). These numerous holes suggest either the presence of a zone where mCD8GFP is excluded from the membrane or a discontinuity of the plasma membrane. Cross-sections of brains doubly stained for PG (PG-Gal4 > UAS-mCD8ChRFP) and SPG (Mdr65-LexA > mCD8GFP) often revealed regions in which the PG and SPG signals are found in the same layer (Fig. 7B). They do not appear to be co-localized since we do not see yellow signals indicative of co-localization. Instead, in these domains, the SPG layer is undetectable and the only PG signal can be detected (Fig. 7B). Such discontinuity of the SPG signal is consistently observed in virtually all brain samples analyzed. To further confirm that this staining pattern is not due to leaky expression of PG-Gal4 in SPG, we used eGFP-tagged Dlg, which is localized to septate junctions formed between neighboring SPGs. When this construct was expressed in SPG using Mdr65-Gal4, eGFP-Dlg highlighted the septate junction structures (Fig. S1E). On the other hand, eGFP-Dlg did not show this pattern when expressed by PG-Gal4 or daw-Gal4. Thus, these two Gal4 drivers are specific to PG and do not drive expressions in SPG. Together, our observations suggest that the PG layer may directly communicate with more interior NBs through the openings in the SPG.

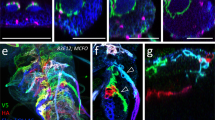
Possible membrane contact between NBs and PG. (A) Distribution of a membrane-bound form of GFP in SPG (SPG > mCD8GFP). (B) Cross-sections of brains doubly stained for PG (PG-Gal4 > UAS-mCD8ChRFP) and SPG (Mdr65-LexA > mCD8GFP). (C–D’) Reconstitution of spGFP fragments. spGFP signal was detected in PG-Gal4/gbb-LexAVP16; UAS-spGFP11/LexAop-spGFP1-10 brains in the glial cell focal plane (C and C’) and NB focal plane (D and D’). Anti-Mira signal is shown in C’ and D’ (magenta). (E) Schematic representation of spGFP reconstitution. spGFP1-10 was expressed in NBs under the control of the gbb promoter through the LexA/LexAop system. spGFP11 expression was driven in the PG by NP Gal4 drivers. (F–G”) shits expression disrupts the PG-NB contact. spGFP signal (green) in PG-ablated brains (PG-Gal4/gbb-LexAVP16; UAS-spGFP11/LexAop-spGFP1-10, UAS-shits) at permissive (F–F”) and restrictive (G–G”) temperatures. Anti-Mira staining marks NBs (magenta). Images for X-Z sections (F’ and G’) and Y-Z sections (F” and G”) at white bars are shown. Arrows show the lack of GFP signal, showing the elimination of physical contact between PG and NBs (G). An example is shown of a large gap in the spGFP signal on the brain surface (H).
To examine whether there is close physical proximity of NBs and PG cell membranes, we employed the split-GFP (spGFP) system. This method is based on the fact that complementation of two split-GFP proteins, spGFP1-10 and spGFP11, reconstitutes green fluorescence while each spGFP fragment alone is not fluorescent61. Membrane-bound forms of spGFP proteins have been successfully used to label areas of close membrane apposition between two cells62. We developed a system to express each spGFP fragment separately in different set of cells in vivo using the Gal4/UAS63 and the LexA/LexAop64 systems. We generated and used gbb-LexA as a NB driver to express a membrane-tethered form of spGFP1-10 in NBs (Fig. 7E). Independent of spGFP1-10, expression of spGFP11 was induced in several different types of glial cells using the Gal4/UAS system. In addition to PG-specific PG-Gal4, we used NP2222-Gal4 and NP3233-Gal4 to induce expression of a membrane bound form of spGFP11 in cortex glia and astrocyte-like glia, respectively38,65.
