
Abstract
Hydrogen sulfide (H2S, 80 ppm) gas in an atmosphere of 17.5% oxygen reportedly induces suspended animation in mice; a state analogous to hibernation that entails hypothermia and hypometabolism. However, exogenous H2S in combination with 17.5% oxygen is able to induce hypoxia, which in itself is a trigger of hypometabolism/hypothermia. Using non-invasive thermographic imaging, we demonstrated that mice exposed to hypoxia (5% oxygen) reduce their body temperature to ambient temperature. In contrast, animals exposed to 80 ppm H2S under normoxic conditions did not exhibit a reduction in body temperature compared to normoxic controls. In conclusion, mice induce hypothermia in response to hypoxia but not H2S gas, which contradicts the reported findings and putative contentions.
Similar content being viewed by others

Introduction
Hibernation is a hypometabolic state characterized by a regulated decrease in core body temperature (Tb) (i.e., hypothermia) towards ambient temperature (Ta) and consequent reduction in oxygen (O2) consumption and carbon dioxide (CO2) production. It is engaged by several mammalian species1 to protect the organism from (environmental) stressors such as extreme cold, hypoxia2,3, and starvation4,5 and ultimately death.
The regulated decrease in Tb, which is termed anapyrexia, encompasses the downmodulation of the ‘internal thermostat’ outside of the thermoneutral zone6,7,8,9,10. The thermoneutral zone constitutes a temperature range in which heat production (from basal metabolism) is in equilibrium with heat loss to the environment. The organism functions best when the Tb resides in the thermoneutral zone, but engages anapyrexia as a coping mechanism. How the anapyrexic signaling is biochemically and physiologically regulated and how the ‘internal thermostat’ is circumvented is largely elusive and hypothetical, but the ultimate outcome is unequivocally a state of hypometabolism. The natural purpose of the hypometabolism is to temporarily realign energy needs with reduced energy/O2 supply under conditions of stress in order to sustain life under circumstances that could otherwise have lethal consequences.
The state of cold hypometabolism is believed to be a result of systematic deviation from homeothermy, which in turn is caused by a reduction in or cessation of metabolism. The resulting hypothermia assists, or propagates, the hypometabolic state in accordance with Arrhenius’ law. This law states that the rate of chemical reactions (i.e., metabolism) decreases when the temperature decreases11,12. Consequently, both the consumption of substrate (in this case O2) and the formation of product (in this case CO2, toxic metabolites such as lactate, and reactive O2 species) are reduced during hypothermia, as has been confirmed in natural hibernators during hibernation in terms of expired CO21. The alignment of metabolic demand with supply as well as the decreased formation of cytotoxic metabolites confer sustenance of life and cytoprotection in the stress-exposed organism.
In line with the above, mimicking these natural phenomena in non-hibernators such as humans by artificially inducing hypometabolism holds tremendous potential in medicine, aviation and space travel, and sports. An artificially induced hypometabolic state has been hypothesized to impart similar protective effects on otherwise stressed cells. Accordingly, numerous studies have focused on identifying agents that are capable of inducing hypometabolism in non-hibernating mammals (i.e., anapyrexic agents), which have yielded 5′-AMP13,14, DADLE15,16, 2-deoxyglucose5,17, thyronamines18,19, and exogenous hydrogen sulfide (H2S)20 as potential anapyrexic agents. Of these, exogenous H2S has received the most attention in the last few years in response to the Science publication by Blackstone et al.20. However, our experiments in mice, which duplicated the experiments by Blackstone et al.20, revealed that exogenous H2S does not induce hypothermia at normoxic conditions. Instead, the hypothermia observed in the experiments emanates from a hypoxia-induced anapyrexic response, which is a natural response in mice to hypoxic stress2,3. The results are described in this paper and addressed in the context of artificial hypometabolism.
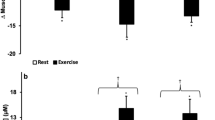
Exogenous H2S has been proposed to induce hypometabolism that is associated with a state of suspended animation20. Mice that were subjected to a gas mixture composed of 17.5% O2, 80% nitrogen (N2), and 80 ppm H2S exhibited a 22 °C reduction in Tb (Fig. 1A) after 4 h of exposure, yielding a Tb that was slightly above the Ta of 13 °C. At this point CO2 production and O2 consumption had decreased by approximately 90%, suggesting that the animals had reached a state of hypometabolism by anapyrexia. Moreover, this state was reversible inasmuch as all metabolic parameters reverted to baseline within 4 h after the exposure to H2S was abrogated. During this recovery period the Tb also gradually restored to baseline at a Ta of 24 °C. In another study by Volpato et al., inhalation of air containing 17.5% O2 and 80 ppm H2S induced similar anapyrexic effects in mice at a Ta of 27 °C as well as 35 °C (Fig. 1B) 21, altogether suggesting that inhaled H2S reduces the Tb to Ta levels.

Previously reported temperature effects of H2S. Temperature effects of inhaled H2S gas (80 ppm) on the Tb of mice as a function of exposure time as reported by Blackstone et al.20 (A) and Volpato et al.21 (B). In (A) mice were exposed to 80 ppm H2S and 17.5% O2 (n = 7) or 17.5% O2 (n = 4) for 6 h, followed by a recovery phase at 17.5% O2 in both groups (6–10 h, right part of red vertical line). The Ta was decreased during the exposure phase (dotted line). In (B) similar experiments were performed as in (A) but at fixed Tas of 27 °C (closed diamonds, n = 3) or 35 °C (open diamonds, n = 4). The 6-h H2S exposure phase was followed by a 3-h recovery phase in air at a Ta of 27 °C (6–9 h, right part of red vertical line). Data modified from20,21.
