
Abstract
Hypertension is the most prevalent health condition worldwide, affecting ~1 billion people. Gordon’s syndrome is a form of secondary hypertension that can arise due to a number of possible mutations in key genes that encode proteins in a pathway containing the With No Lysine [K] (WNK) and its downstream target kinases, SPS/Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress responsive kinase 1 (OSR1). This pathway regulates the activity of the thiazide-sensitive sodium chloride cotransporter (NCC), which is responsible for NaCl reabsorption in the distal nephron. Therefore, mutations in genes encoding proteins that regulate the NCC proteins disrupt ion homeostasis and cause hypertension by increasing NaCl reabsorption. Thiazide diuretics are currently the main treatment option for Gordon’s syndrome. However, they have a number of side effects, and chronic usage can lead to compensatory adaptations in the nephron that counteract their action. Therefore, recent research has focused on developing novel inhibitory molecules that inhibit components of the WNK-SPAK/OSR1-NCC pathway, thereby reducing NaCl reabsorption and restoring normal blood pressure. In this review we provide an overview of the currently reported molecular inhibitors of the WNK-SPAK/OSR1-NCC pathway and discuss their potential as treatment options for Gordon’s syndrome.
Similar content being viewed by others

Hypertension and Gordon’s syndrome
Hypertension (high blood pressure) is the most widespread chronic health condition globally [1]. Between 1980 and 2008, the total number of hypertension cases in adults rose from 600 million to almost 1 billion [2]. This number has been predicted to rise to 1.56 billion by 2025 [3]. Hypertension is a major risk factor for an array of cardiovascular diseases that can lead to disability and premature mortality [1]. Such diseases include ischemic heart disease and stroke, which are the two leading causes of death worldwide [4]. This makes hypertension a pressing global health concern and an important area of research.
Gordon’s syndrome, also known as pseudohypoaldosteronism type II (PHA2) or familial hyperkalemic hypertension (FHHt), is a form of secondary hypertension. It is a rare, inherited form of hypertension that is caused by one of five possible subtypes of mutations mentioned below [5]. The disease shows full penetrance and is characterized by a number of symptoms, including hypertension, hyperkalemia, and metabolic acidosis [6]. The genetic defects that cause Gordon’s syndrome lead to greater sodium chloride reabsorption in the distal tubule. This leads to hypertension and reduced renin levels [6, 7]. Despite the reduced renin secretion, patients display normal or elevated aldosterone levels.
There are five subtypes of Gordon’s syndrome (PHA2 A-E), each of which is characterized by a distinct mutation. The first two subtypes, PHA2A and PHA2B, were discovered in 1997 using linkage studies [8]. PHA2A is caused by a mutation in chromosome 1 (1q31–q42). However, the specific gene has yet to be identified. PHA2B is caused by a mutation in WNK4 (With No Lysine (K)), located on chromosome 17 (17q21.2). PHA2C is caused by a mutation on chromosome 12 (12p13.33); this is a deletion mutation that occurs within WNK1 [9]. The WNK4 and WNK1 genes encode the serine/threonine-protein kinases WNK4 and WNK1, respectively. Through alternative promoters, the WNK1 gene generates the long WNK1 (L-WNK1) isoform and kidney-specific WNK1 (KS-WNK1). L-WNK1 is kinase-active and expressed ubiquitously, whereas KS-WNK1 lacks a functional kinase domain and is expressed in the kidney specifically [10]. These kinases are involved in the regulation of ion reabsorption in the distal renal tubule via the WNK signaling pathway [11, 12]. PHA2D is caused by a mutation in KLHL3, which is located on chromosome 5 (5q31.2) and encodes the protein Kelch-like 3 (KLHL3) [13, 14]. Finally, PHA2E is caused by a mutation in CUL3, which is located on chromosome 2 (2q36.2) and encodes the protein Cullin 3 (CUL3) [13]. CUL3/KLHL3 are components of an E3 ubiquitin ligase complex that promotes WNK1/4 degradation [15].