Using PG-driven spGFP11, we detected complementation of the two spGFP fragments, suggesting that PG cell membranes are in close proximity to NB membranes (Fig. 7C and D). Control brains lacking one of the transgenes showed no GFP signal, proving the specificity of the GFP reconstitution (Fig. S4A–D). Similar experiments with other Gal4 drivers showed that cortex glia have large area of direct contact with NBs (Fig. S4F’), while astrocyte-like glia show very limited contact with NB neuropil (Fig. S4H’). These are expected results based on the anatomy of the larval CNS, further validating the spGFP experiments to detect the membrane contact. Notably, PG and cortex glia showed distinct patterns of spGFP signals: cortex glia contact the entire surface of a NB, while PG contact only the apical membrane of NBs (Figs 7C,D’ and S4), suggesting that each glial cell population may have a unique contribution to NB development.
We also examined NB-PG membrane contact by the spGFP system in PG > shits brain, which shows severely reduced NB proliferation (Fig. 2). At 18 °C, GFP complementation was observed uniformly and continuously on the surface of the brain lobe although the GFP signal is weak due to the low level of Gal4-induced expression at low temperature (Fig. 7F–F”). In contrast, inactivation of shi at the restrictive temperature caused gaps in the spGFP signal, indicating loss of the PG-NB contact on the brain lobe surface (arrowheads in Fig. 7G–G”). Figure 7H shows an example of a large gap in the spGFP signal on the surface of a shits brain lobe at 32 °C. These observations suggest that the NB proliferation defect in shits brain may be caused by loss of contact-dependent communication between NB and PG due to disrupted membrane trafficking in PG.
Taken together, our observations using the spGFP complementation system suggest that PG-NB communication may be mediated through direct membrane contact or transcytotic transport of membrane components across the BBB.
Discussion
Glial cells play essential roles in the Drosophila neural stem cell niche66,67. Our study demonstrated that cellular communication between NBs and surface glia is critical for development of both cell types. Gbb signaling is required autonomously for the proliferation of NBs as well as non-autonomously for the survival of PG. PG is essential for Gbb autocrine signaling. Given that the NB phenotype in PG > shits is associated with aberrant subcellular localization of Dlp in PG (Fig. 2L), one contribution of PG appears to be presenting Dlp on their surface, which likely potentiates Gbb autocrine signaling.
Alignment of amino acid sequences of Drosophila BMPs identified an HS-binding domain in Dpp and Gbb, but not Screw68. A truncated form of Dpp lacking this HS-binding fails to bind to its co-receptor Dally and was quickly degraded by receptor-mediated endocytosis69. This suggested that glypicans can enhance BMP signaling by disrupting receptor-mediated internalization and degradation of the Dpp-receptor complex. We have also shown that both Dally and Dlp can act as a trans co-receptor to enhance Gbb signaling in vitro11.
Expression of p35 by PG-Gal4 in gbb null mutants not only rescued PG but also partially restored NB proliferation. This observation suggests that additional PG-derived factor(s) contribute to NB proliferation. The most likely candidates for these PG factor(s) are activin-like ligands, whose role in NB division has been previously reported6. We identified specific expression of Daw in PG, supporting the idea that activin signaling together with Gbb regulates NB proliferation. Altogether, glia play a “niche-like” role, contributing to the microenvironment required for normal development of contacting NBs. Glia provide Dlp to support Gbb autocrine signaling, and express activin-like ligands as additional promoters of NB proliferation. Thus, we propose a model that bidirectional communication between NBs and glia through TGF-β signaling pathways influences each other’s development in the NSC niche.
Our study raises a fundamental question: how can PG and NBs communicate across the BBB and affect each other’s development? Our experiments using spGFP complementation favor a model in which NBs make direct contact with PG. Thus, it is possible that Dlp acts as a trans co-receptor for Gbb at the contacting sites. However, our study does not exclude other possibilities. For example, one can expect that transcytosis in SPG would allow molecular communications across this layer. Thus, the spGFP complementation signals we observed may result from the transport of membrane components of PG and NBs. It has been shown that lipoprotein particles can cross the Drosophila BBB60. The major Drosophila lipoprotein, lipophorin (Lpp), forms large particles with lipid molecules and transports them between different organs. In addition to lipid, Lpp particles carry sterol-linked and GPI-linked proteins. Dlp is a GPI-linked HSPG and has been shown to be incorporated into the Lpp particles70. This raises an interesting possibility that Dlp is transcytosed by the SPG through a Lpp-mediated mechanism.