The mechanism behind exogenous H2S-induced suspended animation20,21 is generally ascribed to the direct inhibition of oxidative phosphorylation22 and consequent histotoxic hypoxia. Because of is its high membrane permeability, H2S is readily delivered to tissues via the circulation where it transgresses cell membranes and localizes to various intracellular organelles, including mitochondria23. H2S binds cytochrome c oxidase (complex IV) in the electron transport chain in a reversible and noncompetitive fashion. As a result, H2S prevents O2 binding to cytochrome c oxidase and thereby interferes with the reduction of O2 to water. Concurrently, H2S interferes with the production of adenosine triphosphate (ATP) by ATPase due to H2S-induced perturbation of electron transfer and proton gradient over the mitochondrial inner membrane24,25. It should be noted, however, that H2S-mediated histotoxic hypoxia has never been proven to directly translate to H2S-induced hypothermia. Similarly, experimental evidence that H2S triggers a downward shift of the thermoneutral zone directly remains at large.
Although the hypometabolic effects of exogenous H2S seem convincing, the putative mechanism for the hypometabolic state induced by exogenous H2S, i.e., cytochrome c oxidase inhibition22, may not account for the observed effects. As H2S is a toxic, irritant gas22, inhalation is known to provoke epithelial damage in the upper26 and lower respiratory tract27,28 in rats and pulmonary edema in pigs29,30. The pulmonotoxicity of exogenous H2S may therefore be associated with hypoxemic hypoxia.
Hypoxia, on the other hand, is a very potent inducer of anapyrexia, hypothermia, and hypometabolism and, thereby, of suspended animation3. Several hibernating and non-hibernating mammalian species, including mice, exposed to different degrees of hypoxic atmospheres (i.e., FiO2 5–10%) immediately drop their Tb to enter a reversible state of hypometabolism31,32,33,34. The hypothermic effects of hypoxia are known to be caused by downward adjustment of the ‘internal thermostat,’ and involve the preoptic anterior hypothalamus (POAH), as has been demonstrated in thermobehavioral experiments in rodents2. Consequently, we proposed that the hypothermia in exogenous H2S-exposed mice, which constitutes a hallmark feature of hypometabolism, emanated from the combination of mild hypoxia (17.5% O2) and inhalation of H2S gas, and not the exogenous H2S gas per se.
Results
To test the hypothesis that exogenous H2S-induced hypothermia emanates from hypoxia and not H2S, we performed experiments in 48 female C57BL/6 mice using a similar approach as was employed by Blackstone et al.20. The experiments, which are outlined in Fig. 2 and the Materials & Methods section of this paper, encompassed the following groups: (A) 80 ppm H2S in 21% O2 and 79% N2 (H2S in 21% O2 group; N = 12 mice); (B) 80 ppm H2S in 17% O2 and 83% N2 (H2S in 17% O2 group; N = 6 mice); (C) 5% O2 and 95% N2 (5% O2 group; N = 12 mice); (D) 17% O2 and 83% N2 (17% O2 group; N = 6 mice); and (E) 21% O2 and 79% N2 (normoxia group; N = 12 mice). The effects of exogenous H2S and a hypoxic atmosphere on Tb at a Ta of ~21 °C were measured non-invasively with a thermographic camera and the locomotor activity of the animals was quantitated with dedicated motion analysis software.

Schematic illustration of the experimental setup and design. The animals were allocated to one of the following experimental groups: (A) 80 ppm H2S in 21% O2 and 79% N2 (H2S in 21% O2 group, N = 12); (B) 80 ppm H2S in 17% O2 and 83% N2 (H2S in 17% O2 group, N = 6); (C) 5% O2 and 95% N2 (5% O2 group, N = 12); (D) 17% O2 and 83% N2 (17% O2 group, N = 6); and (E) 21% O2 and 79% N2 (normoxia group, N = 12). The experiments were performed at a mean ± SD Ta of 21.2 ± 0.6 °C, measured with a thermistor. During the whole experiment the mice were solitarily housed in a custom-built airtight cage and recorded with a thermographic camera (CAM) for 10 min every hour (red markers) for skin temperature- and locomotor activity analysis. After 1 h of baseline 21:79% O2:N2 exposure, each mouse was exposed to a gas mixture (A–E) for 6 h that was passed through the airtight cage, after which 6 of the animals in each group were allowed to recover at 21:79% O2:N2 for 3 h. The other 6 animals of group (A,C and E) were sacrificed for another study.
