
Abstract
Mutations of the gene encoding WNK1 [With No lysine (K) kinase 1] or WNK4 cause Familial Hyperkalemic Hypertension (FHHt). Previous studies have shown that the activation of SPAK (Ste20-related Proline/Alanine-rich Kinase) plays a dominant role in the development of FHHt caused by WNK4 mutations. The implication of SPAK in FHHt caused by WNK1 mutation has never been investigated. To clarify this issue, we crossed WNK1+/FHHt mice with SPAK knock-in mice in which the T-loop Thr243 residue was mutated to alanine to prevent activation by WNK kinases. We show that WNK1+/FHHT:SPAK243A/243A mice display an intermediate phenotype, between that of control and SPAK243A/243A mice, with normal blood pressure but hypochloremic metabolic alkalosis. NCC abundance and phosphorylation levels also decrease below the wild-type level in the double-mutant mice but remain higher than in SPAK243A/243A mice. This is different from what was observed in WNK4-FHHt mice in which SPAK inactivation completely restored the phenotype and NCC expression to wild-type levels. Although these results confirm that FHHt caused by WNK1 mutations is dependent on the activation of SPAK, they suggest that WNK1 and WNK4 play different roles in the distal nephron.
Similar content being viewed by others

Introduction
Familial Hyperkaliaemic Hypertension (FHHt), also known as Gordon’s syndrome or Pseudohypoaldosteronism type II (PHAII), is a rare genetic form of hypertension associated with hyperkalemia and metabolic hyperchloremic acidosis (OMIM #145260)1. All these symptoms are corrected by thiazide diuretics, which inhibit the activity of the Na+-Cl− transporter NCC, encoded by the SLC12A3 gene1. FHHt is caused by mutations in one of at least four genes: WNK1 [With No lysine (K) 1], WNK4, KLHL3 (Kelch-Like family member 3), and CUL3 (cullin-3)2,3,4. The associated proteins belong to the same regulatory pathway, as WNK1 and WNK4 are recruited by KLHL3 for ubiquitination by the Cul3-E3 RING-type ubiquitin–ligase complex5.
The sensitivity of patients to thiazide diuretics strongly suggested that FHHt is mainly caused by a gain of activity of NCC in the distal nephron. The role of NCC in regulating blood pressure has been established by the discovery of inactivating mutations of the SLC12A3 gene, which cause Gitelman’s syndrome, an inherited disorder that is the mirror image of FHHt, with arterial hypotension, renal salt wasting and hypokalemic metabolic alkalosis (OMIM #263800)6,7.
Therefore, several studies focused on the mechanisms underlying the regulation of NCC by WNK1 and WNK4. In vitro studies have demonstrated that the WNKs activate two kinases, SPAK (Ste20-related Proline/Alanine-rich Kinase) and OSR1 (oxidative stress-responsive kinase 1)8, which can in turn phosphorylate and activate NCC9. SPAK and OSR1 can also activate the Na+-K+-2Cl− cotransporters, NKCC1 and NKCC2, and inhibit the K+-Cl− cotransporters, KCC1–410. In vivo, the expression and phosphorylation of NCC is reduced when SPAK is inactivated11,12. Finally, the fact that the mutation of a key SPAK/OSR1 phosphorylation site on NCC (Thr60) causes Gitelman’s syndrome confirmed the major role of SPAK/OSR1 in the regulation of NCC activity in vivo13.
Several studies have then demonstrated that the activation of SPAK, but not OSR1, by WNK4 is a key step in the pathogenesis of FHHt. First, SPAK abundance and phosphorylation are increased in a mouse model of FHHt caused by a missense mutation of WNK4 (Wnk4D561/+ mice)14. Second, the genetic inactivation of SPAK in the same mouse model corrects all FHHt disorders and allows the restoration of NCC abundance and phosphorylation to the wild-type level15,16. By opposition, Wnk4D561/+ mice bearing a OSR1 inactivation in the nephron (Wnk4D561/+:KSP-Osr1−/−) still display a complete FHHt phenotype16, even though OSR1 deficiency in the nephron causes hypokalemia under normal diet and systolic hypotension when mice are fed a low Na+ diet17.
The implication of SPAK in FHHt caused by a mutation of WNK1 has never been investigated. We previously generated the WNK1+/FHHt mouse model, carrying a heterozygous deletion of WNK1 first intron, corresponding to the mutation found in FHHt patients18. This deletion leads to an increased expression of L-WNK1, the ubiquitous kinase isoform of WNK1, in the DCT and the CNT. WNK1+/FHHt mice display hyperkalemia, hypertension and metabolic acidosis as well as increased NCC abundance and phosphorylation. In this model, SPAK abundance and phosphorylation measured by western blot on renal cortex extracts are similar to those of control mice. However, we showed by immunofluorescence that the abundance of total and phosphorylated SPAK increases near the apical membrane of DCT cells in WNK1+/FHHt mice compared to controls18. To investigate whether or not SPAK is necessary to trigger the FHHt caused by WNK1 mutations, we characterized WNK1+/FHHt mice in which SPAK activation by WNK kinases is prevented by the mutation of the T-loop Thr243 residue into alanine (SPAK243A/243A mice)11. We show here that WNK1+/FHHT:SPAK243A/243A mice display an intermediate phenotype, between that of control and SPAK243A/243A mice. Similarly, NCC abundance and phosphorylation levels decrease below the wild-type level in the double-mutant mice but remain higher than in SPAK243A/243A mice, which is different from what was observed in Wnk4D561/+:SPAK243A/243A 15. These results suggest that WNK1 and WNK4 play different roles in the distal nephron even if in both cases SPAK deficiency is sufficient to preclude the development of FHHt in mice.