WNK-SPAK-NCC kinase pathway
Renal blood pressure regulation
The regulation of salt reabsorption (NaCl) in the kidney is critical for the maintenance of blood pressure [16]. Different amounts of NaCl are reabsorbed at various points in the nephron. In the distal convoluted tubule of the nephron, the sodium chloride cotransporter (NCC) is responsible for the reabsorption of 5%–10% filtered NaCl [17]. NCC is a transmembrane protein between 1002 and 1030 amino acids in length [18]. The NCC protein is encoded by the gene SLC12A3 (solute carrier family 12 member 3), located on chromosome 16 (16q13) [18]. Other members of the SLC12 family include potassium chloride cotransporters (KCC1–4) and sodium potassium chloride cotransporters (NKCC1–2), both of which are also involved in renal ion homeostasis [19]. While the majority of NaCl is reabsorbed in early segments of the nephron, the activity of NCC is crucial for the fine-tuning of the salt concentration in the extracellular fluid [18], which affects blood volume and, therefore, blood pressure.
NCC regulation in the WNK-SPAK/OSR1-NCC pathway
Two opposing processes occur in the distal nephron to control blood pressure: K+ excretion and NaCl reabsorption. WNK signaling is responsible for regulating electrolyte homeostasis and, thus, blood pressure [20]. L-WNK1, WNK3, and WNK4 are responsible for the regulation of a number of cation-chloride cotransporters (CCCs) [21, 22]. These WNK kinases are expressed in the kidney and can activate NCC, KCC1–3–4, NKCC1, and NKCC2 through a cascade of phosphorylation reactions involving SPS/Ste20-related proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase (OSR1) [23, 24]. SPAK and OSR1 are members of the STE protein kinase family and are responsible for the direct phosphorylation and activation of NCC [25]. Upon activation by WNK kinases, SPAK and OSR1 can phosphorylate and activate cation-chloride cotransporters (CCCs) [22, 26].
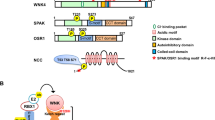
The kinase domain of WNK kinases is found in the N terminus. To phosphorylate SPAK and OSR1, L-WNK1 and WNK4 can bind to the conserved C-terminal (CCT) domains of SPAK and OSR1 [11, 27]. SPAK is phosphorylated by WNK on residues Ser373 and Ser387 of the S-motif and Thr233 of the T-loop. OSR1 is phosphorylated by WNK on residues Ser325 and Ser339 of the S-motif and Thr185 of the T-loop [28]. The phosphorylation of the S-motif of SPAK/OSR1 has recently been found to be important for their stability under osmotic stress [29]. However, the phosphorylation of the T-loop is required for their activation. Once phosphorylation has occurred, SPAK and OSR1 bind to the scaffold protein MO25 (mouse protein 25), which results in 80- and 100-fold activation of SPAK and OSR1, respectively [30]. In complex with MO25, SPAK and OSR1 can subsequently phosphorylate a number of CCCs [31]. For the activation of NCC, the CCT domain of SPAK and OSR1 binds to an RFTI amino acid motif located at its N terminus [27]. Here, SPAK and OSR1 phosphorylate three amino acid residues: Thr46, Thr55, and Thr60 [28]. This activates NCC and allows the cotransport of NaCl from the distal tubule lumen. SPAK CCT domain knock-in mice displayed markedly reduced SPAK activity and the phosphorylation of NCC at the residues phosphorylated by SPAK [32], as has been shown in SPAK kinase-dead knock-in mice [33]. These animals showed typical features of Gitelman syndrome, with mild hypokalemia, hypomagnesemia, and hypocalciuria, and displayed salt wasting upon switching to a low-Na diet [32, 33]. A schematic representation of the WNK-SPAK/OSR1-NCC pathway is shown in Fig. 1.