As shown in Fig. 3 and Supplemental Video S1, hypothermia and reduction in locomotor activity only occurred in mice subjected to hypoxic conditions. 5% O2-exposed animals immediately dropped their Tb to approximately 2 °C above the Ta (Fig. 3B, P < 0.0001) and reduced their locomotor activity to nearly nil compared to the H2S in FiO2 21% and normoxia groups (Fig. 3C, P < 0.0001) during the entire exposure period. The exogenous H2S in 21% FiO2 group did not differ from the normoxia group during 6 h of 80 ppm H2S gas exposure in neither superficial temperature nor locomotor activity. At 3 h of exposure, however, animals in the H2S in 17% FiO2 group started to drop their Tb to approximately 4 °C above Ta, in contrast to FiO2 17%-exposed control animals (Fig. 3B, P < 0.0001). Alleviation of the hypoxic conditions during the restoration phase resulted in complete reversal of the superficial temperature to baseline levels within 1 h in the FiO2 5% group, which is in agreement with previous reports20,21 (Fig. 1). During the 3 h of restoration at normoxic atmosphere, the H2S in FiO2 17% -exposed animals remained hypothermic and only restored Tb to the level of the FiO2 17% and 21% control groups at 9 h (Fig. 3B, P < 0.01). Mice in the H2S groups exhibited some discomfort during H2S exposure, as evidenced by the cringed posture, which occasionally concurred with vigorous locomotion (Supplemental Video S1).

Temperature and locomotor effects of H2S compared to hypoxia. (A) Thermal images of H2S-exposed mice in 21% O2 (top row), in 17% O2 (middle row), and hypoxia-subjected mice (bottom row) before (baseline), during, and after (recovery) exposure. The color of the animals reflects their temperature (scale bar). (B) The difference between the maximum superficial temperature (Δ max. superficial T) of the hypoxia (FiO2 5% and 17%) and H2S (FiO2 17% and 21%) groups versus the normoxia group (FiO2 21%) was plotted as a function of time before exposure (up to 0 h), during exposure (0–6 h), and after exposure (6–9 h). (C) Mouse mean locomotor activity per time point per group plotted as a function of time before exposure (up to 0 h), during exposure (0–6 h), and after exposure (6–9 h). Locomotor activity was derived from temporal changes in pixel grayscale intensity as described in the online supplemental information. In (B) and (C) the means ± SEM are plotted for N = 12/group (group A, C and E) or N = 6/group (group B and D) up to 6 h, and for N = 6/group from 6 to 9 h. Statistically significant intergroup differences are displayed under the corresponding plots.
Peripheral vasodilation is one of the cooling mechanisms that is autonomically regulated in response to a mismatch between the Tb and the internal thermostat (i.e., Tb > thermoneutral zone)3,35,36. Peripheral vasodilation is integral to anapyrexia3, which enables cooling. The cooling process is in turn facilitated by the blockade of thermogenic effectors and the enabling of peripheral vasodilation36,37,38. Therefore, the extent of peripheral vasodilation was determined by measuring the change in tail temperature at baseline and at approximately 4 min after initiation of H2S- or hypoxia exposure.
The tail of 5% O2-exposed animals warmed up right after the start of exposure (+2.1 ± 0.5 °C, N = 3, P < 0.05 versus the H2S group, unpaired student’s t-test), while the tail of H2S in 21% O2-exposed animals (−1.3 ± 1.1 °C, N = 3) and normoxia-exposed animals (+0.1 °C, N = 1) did not exhibit changes in temperature (P > 0.05, unpaired student’s t-test) (Fig. 4). These results provide compelling evidence for the induction of peripheral vasodilation by hypoxia but not exogenous H2S, and hence for hypoxia-mediated anapyrexic signaling. The absence of a vasodilatory response in the exogenous H2S group is in agreement with the surface temperature data, which encompassed an absence of hypothermia (Fig. 3).

Tail temperatures in H2S and hypoxia-exposed mice. Representative thermographic camera images of mice approximately 4 min after initiation of exposure to H2S in 21% O2 (left), 5% O2 (middle), or normoxia (right). Yellow indicates a high surface temperature (31 °C), blue indicates a low surface temperature (20 °C) as indicated by the scale bar. Note the difference in tail temperature (on the basis of yellow intensity) of the hypoxic animal versus the H2S gas-exposed and normoxic animals. The warmer tail in the hypoxic mouse is indicative of peripheral vasodilation; a cooling effector that is induced by anapyrexia.
Discussion
Based on the experimental evidence, namely Tb, tail temperature, and locomotion, it can be concluded that inhalation of H2S gas at 80 ppm in a native atmosphere of 21% O2 and 79% N2 does not induce hypothermia in mice, which contradicts what has been reported previously20,21. Hypoxia, on the other hand, is a very potent inducer of hypothermia that, given the peripheral vasodilation observed in the tail vasculature, may comprise part of an anapyrexic response2,3. The subclinical thermal effects of mild hypoxia, however, are potentiated by combined 80 ppm H2S gas exposure.
One consistent finding in mouse studies on the pharmacological induction of hypothermia is that the animal’s Tb or surface temperature approximates the Ta and subsequently enters a plateau phase that is sustained in the vicinity of Ta. Regardless of what actually caused the hypothermic signaling in the experiments by Blackstone et al.20 and Volpato et al.21, the Tb was in all instances downmodulated to a depth at which the Tb was more or less in equilibrium with the Ta, irrespective of the magnitude of the Ta (i.e., 13 °C, 27 °C, or 35 °C). The same pattern was observed in our experiments (Ta = 21 °C), suggesting that the hypothermia may have been mediated via a common mechanism. Moreover, this decline-plateau pattern suggests that the cooling process is passive once the thermogenic effectors have been shut off. The cooling is halted upon reaching a thermodynamic equilibrium where Tb = Ta, i.e., a point at which the organism is not equipped to cool further. Unlike under normophysiological circumstances, where Tb is tightly regulated via engagement of cooling effectors or thermogenic effectors35,39, the hypothermic state seems to sustain itself through passive heat transfer only.