Results
SPAK inactivation in WNK1 +/FHHt mice results in an intermediate phenotype, between control and SPAK mutant mice
We previously demonstrated an increased blood pressure in WNK1+/FHHt compared to control mice (122.9 ± 4.8 mmHg in WNK1+/FHHt vs 112.0 ± 3.2 mmHg in controls, p = 0.047)18. We measured blood pressure in controls and WNK1+/FHHt:SPAK243A/243A mice by a real-time radiotelemetry approach. WNK1+/FHHt:SPAK243A/243A mice are normotensive (Fig. 1a) compared to control littermates.

SPAK inactivation in WNK1+/FHHt mice restores blood pressure but leads to a mild metabolic hypochloremic alkalosis. (a) Averaged systolic blood pressure during the 12h-day or -night period measured by radiotelemetry in WNK1+/FHHt:SPAK243A/243A and control mice. No difference was observed between the two groups of mice (Student’s test). (b) Plasma electrolyte concentrations in mice of each genotype at baseline. SPAK243A/243A mice display a hypochloremic metabolic alkalosis while WNK1+/FHHt mice exhibit a typical FHHt phenotype. SPAK inactivation in WNK1+/FHHt mice leads to the development of a hypochloremic metabolic alkalosis, which tends to be less severe than in SPAK243A/243A mice. Values are means ± s.e.m. ap < 0.05 vs. controls. bp < 0.05 vs. WNK1+/FHHt (Kruskal-Wallis one-way analysis of variance followed by Mann-Whitney test). Experiments were performed on 9 control, 8 WNK1+/FHHt, 5 WNK1+/FHHt:SPAK243A/243A and 5 SPAK243A/243A male mice.
As previously described18, WNK1+/FHHt mice exhibited hyperkalemia and hyperchloremia while SPAK243A/243A mice exhibited hypochloremic metabolic alkalosis (Fig. 1b). As for blood pressure, the inactivation of SPAK in WNK1+/FHHt mice restores plasma potassium concentration to normal. Surprisingly, it also results in mild hypochloremic metabolic alkalosis (Fig. 1b). However, plasma chloride concentration tends to be higher in WNK1+/FHHt:SPAK243A/243A mice than in SPAK243A/243A mice. In conclusion, the double-mutant mice have an intermediate phenotype, between control and SPAK243A/243A mice, suggesting that the inactivation of SPAK in WNK1+/FHHt mice impairs ion transport in the distal nephron.
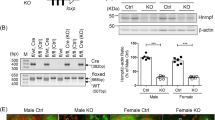
This intermediate phenotype is also observed for NCC abundance and phosphorylation. As previously reported11,18, these parameters significantly increase in WNK1+/FHHt mice compared to control mice, while they decrease in SPAK243A/243A mice (Fig. 2). SPAK inactivation in WNK1+/FHHt mice leads to a significant decreased in NCC abundance and phosphorylation (Fig. 2). Interestingly, these two parameters decrease in WNK1+/FHHT:SPAK243A/243A mice below the levels observed in control mice (about 25% and 50%, respectively), but remain above the levels observed in SPAK243A/243A mice (Fig. 2). This is in contrast with what was observed in Wnk4D561/+:SPAK−/− mice, in which NCC abundance and phosphorylation levels were comparable to control levels16.

SPAK inactivation in WNK1+/FHHt mice decreases NCC abundance and phosphorylation below control levels. Upper panel: Representative immunoblots of NCC and phosphorylated NCC-Thr53 performed on the membrane-enriched fractions of the renal cortex of mice of each genotype. Lower panel: Densitometric analysis. NCC abundance and phosphorylation are increased in WNK1+/FHHt mice compared to control mice. NCC abundance and phosphorylation in WNK1+/FHHt:SPAK243A/243A and SPAK242A/243A mice are corrected below the control levels. Number of animals: 4 control, 5 WNK1+/FHHt, 4 WNK1+/FHHt:SPAK243A:243A and 4 SPAK243A:243A male mice. Values are means ± s.e.m. ap < 0.05 vs. controls. bp < 0.05 vs. WNK1+/FHHt. cp < 0.05 vs. SPAK243A/243A-WNK1+/FHHt FHHt (Kruskal-Wallis one-way analysis of variance followed by Mann-Whitney test). Immunoblot images are cropped images. The full-length images can be found in Supplementary Figure S3.