Recent studies have shown that the surface glia are required to maintain the structural integrity of the neural lamella and to control the shape of the CNS71. A transcriptome study demonstrated that the surface glia express metabolic enzymes and signaling molecules at high levels in addition to various transporters, cell adhesion molecules, and ECM components72. Our study also supports the idea that the surface glia play a role not only as a simple structural insulators but a dynamic regulator of brain physiology development. Further study will be required, to fully elucidate the cellular and molecular mechanisms by which surface glia regulate the microenvironment internal to the BBB.
Methods
Fly stocks
Detailed information for the fly strains used is described in Flybase (http://flybase.bio.indiana.edu/) except where noted. Fly strains used were: dlp1 15, dlpA187 73, gbb1 28, gbb2, gbb4, dallygem 21, ttv524 74, baboFd4 33, pros-Gal4 (a gift from F. Matsuzaki), four Gal4 drivers for glial cells: NP6293-Gal4 (PG-Gal4), NP2276-Gal4 (SPG-Gal4), NP3233-Gal4, and NP2222-Gal438,65, daw-Gal46, gbb-Gal4, Mdr65-LexA (Bloomington Stock Center, BL61562), UAS-lacZ, UAS-dlp39.215, UAS-p35 (Bloomington Stock Center), UAS-dad58, UAS-gbb75, UAS-mCD8GFP76, UAS-ManII-eGFP77, UAS-shits 48, UAS-nlsGFP, UAS-eGFP-Dlg (a gift from V. Budnik), LexAop-rCD2::GFP64, LexAop2-mCD8GFP (Blooming Stock Center, BL32203), vkgG454 (viking::GFP)78, gbb-lacZ, dad-lacZ58, and UAS-mCD8ChRFP. Plasmid DNAs for gbb-LexAVP16, LexAop-spGFP1-10, and UAS-spGFP11 were constructed as described below and transgenic strains bearing these constructs were generated by Rainbow Transgenic Flies, CA.
For overexpression experiments using the Gal4/UAS system, flies were raised at 25 °C for 24 hours AEL and then incubated at 29 °C until the mid-third instar larval stage. To compromise temperature sensitive alleles, flies bearing either UAS-shits or gbb4 were exposed to temperature shift from 18 °C to 32 °C with water-bath until the stage we examined. Larvae were staged by counting the number of teeth on the mouth hook. Homozygotes and trans-heterozygotes were selected based on GFP expression from the CyO-GFP balancer or the Tb dominant marker of TM6B.
Immunostaining and imaging
The following antibodies were used: Guinea pig anti-β-galactosidase (1:500, a gift from Y. Hiromi), rat anti-β-galactosidase (1:500, a gift from Y. Hiromi), rabbit anti-β-galactosidase (1:500, Cappel), mouse anti-Repo (1:200, Developmental Studies Hybridoma Bank (DSHB) 8D12), mouse anti-Chaoptin (1:100, DSHB, 22B10), rat anti-Miranda (Mira) (1:100), guinea pig anti-Deadpan (Dpn) (1:1000), mouse anti-BrdU (1:250, Becton, Dickinson and Company), mouse anti-Dlp (1:4, DSHB, 13G8), rabbit anti-active caspase 3 (1:500, Abcam), mouse anti-Svp (1:4, a gift from Y. Hiromi), rabbit anti-pH3(1:1000, Upstate Biotechnology), and Alexa Fluor 488, 546, 633 conjugated secondary antibodies (1:500, Invitorgen).
Larval brains were dissected in PBS and fixed with 3.7% paraformaldehyde in PBS for 30 min at room temperature. After washing with PBST (0.1% triton X-100 in PBS), tissues were blocked with 5% normal goat serum in PBST for 30 min. The samples were incubated with primary antibodies at 4 °C overnight. After washing with PBS, the samples were incubated with secondary antibodies for 2 hours at room temperature. The tissues were mounted in Vectashield (VectaLabs) and fluorescent signals were observed by confocal laser scanning microscope (Zeiss LSM710) with a 40X water immersion objective.