The main differences between the results of Blackstone et al.20, Volpato et al.21, and our results are the rate of cooling and subsequently the time required to reach the plateau phase (Tb = Ta). The cooling rate was approximately 1.3 °C/h and 4.0 °C/h in the experiments of Volpato et al. and Blackstone et al., respectively, whereas in our experiments the cooling rate was approximately 5.3 °C/h. The convergence of T b with T a required ~6 h in the study of Blackstone et al.20, ~4 h in the study of Volpato et al.21, and 2 h in our study (Fig. 3). The same animal species with similar animal weights were employed in all studies. Hence, it is unlikely that these discrepancies arose from differences related to physical laws such as Galilei’s square-cube law40, the implication of which is that animals with a large body surface:mass ratio (i.e., small animals) cool faster than animals with a small body surface:mass ratio (i.e., large animals)1. The discrepancies in cooling rate also did not emanate from differences in metabolism in accordance with Kleiber’s law, which states that small animals exhibit a relatively higher metabolic rate to maintain euthermia compared to larger animals1,41.
In light of the finding that exogenous H2S is not an inducer of hypothermia, the question that remains to be answered is “why did Blackstone et al. and Volpato et al. observe hypothermia in H2S-exposed mice?” Volpato et al. was able to reproduce the hypothermic effects of 80 ppm H2S of Blackstone et al. Consequently, we do not question the methodology and validity of their results. In our opinion, the answer lies in the hypoxic conditions that were induced by the combination of subatmospheric FiO2 and the various mild forms of exogenous H2S-induced hypoxia. The 3.5% lower FiO2 versus native atmospheric FiO2 (17.5% versus 21%, respectively) is, in itself, not sufficient to trigger anapyrexia in mice, unless such mild hypoxic conditions are exacerbated by exogenous H2S. In line with our results obtained in the 17% FiO2 groups, the exacerbation likely occurred in the experiments by Blackstone et al. and Volpato et al. for four possible reasons. First, as explained in the Introduction section, H2S can induce histotoxic hypoxia by inhibiting cytochrome c oxidase and corollary ATP production, resulting in reduced metabolic supply (energy). Consequently, the organism is forced to adapt its metabolic demand to survive by means of e.g., hypothermia (Arrhenius’ law). Secondly, H2S can limit the binding of O2 to hemoglobin’s O2 binding sites42, thereby causing O2 affinity hypoxia14. Thirdly, H2S reduces cardiac output through its deregulatory and negative chronotropic effects on cardiac rhythm21,28, which leads to circulatory hypoxia43. Fourthly, H2S is pulmonotoxic26,27,28 and may impair pulmonary O2/CO2 exchange and the extent of O2 saturation, which in turn may aggravate the circulatory hypoxia caused by the cardiovascular effects. In addition, based on ex vivo experiments, H2S seems to play an essential role in hypoxic pulmonary vasoconstriction44. Therefore, administration of exogenous H2S to the lungs may further compromise pulmonary blood flow during hypoxic conditions, which can augment hypoxemic hypoxia. Accordingly, all these forms of H2S-mediated hypoxia may add to the mild hypoxia caused by subatmospheric FiO2 levels and culminate in a hypoxic state that is considerable enough to trigger anapyrexia. As addressed in Dirkes et al.45, circulatory hypoxia is sensed through carotid bodies located in the carotid artery46,47 that, under non-hypometabolism-inducing, hypoxic conditions, relay arterial O2 tension (PaO2)-related information to the brain. The brain subsequently (hyper)activates certain physiological functions to remediate the hypoxia48, which include panting49,50,51,52,53 and tachycardia53,54. How this is blocked during the induction of anapyrexia is currently unclear.
Endogenous H2S as well as intracerebrally administered exogenous H2S analogues inhibit the ventilatory and thermal response to hypoxia in the hypothalamus and brain stem. Contrastingly, microinjection of Na2S (H2S precursor) in the anteroventral preoptic hypothalamus of rats potentiates hypothermic signaling by hypoxia, but does not alter Tb under normoxic conditions55. Microinjection of the endogenous H2S production inhibitor amino-oxyacetate in the sympathetic excitatory rostral ventrolateral medulla of rats attenuates hypoxia-induced hypothermia56. As H2S passes the blood-brain barrier freely, central effects of inhaled H2S could have contributed to hypoxia-induced anapyrexia via the hypothalamus or brain stem,22 albeit an unequivocal mechanistic explanation remains warranted in light of the contrasting results.
In the experiments of Blackstone et al. and Volpato et al., Tb was determined by telemetry devices that record the core temperature (i.e., intra-abdominal temperature). In our experiments, the superficial temperature was determined. We believe that this approach is valid for the purpose of this study inasmuch as we were interested in temperature trends as a function of exposure time and gas composition, and not the real Tb per se. Since all groups were thermographically analyzed in the same manner, the resulting data yield credence to our conclusions. Moreover, the use of thermographic imaging has some benefits over intra-abdominal temperature determination, such as the determination of thermoregulatory vasoactivity by tail temperature measurement (Fig. 4).