Grimm et al. previously reported that the expression of the sodium channel ENaC and chloride-bicarbonate exchanger pendrin are increased in SPAK knock-out mice19. We show here that this is also the case in SPAK243A/243A mice (Fig. 3). The abundance of pendrin and γ-subunit of ENaC, as well as its cleavage, increase in WNK1+/FHHt:SPAK243A/243A mice (Fig. 3). The cleaveage of the γ-ENaC subunit is believed to reflect the activation of the channel20. The increased pendrin and ENaC abundance probably compensates for the reduced expression and/or phosphorylation of NCC and NKCC2 resulting from the loss of SPAK in WNK1+/FHHt:SPAK243A/243A mice. Indeed, enhanced expression of pendrin and γENaC has also been reported in Ncc−/− mice21, probably to compensate for NCC inactivation. The increased ENaC abundance, processing and activity could increase the tubular lumen electronegativity, the driving force for H+ secretion, and thus explain the metabolic alkalosis observed in WNK1+/FHHt:SPAK243A/243A mice. As suggested by Grimm and collaborators, pendrin could be stimulated in response to the alkalosis19.

The expression of pendrin and ENaC is increased in the kidney of WNK1+/FHHT:SPAK243A/243A mice. Upper panel: Representative immunoblots performed with antibodies against the γ subunit of the epithelial channel ENaC and pendrin on the membrane-enriched fractions of the renal cortex of mice of each genotype. FL: Full-length and CL: cleaved form of γENaC, respectively. Lower panel: Densitometric analysis. The abundance and cleavage of γ-ENaC are increased in WNK1+/FHHT:SPAK243A/243A mice and SPAK243A/243A mice. The expression of pendrin increases significantly in WNK1+/FHHT:SPAK243A/243A mice. Number of animals: 4 control, 5 WNK1+/FHHt, 4 WNK1+/FHHt:SPAK243A:243A and 4 SPAK243A:243A male mice. Values are means ± s.e.m. ap < 0.05 vs. controls. bp < 0.05 vs. WNK1+/FHHt miceFHHt (Kruskal-Wallis one-way analysis of variance followed by Mann-Whitney test). Immunoblot images are cropped images. The full-length images can be found in Supplementary Figure S4.
Expression of OSR1 and Cab39 in WNK1 +/FHHt:SPAK 243A/243A mice
The fact that NCC phosphorylation is significantly higher in WNK1+/FHHt:SPAK243A/243A mice than in SPAK243A/243A mice strongly suggests that another pathway is activated in the DCT by the overexpressed L-WNK118 to phosphorylate NCC in the absence of SPAK. This hypothesis is indirectly supported by the quantification of the abundance and phosphorylation of NKCC2, another target of SPAK and OSR1. As shown in Supplementary Figure S2. NKCC2 abundance and phosphorylation are decreased in WNK1+/FHHt:SPAK243A/243A and SPAK243A/243A mice. However, in contrast to NCC, there is no significant difference between the two groups of mice. This could be due to the lack of activation of an alternative pathway by L-WNK1 since the deletion of intron 1 does not modify the expression and thus activity of L-WNK1 in the cortical ascending limb of the loop of Henle18, where NKCC2 is expressed.
In addition to SPAK, two other proteins have been implicated in the regulation of cation-chloride cotransporters: OSR1 and Cab39. The calcium-binding protein Cab39 has been shown to activate NCC in combination with WNK kinases in a SPAK-independent manner22. However, Cab39 abundance is similar in all groups of mice (Fig. 4a), suggesting that Cab39 may not be involved in the remaining phosphorylation of NCC observed in WNK1+/FHHt:SPAK243A/243A kidneys.

OSR1 remains at the apical membrane of SPAK243A/243A and WNK1+/FHHT:SPAK243A/243A DCT, where its phosphorylation tends to increase. Upper panel: Representative Immunoblots performed on total cortex protein extract with antibodies against total SPAK, total OSR1, pSPAK/pOSR1 (S-motif) and Cab39. Lower panel: Densitometric analysis. SPAK inactivation mutation does not affect SPAK expression and S-motif phosphorylation, while it tends to slightly increase OSR1 expression and phosphorylation. Number of animals: 4 control, 5 WNK1+/FHHt, 4 WNK1+/FHHt:SPAK243A:243A and 4 SPAK243A:243A male mice. Values are means ± s.e.m. ap < 0.05 vs. controls. bp < 0.05 vs. WNK1+/FHHt miceFHHt (Kruskal-Wallis one-way analysis of variance followed by Mann-Whitney test). Immunoblot images are cropped images. The full-length images can be found in Supplementary Figure S5.
We thus hypothesized that OSR1 could be the alternative kinase activated by L-WNK1. L-WNK1 phosphorylates SPAK and OSR1 at two conserved residues, a Thr residue in the T-loop and a Ser residue in the S-motif8. While T-loop phosphorylation triggers the activation of SPAK and OSR1, the role of the S-motif phosphorylation is unclear, as its mutation does not affect activation8. We could not quantify the phosphorylation of the T loop as it is too low in kidney homogenates11. OSR1 abundance is increased in the kidney of WNK1+/FHHt:SPAK243A/243A and SPAK243A/243A mice but not of WNK1+/FHHt mice (Fig. 4). The phosphorylation of the S-motif of OSR1 is increased, albeit not significantly, in WNK1+/FHHt:SPAK243A/243A mouse kidneys compared to controls (Fig. 4). It also increases in WNK1+/FHHt but not in SPAK243A/243A kidneys. The phosphorylation of SPAK S-motif is also increased in WNK1+/FHHt:SPAK243A/243A mice (Fig. 4). This increase is probably a consequence of the increased L-WNK1 expression18.