BrdU incorporation
Larval brains were dissected in PBS and incubated with Schneider’s Drosophila medium (Invitrogen) containing 40 ng/ml of BrdU (Roche) for 30 min at room temperature, and subsequently with Schneider’s Drosophila medium without BrdU for 24 hours. Tissues were washed in PBST and fixed with 3.7% paraformaldehyde in PBT for 30 min at room temperature. After tissues were treated with 1 N HCl for 30 min at room temperature, immunostaining was performed as described above. Proliferation Index (PI) was calculated as follows. The number of BrdU positive neurons from NBs in experimental brains was divided by that of wild-type control brains (yw or Oregon R). The control brains were labeled with BrdU in the same tube with experimental brain samples and the genotypes were distinguished by y+ or y− mouth hook.
In vitro characterization of Gbb
Drosophila S2 cells were grown in Schneider’s medium (Invitrogen) supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin. Transient transfections were performed using Effectene (Qiagen) as per manufacturer’s instructions.
There are multiple proteolytic processing sites in Gbb, which produce distinct mature ligand forms, including gbb15 and Gbb38, and the amount of each product expressed in S2 cells is generally low. When a few cleavage sites are blocked, a larger amount of Gbb38 is produced59. S2 cells were transfected with pAW-Gbb[mS1mS0]-HA and incubated for 72 hours. The conditioned medium containing Gbb38-HA was incubated with heparin sepharose CL-6B (Amersham Biosciences) for 4 hours. After extensive washing of unbound proteins, a heparin-bound fraction was eluted by 0.25 M NaCl and analyzed by immunoblotting using anti-HA antibody.
For cellular immuno-colocalization analysis, S2 cells expressing Dlp-Myc and Gbb-HA were incubated with anti-HA and anti-Myc in a Concanavalin A-coated coverslip for 30 minutes at 25 °C, washed, and fixed with 3.7% formaldehyde. Dlp-Myc and Gbb-HA were detected as described before79.
Plasmid construction
The original plasmids of spGFP fragments, ace-4p CD4-2 spGFP1-10 and rig-3p CD4-2 spGFP11, were provided by C. Bargmann62. To optimize the spGFP constructs for Drosophila, the original signal peptide (Par3) was replaced with the Drosophila myospheroid signal peptide (1-28). The spGFP11 and spGFP1-10 fragments were amplified by PCR using primers 1 and 3, and primers 2 and 3, respectively (see below), and subcloned into the pENTR/D-TOPO Gateway entry vector (Invitrogen). PCR fragments were verified by DNA sequencing. The destination vectors used for germline transformation were pTW from the Drosophila Gateway Vector Collection and pLot W from S. Diegelmann80.
gbb-gal4 and gbb-lacZ constructs were made by ligating gal4 and lacZ sequences downstream of the 5′ genomic sequences of the gbb gene, respectively, and subcloned into pCaSpeR3. gbb-LexAVP16 was constructed in the same manner after the LexAVP16 fragment was amplified by PCR using primers 4 and 5.
Primer 1: 5′-CACCATGATCCTCGAGAGAAACCGGAGGTGCCAGCTGGCCCTCCTCATGATCGCAATACTGGCCGCCATCGCTGGACAAACGGATGCCCAGCGTGACCACATGGTCCTTCATGAGTATGTAAATGC-3′
Primer 2: 5′-CACCATGATCCTCGAGAGAAACCGGAGGTGCCAGCTGGCCCTCCTCATGATCGCAATACTGGCCGCCATCGCTGGACAAACGGATGCCCAGATGTCCAAAGGAGAAGAACTGTTTACC-3′
Primer 3: 5′-CTAGCGCCTTCGGTGCCGG-3′
Primer 4: 5′-GTGGCGGCCGCTCCGTCCAAAACACGAAATAATGAAAGCGTTAACGGCCAGGCAACAAGAG-3′
Primer 5: 5′-GTGGCGGCCGCCTACCCACCGTACTCGTCAATTCCAAGGGC-3′
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files.
References
Sousa-Nunes, R., Cheng, L. Y. & Gould, A. P. Regulating neural proliferation in the Drosophila CNS. Curr Opin Neurobiol 20, 50–57, https://doi.org/10.1016/j.conb.2009.12.005 (2010).