Although this paper focused on the hypometabolic properties of H2S gas, several animal studies on the effects of liquid H2S analogues NaHS and Na2S have been published. After inhalation, H2S gas diffuses freely across the alveolar membrane and enters the blood as predominantly HS− and H2S22. Accordingly, intravenous administration of solubilized H2S precursors/analogues is believed to follow the same pharmacodynamics as administration through inhalation, only without the detrimental effects on local pulmonary physiology and toxicity. The hypothermic effects of NaHS and Na2S in small as well as in large animals have been reviewed before57. Continuous administration of NaHS is assumed to induce hypothermia in anesthetized rats, although these studies lack essential control groups58,59. The evidence considering the hypothermic and hypometabolic effects of NaHS in large animals has been conflicting: in a pig study a small hypothermic effect was observed following 8 continuous hours of NaHS administration29, whereas in several other studies in pigs45,60 and sheep61 such hypothermic effects were not reproducible. The differences between the effects of H2S in small and large animals have been contemplated by Dirkes et al. and are explained by the inability of large animals to lose heat sufficiently due to the low body surface:mass ratio45.
In this paper, the tail temperature was used as a measure of central activation of peripheral cooling mechanisms (i.e., peripheral vasodilation), as has been used before in the determination of thermoregulatory peripheral vasoactivity in pyrexic mice38. However, as reviewed by Liu et al., H2S has biphasic effects on the vascular tone: at low concentrations H2S induces vasoconstriction and at higher doses vasodilation is induced, as evidenced in mouse and rat aortic tissue62,63,64. Consequently, the absence of thermoregulatory vasodilation and a consequent increase in the tail temperature of 3 animals (Fig. 4) could also be a direct vasoconstrictive effect of low-dose H2S. Nevertheless, H2S-induced vasoconstriction is unlikely to be responsible for the absence of H2S-induced hypothermia in our experiments. A ‘masked’ thermoregulatory vasodilative response would be accompanied by deactivation of brown adipose tissue (BAT) and shivering thermogenesis (i.e., major source of heat in mice at a Ta of 21 °C)1,39. Subsequently, the cessation of thermogenesis would be reflected in the Tb/superficial temperature of H2S-exposed animals, which was not observed (Fig. 3).
In conclusion, exogenous H2S is not a hypometabolism-inducing agent. The hypometabolism induced in mice that were subjected to exogenous H2S was caused by hypoxia. At subatmospheric FiO2 levels, exogenous H2S exacerbates the hypoxic conditions to such a degree that anapyrexia and hypothermia are triggered. Accordingly, exogenous H2S is a hypometabolic adjuvant rather than a hypometabolism-inducing agent.
Materials and Methods
Animals
Forty-eight female C57Bl/6 mice (Charles River, L’Arbresle, France; 10–12 weeks of age) were acclimated for 2 weeks under standardized laboratory conditions with a 12 h light/dark cycle, a constant ambient temperature (Ta) of approximately 21 °C, and ad libitum access to standard chow and drinking water. The experimental protocol was evaluated and approved by the animal ethics and welfare committee of the Academic Medical Center, University of Amsterdam under protocol number BEX 102753. Animals were treated in compliance with institutional guidelines and the National Institute of Health Guidelines for the Care and Use of Laboratory Animals (NIH publication No. 86–23, revised 2011).
Experimental setup and gas mixtures
The hydrogen sulfide (H2S) and 17% oxygen (O2) gas mixtures were obtained from Westfalen (Münster, Germany) and consisted of (1) 80 ppm H2S, 21% O2, and 79% nitrogen (N2); (2) 80 ppm H2S, 17% O2, and 83% N2; or (3) 17% O2, and 83% N2. The 5% O2 gas mixture was obtained from Linde Gas (The Linde Group, Munich, Germany) and consisted of 5% O2 and 95% N2. Normo-atmospheric air (21% O2 and 79% N2) was used as control.
An experimental setup was custom-built to allow controlled gas exposure while unobtrusively assessing body temperature (Tb) with a thermographic camera (ThermaCAM SC2000, FLIR Systems, Wilsonville, OR) in non-anesthetized mice. The setup consisted of gas-tight polypropylene chambers (Fig. 5A, length × depth × height of 109 mm × 109 mm × 61 mm) that were sealed at the imaging end with a thin, infrared light-permeable polyethylene sheet to permit thermal imaging from outside (Fig. 5B). Metal wires were secured longitudinally so that the animals could not reach the polyethylene sheet (Fig. 5C). Gas inflow and outflow tubes were connected to each box at the posterior end for modulation of experimental conditions (Fig. 5E). The gas permeability of the chambers was tested by air pressure decline experiments. Also, a thermistor (Fluke 51 II, Fluke Corporation, Everett, WA) was secured in the posterior wall (Fig. 5D) to facilitate the measurement of the temperature in the chamber. The thermistor was used as a calibrator for the thermographic camera images, as the thermographic images display the temperature of the copper bolt retaining the thermistor.