We previously showed that SPAK abundance near the apical membrane of DCT cells increases in WNK1+/FHHt mice18, suggesting that the activation of SPAK by WNK kinases stimulates its apical localisation. Similarly, OSR1 phosphorylation and apical localisation increase in mice subjected to potassium depletion23,24, a situation in which WNKs are activated23. Therefore, we hypothesized that OSR1 localisation near the apical membrane of DCT cells should increase in WNK1+/FHHt:SPAK243A/243A if OSR1 is indeed stimulated by L-WNK1 to compensate for the absence of functional SPAK. In order to test this hypothesis, we studied OSR1 expression in the DCT by double immunofluorescence staining, with antibodies directed against NCC and OSR1. As shown in Fig. 5, OSR1 is located both in cytoplasmic punctate structures and near the apical membrane in the DCT cells of control mice. In contrast, OSR1 signal is significantly increased near the apical membrane in the DCT of WNK1+/FHHt:SPAK243A/243A mice, where a stronger colocalisation with NCC can be observed compared to control mice (Fig. 5). In this situation, it is expected that OSR1 could phosphorylate NCC as the phosphorylation of the cotransporter occurs near or within the apical membrane25. Taken together, these observations suggest that OSR1 could be activated by L-WNK1 and partially compensate for the lack of SPAK in the DCT WNK1+/FHHt:SPAK243A/243A mice, in the setting of overexpressed L-WNK1.

Representative double immunostainingstaining for total OSR1 (in green) and NCC (in red) in the renal cortex of control, WNK1+/FHHt, WNK1+/FHHt:SPAK243A/243A and SPAK243A/243A mice. OSR1 colocalize with NCC in distal convoluted tubule cells. Lower panel: quantification of apical OSR1 staining in distal convoluted tubule cells. OSR1 staining is significantly more apical in WNK1+/FHHt:SPAK243A:243A and SPAK243A:243A mice. Number of animals: 4 control, 2 WNK1+/FHHt, 4 WNK1+/FHHt:SPAK243A:243A and 4 SPAK243A:243A male mice. Values are means ± s.e.m. ap < 0.05 vs. controls.
Our observations also support the hypothesis that SPAK could be required for OSR1 redistribution to the apical membrane of DCT cells. Indeed, Grimm et al. showed that OSR1 is found in dense punctate structures in DCT cells of SPAK−/− mice26 whereas OSR1 apical localisation is increased in SPAK243A/243A DCT (Fig. 5).
Increased WNK1 is localised near the apical membrane of DCT cells
In order to understand how OSR1 can be activated in WNK1+/FHHt:SPAK243A/243A mice, we characterized the expression of WNK1 and WNK4 in the different groups of mice. While WNK4 expression level is unaffected in WNK1+/FHHt mice, it significantly decreases in the kidney of WNK1+/FHHt:SPAK243A/243A and SPAK243A/243A mice (Supplementary Fig. S2). This decreased expression of WNK4 may result from the DCT hypotrophy caused by the drastic decreased NCC expression, as observed in Ncc−/− or SPAK−/− mice26,27.
We then characterised the expression of WNK1 in the DCT by immunostaining using an antibody directed against a C-terminal fragment of the protein, which thus recognizes both L-WNK1 and KS-WNK1 (Fig. 6). In control and SPAK243A/243A mice, WNK1 signal is located in cytoplasmic punctate structures in the DCT. In contrast, WNK1 signal is found near the apical membrane in the DCT of WNK1+/FHHt and WNK1+/FHHt:SPAK243A/243A mice. In these two groups of mice, L-WNK1 transcription is increased following the deletion of WNK1 first intron. These observations support our hypothesis that overexpressed L-WNK1 could contribute to a stronger activation of OSR1 and thus NCC in WNK1+/FHHt:SPAK234A/243A mice compared to SPAK243A/243A.

WNK1 cellular localisation is modified when overexpressed. Representative double immunostainingstaining for WNK1 (in red) and NCC (in green) in control, WNK1+/FHHt, WNK1+/FHHt:SPAK243A/243A and SPAK243A/243A mice. While WNK1 is located in cytoplasmic puncta in DCT cells of control and SPAK243A/243A mice, it is observed near the apical membrane of DCT cells in WNK1+/FHHt and WNK1+/FHHt:SPAK243A/243A mice, in which L-WNK1 transcription is increased.
Discussion
In this study, we used a mouse model to investigate the consequences of SPAK inactivation in the pathogenesis of FHHt caused by WNK1 mutation. Indeed, several studies have demonstrated the essential role of SPAK in the pathogenesis of FHHt caused by WNK4 missense mutations15,16 but the implication of SPAK downstream of L-WNK1 in FHHt remained to be formally demonstrated.