Homem, C. C., Repic, M. & Knoblich, J. A. Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 16, 647–659, https://doi.org/10.1038/nrn4021 (2015).
Homem, C. C. & Knoblich, J. A. Drosophila neuroblasts: a model for stem cell biology. Development 139, 4297–4310, https://doi.org/10.1242/dev.080515 (2012).
Egger, B., Chell, J. M. & Brand, A. H. Insights into neural stem cell biology from flies. Philos Trans R Soc Lond B Biol Sci 363, 39–56, 90045L4172216858 https://doi.org/10.1098/rstb.2006.2011 (2008).
Siegrist, S. E. & Doe, C. Q. Extrinsic cues orient the cell division axis in Drosophila embryonic neuroblasts. Development 133, 529–536, 10.1242/dev.02211 (2006).
Zhu, C. C. et al. Drosophila Activin- and the Activin-like product Dawdle function redundantly to regulate proliferation in the larval brain. Development 135, 513–521 (2008).
Dumstrei, K., Wang, F. & Hartenstein, V. Role of DE-cadherin in neuroblast proliferation, neural morphogenesis, and axon tract formation in Drosophila larval brain development. J Neurosci 23, 3325–3335 (2003).
Ebens, A. J., Garren, H., Cheyette, B. N. & Zipursky, S. L. The Drosophila anachronism locus: a glycoprotein secreted by glia inhibits neuroblast proliferation. Cell 74, 15–27, 0092-8674(93)90291-W [pii] (1993).
Hayashi, Y., Kobayashi, S. & Nakato, H. Drosophila glypicans regulate the germline stem cell niche. J Cell Biol 187, 473–480 (2009).
Guo, Z. & Wang, Z. The glypican Dally is required in the niche for the maintenance of germline stem cells and short-range BMP signaling in the Drosophila ovary. Development 136, 3627–3635, https://doi.org/10.1242/dev.036939 (2009).
Dejima, K., Kanai, M. I., Akiyama, T., Levings, D. C. & Nakato, H. Novel contact-dependent bone morphogenetic protein (BMP) signaling mediated by heparan sulfate proteoglycans. J Biol Chem 286, 17103–17111, https://doi.org/10.1074/jbc.M110.208082 (2011).
Levings, D. C., Arashiro, T. & Nakato, H. Heparan sulfate regulates the number and centrosome positioning of Drosophila male germline stem cells. Mol Biol Cell 27, 888–896, https://doi.org/10.1091/mbc.E15-07-0528 (2016).
Takemura, M. & Nakato, H. Drosophila Sulf1 is required for the termination of intestinal stem cell division during regeneration. J Cell Sci 130, 332–343, https://doi.org/10.1242/jcs.195305 (2017).
Levings, D. C. & Nakato, H. Loss of heparan sulfate in the niche leads to tumor-like germ cell growth in the Drosophila testis. Glycobiology, https://doi.org/10.1093/glycob/cwx090 (2017).
Kirkpatrick, C. A., Dimitroff, B. D., Rawson, J. M. & Selleck, S. B. Spatial regulation of Wingless morphogen distribution and signaling by Dally-like protein. Dev Cell 7, 513–523 (2004).
Bishop, J. R., Schuksz, M. & Esko, J. D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 (2007).
Xu, D. & Esko, J. D. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem 83, 129–157, https://doi.org/10.1146/annurev-biochem-060713-035314 (2014).
Nakato, H. & Kimata, K. Heparan sulfate fine structure and specificity of proteoglycan functions. Biochim Biophys Acta 1573, 312–318 (2002).
Nakato, H. & Li, J. P. Functions of Heparan Sulfate Proteoglycans in Development: Insights From Drosophila Models. Int Rev Cell Mol Biol 325, 275–293, https://doi.org/10.1016/bs.ircmb.2016.02.008 (2016).
Fujise, M. et al. Dally regulates Dpp morphogen gradient formation in the Drosophila wing. Development 130, 1515–1522 (2003).
Tsuda, M. et al. The cell-surface proteoglycan Dally regulates Wingless signalling in Drosophila. Nature 400, 276–280, https://doi.org/10.1038/22336 (1999).