Components of the experimental setup. (A) The bare polypropylene chamber, consisting of the main chamber and the lid (back part of the chamber). (B) The bottom of the main chamber was removed and replaced with a polyethylene sheet. (C) Metal wires were inserted in front of the polyethylene sheet at a distance at which the mice could not pass through. (D) A bolt was inserted into the back panel to measure the actual temperature in the chamber. The bolt was connected to a thermistor. (E) Configuration of the tubing that was connected to the different gas-containing cylinders and used to modulate the chamber atmosphere during the experiments.
To ascertain sufficient inflow of gas in all experiments and prevent CO2 accumulation, the flow rates were controlled on the basis of CO2 outflow concentrations (<600 ppm, CO2 Meter, Ormond Beach, FL). The system was also connected to an O2 and H2S meter (model OdaLog 7000, App-Tek International, Brendale, Australia), which was calibrated by a certified company prior to the experiments (Carltech, Maarheeze, the Netherlands). The O2 and H2S meter was post hoc tested for measurement accuracy. The experiments were performed at a mean ± SD Ta of 21.2 ± 0.6 °C.
Experimental procedure
To test the hypothesis that H2S-induced hypothermia emanates from hypoxia and not H2S, all 48 animals were randomly divided among 5 experimental groups. Group A was exposed to 80 ppm H2S in 21% O2 and 79% N2 (H2S in 21% O2 group, N = 12), group B was exposed to 80 ppm H2S in 17% O2 and 83% N2 (H2S in 17% O2 group, N = 6), group C was exposed to 5% O2 and 95% N2 (5% O2 group, N = 12), group D was exposed to 17% O2 and 83% N2 (17% O2 group, N = 6), and group E was exposed to 21% O2 and 79% N2 (normoxia group, N = 12).
Mice were placed in the chambers individually. After 1 h of exposure to normoxia (21% O2 and 79% N2), the mice were exposed to one of the gas mixtures (A – E) for 6 h, after which 6 of the animals per group were allowed to recover at normoxic conditions for 3 h before being terminated. The other 6 animals of group A, C and E were terminated immediately after the 6 h of exposure for another study.
No anesthetics were used before or during the experimental procedure.
Thermal imaging and data processing
Animals were filmed every hour for 10 min with a thermographic camera (ThermaCAM SC2000, FLIR Systems, Wilsonville, OR)(Fig. 2). Thermographic camera images (3 images per second) were processed and analyzed in ThermaCAM Researcher 2001 (FLIR Systems). The mean maximum superficial temperature was calculated per time point per group.
The tail temperatures of animals in group A (N = 3), C (N = 3), and E (N = 1) were obtained from the thermographic camera images at 0 h, just before the start of exposure, and approximately 4 min after the start of exposure. We noticed the intergroup differences in tail temperature during the experiments, as a result of which the tail temperature was measured in only 7 animals. The mean difference in tail temperature between both time points was calculated and compared for group A and C.
Locomotor activity was assessed per time point using the same thermal images as were used for the calculation of superficial temperature. An analytics program was written in LabVIEW (LabVIEW, National Instruments, Austin, TX). The thermographic camera images were converted to grayscale images and loaded into LabVIEW. Locomotor activity was calculated per animal per time point (−1 up to 9 h) on the basis of fluctuations in pixel intensity. A pixel was considered to reflect ‘motion’ when the grayscale intensity difference between direct temporally consecutive pixels exceeded 7 on a scale of 0 to 255. The intensity difference of at least 7 was based on the disappearance of background scatter present as intensity differences between 1 and 6. Values were expressed as the mean ± SEM amount of pixels with ‘motion’ per group per time point.
Statistical analysis
Statistical analyses were performed using MatLab 2013a (MathWorks, Natick, MA). Homogeneity of variance in each group was tested using the Bartlett’s test. Based on equality of variances, either a one-way ANOVA or a Kruskal-Wallis test was performed, followed by a Tukey’s range test or Dunn’s test, respectively, to compare ordinal variables related to maximum superficial temperature and locomotor activity between groups. Tail temperature values were compared using an unpaired student’s t-test. P-values less than 0.05 were considered significant. All values were presented as mean ± SEM, unless otherwise mentioned.
References
Heldmaier, G., Ortmann, S. & Elvert, R. Natural hypometabolism during hibernation and daily torpor in mammals. Respir. Physiol. Neurobiol. 141, 317–29 (2004).
Gordon, C. J. & Fogelson, L. Comparative effects of hypoxia on behavioral thermoregulation in rats, hamsters, and mice. Am. J. Physiol. 260, R120–5 (1991).
Steiner, A. A. & Branco, L. G. S. Hypoxia-induced anapyrexia: implications and putative mediators. Annu. Rev. Physiol. 64, 263–88 (2002).
Dark, J., Miller, D. R. & Zucker, I. Reduced glucose availability induces torpor in Siberian hamsters. Am. J. Physiol. 267, R496–501 (1994).
Planel, E. et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J. Neurosci. 24, 2401–11 (2004).
Heller, H. C. & Colliver, G. W. CNS regulation of body temperature during hibernation. Am. J. Physiol. 227, 583–9 (1974).
Heller, H. C., Colliver, G. W. & Bread, J. Thermoregulation during entrance into hibernation. Pflugers Arch. 369, 55–9 (1977).
Song, X., Körtner, G. & Geiser, F. Reduction of metabolic rate and thermoregulation during daily torpor. J. Comp. Physiol. B. 165, 291–7 (1995).