We have previously reported the characterisation of WNK1+/FHHt mice, which we generated by deleting WNK1 first intron to mimic the large intronic deletions identified in FHHt patients18. In these mice, the expression of the ubiquitous catalytic WNK1 isoform (L-WNK1) increases in the DCT and CNT, whereas the expression of the kidney-specific and kinase-defective isoform (KS-WNK1) is unchanged. L-WNK1 overexpression augments the abundance and phosphorylation of NCC, probably via the activation of SPAK. We indeed observed an increased abundance of phosphorylated SPAK near the apical membrane of DCT cells in WNK1+/FHHt mice18. In order to demonstrate the role played by SPAK in the development of FHHt caused by WNK1 mutation, we crossed WNK1+/FHHt mice with SPAK243A/243A mice. The latter carry a Thr243Ala mutation, which precludes the phosphorylation of SPAK T-loop by WNK kinases11. WNK1+/FHHT:SPAK243A/243A mice exhibit normal blood pressure and plasma K+ concentration compared to control mice. However, they develop a hypochloremic metabolic alkalosis. Furthermore, the plasma chloride concentration tends to be significantly higher in WNK1+/FHHT:SPAK243A/243A than in SPAK243A/243A mice. The same result was obtained for NCC abundance and phosphorylation, which are lower than in control mice but higher than in SPAK243A/243A mice. The inhibition of SPAK in WNK1+/FHHt mice therefore results in an intermediate phenotype, between control and SPAK243A/243A mice.
These results differ from what has been observed in mouse models carrying both a missense FHHt mutation in WNK4 and an inactivating mutation of SPAK (Wnk4D561A/+:SPAK243A/243A or Wnk4D561A/+:SPAK−/− mice)15,16. Indeed, the inhibition of SPAK in Wnk4D561A/+ mice restored blood pressure, potassium and acid-base balance as well as NCC abundance and phosphorylation to control levels. Taken together, these observations suggest that, even though they both can activate SPAK and OSR1 in vitro, L-WNK1 and WNK4 have different functions in the distal nephron, more particularly in the DCT. This is supported by the fact that NCC abundance and phosphorylation are drastically reduced in mice carrying a null mutation of WNK4 (WNK4−/−)28. Therefore, the absence of WNK4 cannot be compensated for by L-WNK1. This could be due to the low level of physiological expression of L-WNK1 in the DCT29 since an overexpression of L-WNK1 in this segment can compensate for and even induce hyperkalemic and acidotic hypertension in the absence of WNK430. One could therefore wonder if L-WNK1 plays a physiological role in the DCT. It is interesting to note that, whereas KS-WNK1 and WNK4 transcription in the mouse kidney is modulated by changes in Na+ or K+ intake and aldosterone, it is not the case for L-WNK131. Recently, Roy et al. reported that L-WNK1 protein abundance is increased by aldosterone through the inhibition of its ubiquitination by Nedd4-232. The precise characterisation of the roles played by L-WNK1 in the distal nephron is complicated by the fact that L-WNK1 knock-out impairs cardiovascular development and thus results in embryonic lethality33.
We also show for the first time that WNK1 cellular localisation is modified in the DCT of WNK1+/FHHt and WNK1+/FHHT:SPAK243A/243A mice. While it is located in cytoplasmic punctate structures in control mice, the WNK1 signal is found near the apical membrane in the mutant mice (Fig. 6). The antibody we used recognized both L-WNK1 and KS-WNK1. Boyd-Shiwarski and collaborators showed that, in vitro, transfected KS-WNK1 localises to cytoplasmic puncta, similar to those observed in vivo in DCT cells, while transfected L-WNK1 was distributed diffusively throughout the cytoplasm34. Interestingly, both isoforms were found in the puncta when cotransfected. The deletion of WNK1 first intron, which corresponds to the FHHt mutation carried by the two mouse models, stimulates L-WNK1 transcription18. Taken together, these results suggest that, upon overexpression and probably activation, L-WNK1 translocates from the puncta to the apical membrane of DCT cells. Again, this result is different from what has been described for WNK4. Indeed, Terker and collaborators showed that WNK4 remains located in cytoplasmic punctate structures in the DCT of mice subjected to a potassium depletion, a situation in which WNK4 is overexpressed and activated by phosphorylation23. Whether the WNK4-positive puncta also contain WNK1 isoforms or whether there are two different structures remains to be defined.