Kramer, K. L. & Yost, H. J. Ectodermal syndecan-2 mediates left-right axis formation in migrating mesoderm as a cell-nonautonomous Vg1 cofactor. Dev Cell 2, 115–124 (2002).
Jakobsson, L. et al. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell 10, 625–634 (2006).
Peterson, A. J. & O’Connor, M. B. Strategies for exploring TGF-beta signaling in Drosophila. Methods 68, 183–193, https://doi.org/10.1016/j.ymeth.2014.03.016 (2014).
Irish, V. F. & Gelbart, W. M. The decapentaplegic gene is required for dorsal-ventral patterning of the Drosophila embryo. Genes Dev 1, 868–879 (1987).
Arora, K., Levine, M. S. & O’Connor, M. B. The screw gene encodes a ubiquitously expressed member of the TGF-beta family required for specification of dorsal cell fates in the Drosophila embryo. Genes Dev 8, 2588–2601 (1994).
Spencer, F. A., Hoffmann, F. M. & Gelbart, W. M. Decapentaplegic: a gene complex affecting morphogenesis in Drosophila melanogaster. Cell 28, 451–461 (1982).
Wharton, K. A. et al. Genetic analysis of the bone morphogenetic protein-related gene, gbb, identifies multiple requirements during Drosophila development. Genetics 152, 629–640 (1999).
Chen, S., Wang, S. & Xie, T. Restricting self-renewal signals within the stem cell niche: multiple levels of control. Curr Opin Genet Dev 21, 684–689, https://doi.org/10.1016/j.gde.2011.07.008 (2011).
Peterson, A. J. & O’Connor, M. B. Activin receptor inhibition by Smad2 regulates Drosophila wing disc patterning through BMP-response elements. Development 140, 649–659, https://doi.org/10.1242/dev.085605 (2013).
Peterson, A. J. et al. R-Smad competition controls activin receptor output in Drosophila. PLoS One 7, e36548, https://doi.org/10.1371/journal.pone.0036548 (2012).
Brummel, T. et al. The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev 13, 98–111 (1999).
Zheng, X. et al. TGF-beta signaling activates steroid hormone receptor expression during neuronal remodeling in the Drosophila brain. Cell 112, 303–315 (2003).
Awasaki, T., Huang, Y., O’Connor, M. B. & Lee, T. Glia instruct developmental neuronal remodeling through TGF-beta signaling. Nat Neurosci 14, 821–823, https://doi.org/10.1038/nn.2833 (2011).
Parker, L., Ellis, J. E., Nguyen, M. Q. & Arora, K. The divergent TGF-beta ligand Dawdle utilizes an activin pathway to influence axon guidance in Drosophila. Development 133, 4981–4991, https://doi.org/10.1242/dev.02673 (2006).
Serpe, M. & O’Connor, M. B. The metalloprotease tolloid-related and its TGF-beta-like substrate Dawdle regulate Drosophila motoneuron axon guidance. Development 133, 4969–4979, https://doi.org/10.1242/dev.02711 (2006).
Ghosh, A. C. & O’Connor, M. B. Systemic Activin signaling independently regulates sugar homeostasis, cellular metabolism, and pH balance in Drosophila melanogaster. Proc Natl Acad Sci USA 111, 5729–5734, https://doi.org/10.1073/pnas.1319116111 (2014).
Awasaki, T., Lai, S. L., Ito, K. & Lee, T. Organization and postembryonic development of glial cells in the adult central brain of Drosophila. J Neurosci 28, 13742–13753 (2008).
Ito, K., Urban, J. & Technau, G. Distribution, classification, and development ofDrosophila glial cells in the late embryonic and early larval ventral nerve cord. Development Genes and Evolution 204, 284–307 (1995).
Sasse, S., Neuert, H. & Klambt, C. Differentiation of Drosophila glial cells. Wiley Interdiscip Rev Dev Biol 4, 623–636, https://doi.org/10.1002/wdev.198 (2015).
Banerjee, S. & Bhat, M. A. Neuron-glial interactions in blood-brain barrier formation. Annu Rev Neurosci 30, 235–258 (2007).
Stork, T. et al. Organization and function of the blood-brain barrier in Drosophila. J Neurosci 28, 587–597 (2008).