Song, X., Körtner, G. & Geiser, F. Thermal relations of metabolic rate reduction in a hibernating marsupial. Am. J. Physiol. 273, R2097–104 (1997).
Cannon, B. & Nedergaard, J. Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 (2004).
Arrhenius, S. A. Über die Dissociationswärme und den Einfluß der Temperatur auf den Dissociationsgrad der Elektrolyte [Concerning the heat of dissociation and the influence of the temperature on the degree of dissociation of electrolytes]. Zeitschrift für Phys. Chemie 4, 96–116 (1889).
Peleg, M., Normand, M. D. & Corradini, M. G. The Arrhenius equation revisited. Crit. Rev. Food Sci. Nutr. 52, 830–51 (2012).
Zhang, J., Kaasik, K., Blackburn, M. R. & Lee, C. C. Constant darkness is a circadian metabolic signal in mammals. Nature 439, 340–3 (2006).
Daniels, I. S. et al. A role of erythrocytes in adenosine monophosphate initiation of hypometabolism in mammals. J. Biol. Chem. 285, 20716–23 (2010).
Dawe, A. R. & Spurrier, W. A. Hibernation induced in ground squirrels by blood transfusion. Science 163, 298–9 (1969).
Horton, N. D. et al. Isolation and partial characterization of an opioid-like 88 kDa hibernation-related protein. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 119, 787–805 (1998).
Bechtold, D. A. et al. A role for the melatonin-related receptor GPR50 in leptin signaling, adaptive thermogenesis, and torpor. Curr. Biol. 22, 70–7 (2012).
Scanlan, T. S. et al. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat. Med. 10, 638–42 (2004).
Braulke, L. J. et al. 3-Iodothyronamine: a novel hormone controlling the balance between glucose and lipid utilisation. J. Comp. Physiol. B. 178, 167–77 (2008).
Blackstone, E., Morrison, M. & Roth, M. B. H2S induces a suspended animation-like state in mice. Science 308, 518 (2005).
Volpato, G. P. et al. Inhaled hydrogen sulfide: a rapidly reversible inhibitor of cardiac and metabolic function in the mouse. Anesthesiology 108, 659–68 (2008).
Beauchamp, R. O., Bus, J. S., Popp, J. A., Boreiko, C. J. & Andjelkovich, D. A. A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 13, 25–97 (1984).
Cuevasanta, E., Denicola, A., Alvarez, B. & Möller, M. N. Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS One 7, e34562 (2012).
Cooper, C. E. & Brown, G. C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 40, 533–9 (2008).
Nicholls, P., Marshall, D. C., Cooper, C. E. & Wilson, M. T. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 41, 1312–6 (2013).
Lopez, A., Prior, M., Yong, S., Albassam, M. & Lillie, L. E. Biochemical and cytologic alterations in the respiratory tract of rats exposed for 4 hours to hydrogen sulfide. Fundam. Appl. Toxicol. 9, 753–62 (1987).
Prior, M. G., Sharma, A. K., Yong, S. & Lopez, A. Concentration-time interactions in hydrogen sulphide toxicity in rats. Can. J. Vet. Res. 52, 375–9 (1988).
Stein, A. et al. Metabolic and cardiac signaling effects of inhaled hydrogen sulfide and low oxygen in male rats. J. Appl. Physiol. 112, 1659–69 (2012).
Simon, F. et al. Hemodynamic and metabolic effects of hydrogen sulfide during porcine ischemia/reperfusion injury. Shock 30, 359–64 (2008).
Althaus, M., Urness, K. D., Clauss, W. G., Baines, D. L. & Fronius, M. The gasotransmitter hydrogen sulphide decreases Na+ transport across pulmonary epithelial cells. Br. J. Pharmacol. 166, 1946–63 (2012).
Frappell, P., Lanthier, C., Baudinette, R. V. & Mortola, J. P. Metabolism and ventilation in acute hypoxia: a comparative analysis in small mammalian species. Am. J. Physiol. 262, R1040–6 (1992).
Gautier, H. Interactions among metabolic rate, hypoxia, and control of breathing. J. Appl. Physiol. 81, 521–7 (1996).
Branco, L. G., Carnio, E. C. & Barros, R. C. Role of the nitric oxide pathway in hypoxia-induced hypothermia of rats. Am. J. Physiol. 273, R967–71 (1997).
Hinrichsen, C. F., Maskrey, M. & Mortola, J. P. Ventilatory and metabolic responses to cold and hypoxia in conscious rats with discrete hypothalamic lesions. Respir. Physiol. 111, 247–56 (1998).
Nakamura, K. Central circuitries for body temperature regulation and fever. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1207–28 (2011).
Kanosue, K., Yanase-Fujiwara, M. & Hosono, T. Hypothalamic network for thermoregulatory vasomotor control. Am. J. Physiol. 267, R283–8 (1994).
Owens, N. C., Ootsuka, Y., Kanosue, K. & McAllen, R. M. Thermoregulatory control of sympathetic fibres supplying the rat’s tail. J. Physiol. 543, 849–58 (2002).
Rudaya, A. Y., Steiner, A. A., Robbins, J. R., Dragic, A. S. & Romanovsky, A. A. Thermoregulatory responses to lipopolysaccharide in the mouse: dependence on the dose and ambient temperature. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1244–52 (2005).
Clapham, J. C. Central control of thermogenesis. Neuropharmacology 63, 111–23 (2012).