The fact that NCC is still phosphorylated in WNK1+/FHHT:SPAK243A/243A mice at a higher level than in SPAK243A/243A mice suggest that NCC can be phosphorylated by another WNK1-activated kinase in vivo. The obvious candidate is OSR1, the phosphorylation of which tends to increase in WNK1+/FHHT:SPAK243A/243A mice (α. 4). Grimm et al. showed that OSR1 is retained in cytoplasmic punctate structures in DCT cells in the absence of SPAK26, where it would not be able to phosphorylate NCC. The authors suggested that an interaction between SPAK and OSR1 could be necessary for the proper localisation of OSR1 near the apical membrane of DCT cells. This hypothesis is supported by the fact that OSR1 is still located near the apical membrane of SPAK243A/243A DCTs (Fig. 5). Indeed, the SPAK protein is still present in these mice, the threonine to alanine mutation only precluding the activation by WNKs (Fig. 4 and11). Tsutsumi et al. showed that SPAK can translocate from the cytoplasm to the cytoskeleton, where it associates with F-actin, upon stress such as an incubation in a hypertonic solution35. It would be interesting to determine if the phosphorylation of the S-motif of SPAK is involved in the regulation of the intracellular localisation of the kinase. Indeed, if it is known that this residue is phosphorylated by the WNKs and that it is not required for the activation of SPAK8, its precise role remains to be characterized. Taken together, these observations suggest that OSR1 could be activated by L-WNK1 and partially compensate for the absence of SPAK in the setting of FHHt. However, at least a third protein could also phosphorylate NCC in vivo since the cotransporter is still phosphorylated and activated by potassium depletion, albeit at a very low level, in a mouse model carrying an inactivation of both SPAK and OSR123. A potential candidate is the calcium-binding protein Cab39, which promotes the binding of WNK4 to NKCC1 and thus enables the activation of the cotransporter in a SPAK-independent manner22. Whether or not Cab39 is involved in the activation of NCC in WNK1+/FHHT:SPAK243A/243A mice remains to be determined as we did not observe a change in Cab39 in the different mouse models.
In conclusion, the present study shows that SPAK activation is required downstream of L-WNK1 to stimulate NCC and provokes FHHt. It also suggests that L-WNK1 does not play the same role than WNK4 in physiological conditions, even though both kinases can activate SPAK in a similar manner in vitro. The physiological role played by L-WNK1 in the DCT, and more particularly the conditions in which it may be essential, remain to be defined.
Material and Methods
Generation of mice
SPAK243A/243A and WNK1+/FHHt mice (C57BL/6 J background) were described previously11,18. SPAK243A/243A mice were first crossed with WNK1+/FHHt to obtain WNK1+/FHHt:SPAK+/243A. Double heterozygous mice were then crossed to produce WNK1+/+:SPAK+/+ (controls), WNK1+/FHHt:SPAK+/+ (WNK1+/FHHt), WNK1+/FHHt:SPAK243/243A and SPAK243/243A mice. All studies were conducted on 3- to 5-month-old male mice and performed in accordance with the European Communities Council Directive. The project has been approved by the French Ministry of Research (#02650.02).
Blood analysis
Blood was collected by retro-orbital puncture on anesthetized mice. Blood gas and electrolytes were analysed on an ABL80Flex analyser (Radiometer).
Telemetry
Experiments were performed on 5 control and 5 WNK1+/FHHt:SPAK243A:243A male mice. The catheter of the BP telemeter (model TA11PA-C10; Data Sciences International) was inserted into the right femoral artery. The telemetric transmitter probe was positioned subcutaneously on the flank. To reduce any infection and pain, the mice received one dose (20 mg/kg ip) of amoxicillin (Clamoxyl; SmithKlineBeecham Laboratories, Nanterre, France) and one dose (5 mg/kg ip) of ketoprofen (Profenid; Aventis, Paris, France). After the mice had recovered from the anesthesia in a warm (36 °C) box, they were housed in individual cages placed on top of the telemetric receivers in a light-dark cycled recording room (7:00–19:00). After a 1-wk recovery period, recordings of cardiovascular parameters and locomotor activity were obtained continuously during 24 h in the freely moving mice in their home cages every 15 minutes for 60 seconds. Each recording was visualized to select one segment without erratic fluctuations of enough duration (51.2 s) every 15 min (4 segments per hour) for the 24 hours i.e. 96 segments for each animal per day. The values for each recording segment were averaged over a 12-hour period (7:00–19:00 for day-time and 19:00–7:00 for night-time. Mice were fed standard A04 mouse chow (UAR, Epinay-sur-Orge, France) with water ad libitum.
Immunoblot analysis
Renal cortex samples were homogenized in a cold extraction buffer containing 0.25 M sucrose, 20 mM tris-Hepes pH 7.4, proteases and phosphatase inhibitors (Complete and PhosSTOP tablets; Roche Diagnostics). The homogenates were then subjected to a first centrifugation (4,000 g for 15 min) to obtain post-nuclear fractions. The supernatant was centrifuged at 17,000 g for 20 min: the resulting pellet corresponds to the plasma membranes-enriched fraction. These fractions were then submitted to SDS/PAGE electrophoresis, and immunoblotting was performed as described36. The following antibodies were used: NCC (gift from D. Ellison), ENaC γ-subunit (gift from J. Loffing), pendrin (gift from S.Wall), NKCC2 phospho-Thr96-T101 (gift from K. Mutig), Cab39 (Cell signalling Technology®), NCC phospho-Thr55, SPAK, OSR1, SPAK/OSR1 phospho-S motif (S383/325) and NKCC2. The last five antibodies were obtained from the Division of Signal Transduction Therapy of the University of Dundee. As cropped images of the immunoblots are presented in the Figures, the full-length images are provided in the Supplementary Figures S3 to S5.