Schirmeier, S. & Klambt, C. The Drosophila blood-brain barrier as interface between neurons and hemolymph. Mech Dev 138(Pt 1), 50–55, https://doi.org/10.1016/j.mod.2015.06.002 (2015).
Hindle, S. J. & Bainton, R. J. Barrier mechanisms in the Drosophila blood-brain barrier. Front Neurosci 8, 414, https://doi.org/10.3389/fnins.2014.00414 (2014).
Limmer, S., Weiler, A., Volkenhoff, A., Babatz, F. & Klambt, C. The Drosophila blood-brain barrier: development and function of a glial endothelium. Front Neurosci 8, 365, https://doi.org/10.3389/fnins.2014.00365 (2014).
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R. & Begley, D. J. Structure and function of the blood-brain barrier. Neurobiol Dis 37, 13–25, https://doi.org/10.1016/j.nbd.2009.07.030 (2010).
Abbott, N. J. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol 25, 5–23 (2005).
Kitamoto, T. Conditional modification of behavior in Drosophila by targeted expression of a temperature-sensitive shibire allele in defined neurons. Journal of Neurobiology 47, 81–92 (2001).
Pelissier, A., Chauvin, J. P. & Lecuit, T. Trafficking through Rab11 endosomes is required for cellularization during Drosophila embryogenesis. Curr Biol 13, 1848–1857 (2003).
Awasaki, T. & Ito, K. Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr Biol 14, 668–677 (2004).
Chell, J. M. & Brand, A. H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell 143, 1161–1173, S0092-8674(10)01425-X [pii]https://doi.org/10.1016/j.cell.2010.12.007 (2010).
Awasaki, T. & Lee, T. New tools for the analysis of glial cell biology in Drosophila. Glia, https://doi.org/10.1002/glia.21133 (2011).
Akong, K. et al. Drosophila APC2 and APC1 play overlapping roles in wingless signaling in the embryo and imaginal discs. Dev Biol 250, 91–100 (2002).
Barrett, A. L., Krueger, S. & Datta, S. Branchless and Hedgehog operate in a positive feedback loop to regulate the initiation of neuroblast division in the Drosophila larval brain. Dev Biol 317, 234–245 (2008).
Park, Y. et al. Drosophila perlecan modulates FGF and hedgehog signals to activate neural stem cell division. Dev Biol 253, 247–257 (2003).
Yoshida, S. et al. DPP signaling controls development of the lamina glia required for retinal axon targeting in the visual system of Drosophila. Development 132, 4587–4598 (2005).
Marques, G. et al. Retrograde Gbb signaling through the Bmp type 2 receptor wishful thinking regulates systemic FMRFa expression in Drosophila. Development 130, 5457–5470 (2003).
Tsuneizumi, K. et al. Daughters against dpp modulates dpp organizing activity in Drosophila wing development. Nature 389, 627–631 (1997).
Anderson, E. N. & Wharton, K. A. Alternative cleavage of the bone morphogenetic protein (BMP), Gbb, produces ligands with distinct developmental functions and receptor preferences. J Biol Chem 292, 19160–19178, https://doi.org/10.1074/jbc.M117.793513 (2017).
Brankatschk, M. & Eaton, S. Lipoprotein particles cross the blood-brain barrier in Drosophila. J Neurosci 30, 10441–10447, https://doi.org/10.1523/JNEUROSCI.5943-09.2010 (2010).
Ghosh, I., Hamilton, A. D. & Regan, L. Antiparallel Leucine Zipper-Directed Protein Reassembly:*≠* Application to the Green Fluorescent Protein. Journal of the American Chemical Society 122, 5658–5659 (2000).
Feinberg, E. H. et al. GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353–363 (2008).
Brand, A. H. & Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 (1993).
Lai, S. L. & Lee, T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat Neurosci 9, 703–709 (2006).
Hayashi, S. et al. GETDB, a database compiling expression patterns and molecular locations of a collection of Gal4 enhancer traps. Genesis 34, 58–61 (2002).
Bailey, A. P. et al. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 163, 340–353, https://doi.org/10.1016/j.cell.2015.09.020 (2015).