Galilei, G. Discorsi e dimostrazioni matematiche intorno a due nuove scienze (Lodewijk Elsevier, 1638).
Kleiber, M. Body size and metabolic rate. Physiol. Rev. 27, 511–41 (1947).
Ríos-González, B. B., Román-Morales, E. M., Pietri, R. & López-Garriga, J. Hydrogen sulfide activation in hemeproteins: the sulfheme scenario. J. Inorg. Biochem. 133, 78–86 (2014).
Common Surgical Diseases., https://doi.org/10.1007/978-0-387-75246-4 (Springer New York, 2008).
Madden, J. A., Ahlf, S. B., Dantuma, M. W., Olson, K. R. & Roerig, D. L. Precursors and inhibitors of hydrogen sulfide synthesis affect acute hypoxic pulmonary vasoconstriction in the intact lung. J. Appl. Physiol. 112, 411–8 (2012).
Dirkes, M. C., Milstein, D. M. J., Heger, M. & van Gulik, T. M. Absence of Hydrogen Sulfide-Induced Hypometabolism in Pigs: A Mechanistic Explanation in Relation to Small Nonhibernating Mammals. Eur. Surg. Res. 54, 178–191 (2015).
Milsom, W. K. & Burleson, M. L. Peripheral arterial chemoreceptors and the evolution of the carotid body. Respir. Physiol. Neurobiol. 157, 4–11 (2007).
Prabhakar, N. R. & Semenza, G. L. Gaseous messengers in oxygen sensing. J. Mol. Med. (Berl). 90, 265–72 (2012).
Marshall, J. M. Peripheral chemoreceptors and cardiovascular regulation. Physiol. Rev. 74, 543–94 (1994).
Izumizaki, M., Pokorski, M. & Homma, I. Role of the carotid bodies in chemosensory ventilatory responses in the anesthetized mouse. J. Appl. Physiol. 97, 1401–7 (2004).
Peng, Y.-J. et al. H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. USA 107, 10719–24 (2010).
Li, Q. et al. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid. Redox Signal. 12, 1179–89 (2010).
Teppema, L. J. & Dahan, A. The ventilatory response to hypoxia in mammals: Mechanisms, Measurement, and Analysis. Physiol. Rev. 90, 675–754 (2010).
Sugimura, M. et al. Influence of acute hypoxia combined with nitrous oxide on cardiovascular variability in conscious hypertensive rats. Auton. Neurosci. 156, 73–81 (2010).
Lifson, J. D., Rubinstein, E. H. & Sonnenschein, R. R. Hypoxic tachycardia in the rat. Experientia 33, 476–7 (1977).
Kwiatkoski, M. et al. Hydrogen sulfide as a cryogenic mediator of hypoxia-induced anapyrexia. Neuroscience 201, 146–56 (2012).
Donatti, A. F., Soriano, R. N., Sabino, J. P. & Branco, L. G. S. Involvement of endogenous hydrogen sulfide (H2S) in the rostral ventrolateral medulla (RVLM) in hypoxia-induced hypothermia. Brain Res. Bull. 108, 94–9 (2014).
Asfar, P., Calzia, E. & Radermacher, P. Is pharmacological, H2S-induced ‘suspended animation’ feasible in the ICU? Crit. Care 18, 215 (2014).
Aslami, H. et al. Suspended animation inducer hydrogen sulfide is protective in an in vivo model of ventilator-induced lung injury. Intensive Care Med. 36, 1946–52 (2010).
Aslami, H. et al. A short course of infusion of a hydrogen sulfide-donor attenuates endotoxemia induced organ injury via stimulation of anti-inflammatory pathways, with no additional protection from prolonged infusion. Cytokine 61, 614–21 (2013).
Drabek, T. et al. Intravenous hydrogen sulfide does not induce hypothermia or improve survival from hemorrhagic shock in pigs. Shock 35, 67–73 (2011).
Haouzi, P. et al. H2S induced hypometabolism in mice is missing in sedated sheep. Respir. Physiol. Neurobiol. 160, 109–15 (2008).
Kubo, S., Doe, I., Kurokawa, Y., Nishikawa, H. & Kawabata, A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: contribution to dual modulation of vascular tension. Toxicology 232, 138–46 (2007).
Ali, M. Y. et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 149, 625–34 (2006).
Lim, J. J., Liu, Y.-H., Khin, E. S. W. & Bian, J.-S. Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 295, C1261–70 (2008).
Acknowledgements
Ronald Oude-Elferink is acknowledged for introducing the concept of hibernation for medical applications to us. We are grateful to Adrie Maas for technical assistance.
Author information
Authors and Affiliations
Contributions
S.D.H. prepared and performed the experiments, analyzed the data, and co-wrote the manuscript. M.C.D. is responsible for the conceptual design of the experiments and supervised the experiments. M.H.N.V. analyzed the thermographic data and RB designed the motion analysis software. T.M.G. supervised the project. M.H. supervised the project and wrote the manuscript. All authors critically read the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hemelrijk, S.D., Dirkes, M.C., van Velzen, M.H.N. et al. Exogenous hydrogen sulfide gas does not induce hypothermia in normoxic mice. Sci Rep 8, 3855 (2018). https://doi.org/10.1038/s41598-018-21729-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21729-8
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