For all experiments, control gels were run prior to western blotting and were stained with Coomassie. An example of such a gel is provided in Supplementary Fig. S6. Several representative bands were then quantified by densitometry to assure equality of loading between samples37.
Quantification of the band(s) corresponding to the protein of interest was performed by densitometry using the Image J software. Densitometric values were normalized to the mean of the control group that was defined as 100%, and results were expressed as mean ± s.e.m.
Immunofluorescence on frozen kidney sections
Kidneys were fixed by perfusion in the aorta with 10% formol - pH 7.0 (Microm-Microtech, France). Dissected kidneys were washed in cold PBS and frozen in isopentane cooled with liquid nitrogen. 4 µm thick cryosections were blocked with PBS-0.02% BSA and then incubated overnight at 4 °C with the primary antibodies [sheep anti-OSR1 and sheep anti-NCC (Division of Signal Transduction Therapy of the University of Dundee), rabbit anti-NCC (Millipore), and rabbit anti-WNK1 (Bethyl)]. After three 5-min washes with PBS, the sections were incubated with the two fluorophore-conjugated secondary antibodies [AlexaFluor™ 488 Donkey-anti Sheep, AlexaFluor™ 546 Goat-anti Rabbit (Invitrogen™, ThermoFisher Scientific)] for two hours at room temperature and then washed with PBS. Sections were mounted with the Mountant Permafluor medium (ThermoFisher Scientific). Representative images were acquired with an inverted Olympus IX83 microscope.
Quantification of the OSR1 signal was performed using the Image J software. Regions of interest (ROI) were set on individual DCTs as lines across the height of a single epithelial cell, avoiding the nucleus. Five ROIs, in average, were traced for each DCT and at least 7 tubules were analysed per animal. The intensity of each pixel of the ROI was measured. The background, measured of each section, was measured and subtracted. We then calculated two means: (1) the mean of the intensities along the whole ROI and (2) the mean of the intensities of the pixels located in the first quarter of the ROI, near the apical membrane (Supplementary Figure S7). The ratio between these two values allowed us to evaluate the fraction of OSR1 protein located near the apical membrane.
Statistical analysis
When analysing two groups of mice, we used an unpaired student t-test. An one-way ANOVA followed by Sidak’s multiple comparisons test was used to analyse more than two groups with n ≥5 while a Kruskal-Wallis followed by a Mann-Whitney test was used to analyse more than two groups with n <5. Data are given as mean ± s.e.m. A difference between groups was considered significant when P < 0.05.
Materials and Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files). All the materials and protocols, except those generated by other laboratories as stated above, are available from the corresponding author.
References
Gordon, R. D. Syndrome of hypertension and hyperkalemia with normal glomerular filtration rate. Hypertension 8, 93–102 (1986).
Wilson, F. H. et al. Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112 (2001).
Boyden, L. M. et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482, 98–102 (2012).
Louis-Dit-Picard, H. et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 44, 456–460, S1–3 (2012).
Uchida, S., Sohara, E., Rai, T. & Sasaki, S. Regulation of with-no-lysine kinase signaling by Kelch-like proteins. Biol. Cell 106, 45–56 (2014).
Mastroianni, N. et al. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome. Am. J. Hum. Genet. 59, 1019–1026 (1996).
Simon, D. B. et al. Gitelman’s variant of Barter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat. Genet. 12, 24–30 (1996).
Vitari, A. C., Deak, M., Morrice, N. A. & Alessi, D. R. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem. J. 391, 17–24 (2005).
Moriguchi, T. et al. WNK1 Regulates Phosphorylation of Cation-Chloride-coupled Cotransporters via the STE20-related Kinases, SPAK and OSR1. J. Biol. Chem. 280, 42685–42693 (2005).
Piechotta, K., Lu, J. & Delpire, E. Cation Chloride Cotransporters Interact with the Stress-related Kinases Ste20-related Proline-Alanine-rich Kinase (SPAK) and Oxidative Stress Response 1 (OSR1). J. Biol. Chem. 277, 50812–50819 (2002).
Rafiqi, F. H. et al. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2, 63–75 (2010).
Yang, S.-S. et al. SPAK-Knockout Mice Manifest Gitelman Syndrome and Impaired Vasoconstriction. J. Am. Soc. Nephrol. JASN 21, 1868–1877 (2010).
Yang, S.-S. et al. Phosphorylation Regulates NCC Stability and Transporter Activity In Vivo. J. Am. Soc. Nephrol. JASN 24, 1587–1597 (2013).
Yang, S.-S. et al. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 5, 331–344 (2007).
Chiga, M. et al. Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J Cell Sci 124, 1391–1395 (2011).
Chu, P.-Y. et al. SPAK Deficiency Corrects Pseudohypoaldosteronism II Caused by WNK4 Mutation. Plos One 8 (2013).
Lin, S.-H. et al. Impaired phosphorylation of Na+-K+-2Cl− cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc. Natl. Acad. Sci. USA 108, 17538–17543 (2011).