Volkenhoff, A. et al. Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab 22, 437–447, https://doi.org/10.1016/j.cmet.2015.07.006 (2015).
Kunnapuu, J., Bjorkgren, I. & Shimmi, O. The Drosophila DPP signal is produced by cleavage of its proprotein at evolutionary diversified furin-recognition sites. Proc Natl Acad Sci USA 106, 8501–8506, https://doi.org/10.1073/pnas.0809885106 (2009).
Akiyama, T. et al. Dally regulates Dpp morphogen gradient formation by stabilizing Dpp on the cell surface. Dev Biol 313, 408–419, https://doi.org/10.1016/j.ydbio.2007.10.035 (2008).
Eugster, C., Panakova, D., Mahmoud, A. & Eaton, S. Lipoprotein-heparan sulfate interactions in the Hh pathway. Dev Cell 13, 57–71, https://doi.org/10.1016/j.devcel.2007.04.019 (2007).
Meyer, S., Schmidt, I. & Klambt, C. Glia ECM interactions are required to shape the Drosophila nervous system. Mech Dev 133, 105–116, https://doi.org/10.1016/j.mod.2014.05.003 (2014).
DeSalvo, M. K., Mayer, N., Mayer, F. & Bainton, R. J. Physiologic and anatomic characterization of the brain surface glia barrier of Drosophila. Glia 59, 1322–1340, https://doi.org/10.1002/glia.21147 (2011).
Han, C., Belenkaya, T. Y., Wang, B. & Lin, X. Drosophila glypicans control the cell-to-cell movement of Hedgehog by a dynamin-independent process. Development 131, 601–611 (2004).
Takei, Y., Ozawa, Y., Sato, M., Watanabe, A. & Tabata, T. Three Drosophila EXT genes shape morphogen gradients through synthesis of heparan sulfate proteoglycans. Development 131, 73–82, https://doi.org/10.1242/dev.00913 (2004).
Khalsa, O., Yoon, J. W., Torres-Schumann, S. & Wharton, K. A. TGF-beta/BMP superfamily members, Gbb-60A and Dpp, cooperate to provide pattern information and establish cell identity in the Drosophila wing. Development 125, 2723–2734 (1998).
Lee, T. & Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461 (1999).
Zheng, Y. et al. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol 10, 1172–1180 (2008).
Morin, X., Daneman, R., Zavortink, M. & Chia, W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci USA 98, 15050–15055 (2001).
Kleinschmit, A. et al. Drosophila heparan sulfate 6-O endosulfatase regulates Wingless morphogen gradient formation. Dev Biol 345, 204–214, https://doi.org/10.1016/j.ydbio.2010.07.006 (2010).
Diegelmann, S., Bate, M. & Landgraf, M. Gateway cloning vectors for the LexA-based binary expression system in Drosophila. Fly (Austin) 2 (2008).
Acknowledgements
We thank C. Kirkpatrick, F. Matsuzaki, Y. Jan, T. Lee, Y. Hiromi, C. Bargmann, S. Diegelmann, V. Budnik, the Developmental Studies Hybridoma Bank, and the Bloomington Stock Center for fly stocks and reagents. We are grateful to members of Nakato lab and O’Connor lab for valuable comments on the manuscript. This work was partly supported by the National Institute of Health (R01 GM115099) and Mizutani Foundation for Glycoscience to H.N.
Author information
Authors and Affiliations
Contributions
H.N. and M.O. designed the project. M.I.K., and M.K. conducted the experiments. M.T. performed in vitro assays. M.I.K., M.K., and H.N. analyzed the results. T.A. and K.W. provided critical unpublished materials. M.I.K. and H.N. wrote and all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kanai, M.I., Kim, MJ., Akiyama, T. et al. Regulation of neuroblast proliferation by surface glia in the Drosophila larval brain. Sci Rep 8, 3730 (2018). https://doi.org/10.1038/s41598-018-22028-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22028-y
This article is cited by
-
An original infection model identifies host lipoprotein import as a route for blood-brain barrier crossing
Nature Communications (2020)
-
Functional analysis of glycosylation using Drosophila melanogaster
Glycoconjugate Journal (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