Vidal-Petiot, E. et al. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc. Natl. Acad. Sci. USA 110, 14366–14371 (2013).
Grimm, P. R. et al. Integrated compensatory network is activated in the absence of NCC phosphorylation. J. Clin. Invest. 125, 2136–2150 (2015).
Masilamani, S. et al. Time course of renal Na-K-ATPase, NHE3, NKCC2, NCC, and ENaC abundance changes with dietary NaCl restriction. Am. J. Physiol. - Ren. Physiol. 283, F648–F657 (2002).
Vallet, M. et al. Pendrin Regulation in Mouse Kidney Primarily Is Chloride-Dependent. J. Am. Soc. Nephrol. 17, 2153–2163 (2006).
Ponce-Coria, J. et al. A Novel Ste20-related Proline/Alanine-rich Kinase (SPAK)-independent Pathway Involving Calcium-binding Protein 39 (Cab39) and Serine Threonine Kinase with No Lysine Member 4 (WNK4) in the Activation of Na-K-Cl Cotransporters. J. Biol. Chem. 289, 17680–17688 (2014).
Terker, A. S. et al. Potassium Modulates Electrolyte Balance and Blood Pressure through Effects on Distal Cell Voltage and Chloride. Cell Metab. 21, 39–50 (2015).
Wade, J. B. et al. SPAK-mediated NCC regulation in response to low-K+ diet. Am. J. Physiol. - Ren. Physiol. 308, F923–F931 (2015).
Lee, D. H. et al. Effects of ACE inhibition and ANG II stimulation on renal Na-Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am. J. Physiol. - Cell Physiol. 304, C147–C163 (2013).
Grimm, P. R. et al. SPAK Isoforms and OSR1 Regulate Sodium-Chloride Co-transporters in a Nephron-specific Manner. J. Biol. Chem. 287, 37673–37690 (2012).
Schultheis, P. J. et al. Phenotype Resembling Gitelman’s Syndrome in Mice Lacking the Apical Na+-Cl− Cotransporter of the Distal Convoluted Tubule. J. Biol. Chem. 273, 29150–29155 (1998).
Castañeda-Bueno, M. et al. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc. Natl. Acad. Sci. USA 109, 7929–7934 (2012).
Vidal-Petiot, E. et al. A New Methodology for Quantification of Alternatively Spliced Exons Reveals a Highly Tissue-Specific Expression Pattern of WNK1 Isoforms. Plos One 7 (2012).
Chávez-Canales, M. et al. WNK-SPAK-NCC Cascade RevisitedNovelty and Significance. Hypertension 64, 1047–1053 (2014).
O’Reilly, M. et al. Dietary Electrolyte–Driven Responses in the Renal WNK Kinase Pathway In Vivo. J. Am. Soc. Nephrol. 17, 2402–2413 (2006).
Roy, A. et al. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J. Clin. Invest. 125, 3433–3448 (2015).
Zambrowicz, B. P. et al. Wnk1 kinase deficiency lowers blood pressure in mice: A gene-trap screen to identify potential targets for therapeutic intervention. Proc. Natl. Acad. Sci. USA 100, 14109–14114 (2003).
Boyd-Shiwarski, C. R. et al. Potassium-Regulated Distal Tubule WNK Bodies are Kidney-Specific WNK1 Dependent. Mol. Biol. Cell https://doi.org/10.1091/mbc.E17-08-0529 (2017).
Tsutsumi, T., Ushiro, H., Kosaka, T., Kayahara, T. & Nakano, K. Proline- and Alanine-rich Ste20-related Kinase Associates with F-actin and Translocates from the Cytosol to Cytoskeleton upon Cellular Stresses. J. Biol. Chem. 275, 9157–9162 (2000).
Picard, N. et al. Defective ENaC Processing and Function in Tissue Kallikrein-deficient Mice. J. Biol. Chem. 283, 4602–4611 (2008).
Terris, J., Ecelbarger, C. A., Nielsen, S. & Knepper, M. A. Long-term regulation of four renal aquaporins in rats. Am. J. Physiol 271, F414–422 (1996).
Acknowledgements
The authors thank all the technicians of the Elevage Rongeurs Inserm for the care given to the mice. This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), the Fondation pour la Recherche pour l’Hypertension Artérielle, the Agence Nationale pour la Recherche (ANR-12-ISVS1-0001-01/New-BP-KIT). CR received a fellowship from the Ministère de l’Enseignement Supérieur et de la Recherche.
Author information
Authors and Affiliations
Contributions
C.R. designed the study, performed and analysed the experiments and wrote the manuscript. C.S. and V.B. performed and analysed the experiments. P.F. analysed the data. J.H. designed the study, analysed the experiments and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rafael, C., Soukaseum, C., Baudrie, V. et al. Consequences of SPAK inactivation on Hyperkalemic Hypertension caused by WNK1 mutations: evidence for differential roles of WNK1 and WNK4. Sci Rep 8, 3249 (2018). https://doi.org/10.1038/s41598-018-21405-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-21405-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