a WNK-SPAK/OSR1-NCC pathway: L-WNK1 or WNK4 binds to the CCT domain of SPAK/OSR1 and phosphorylates its T-loop kinase domain. SPAK is phosphorylated at Thr233 and OSR1 is phosphorylated at Thr185. The scaffold protein MO25 binds to phosphorylated SPAK/OSR1, activating it further. The CCT domain of activated SPAK/OSR1 binds to the RFTI motif of NCC. The T-loop kinase domain of SPAK/OSR1 phosphorylates NCC at three amino acid residues: Thr46, Thr55, and Thr60. This activates NCC, allowing the cotransport of NaCl from the tubule lumen. b WNK ubiquitination by the CUL3-KLHL3 E3 ubiquitin ligase complex: L-WNK1 or WNK4 is bound to the CUL3-KLHL3 E3 ubiquitin ligase complex via the C terminus domain of KLHL3. RING-box protein 1 is bound to the BTB domain of CUL3 and recruits an E2 ubiquitin-conjugating enzyme that ubiquitinates L-WNK1 or WNK4. This targets WNK1/4 for proteasomal degradation, thereby reducing the amount of available L-WNK1 or WNK4.
Regulation of the WNK-SPAK/OSR1-NCC pathway by the CUL3-KLHL3-E3 ubiquitin ligase complex
The WNK-SPAK/OSR1-NCC pathway can be downregulated by enhancing the degradation of WNK kinases. This is achieved by the CUL3-KLHL3 E3 ubiquitin ligase complex [34]. This complex includes the proteins KLHL3 and CUL3. KLHL3 is expressed primarily in the distal tubule of the nephron on the apical membrane, and CUL3 is expressed ubiquitously throughout the nephron [35].
The CUL3-KLHL3 E3 ubiquitin ligase complex stems from a Cullin 3-Ring E3 ubiquitin ligase complex. The Cullin 3-Ring complex contains CUL3 bound to a RING ubiquitin ligase. The Cullin 3-Ring complex interacts with its substrate via an adapter protein, KLHL3. The KLHL3 protein contains three domains: a BTB domain in its N terminus, a BACK domain, and a C terminal domain containing Kelch-like repeats in a β-propeller structure [35, 36]. The BTB domain of KHL3 interacts with CUL3. The C terminal domain of KLHL3 binds substrate proteins, allowing KLHL3 to recruit substrates for the Cullin3-based ubiquitin ligase. The RING protein of the Cullin3-based ubiquitin ligase then transfers ubiquitin to the substrate bound to the C terminal domain of KLHL3, making the substrate for proteasome degradation.
L-WNK1 and WNK4 are both substrates of the CUL3-KLHL3 E3 ubiquitin ligase [34, 37]. L-WNK1 and WNK4 contain an acid motif that is the binding site for KLHL3 [9]. Once bound to the ligase, via the C terminus of KLHL3, L-WNK1 and WNK4 can be ubiquitinated by the RING ubiquitin ligase. This ubiquitination targets WNKs for proteasomal degradation [34, 36]. A graphic representation of the CUL3-KLHL3 E3 ubiquitin ligase complex interacting with WNKs is shown in Fig. 1.
WNK-SPAK/OSR1-NCC pathway in Gordon’s syndrome
Mutations causing Gordon’s syndrome affect the function of the WNK-SPAK/OSR1-NCC pathway. This results in disrupted ion homeostasis, thus leading to hypertension. The mutations that occur in the WNK4 and WNK1 kinases give rise to Gordon’s syndrome (PHA2B and PHA2C, respectively) due to an increase in the abundance of these kinases [38]. In turn, this leads to the overactivation of NCC. The mutations that occur in KLHL3 and CUL3 give rise to Gordon’s syndrome (PHA2D and PHA2E, respectively) due to the insufficient degradation of WNK kinases. This leads to an accumulation of WNKs, thus causing the overactivation of NCC. Mutations in KLHL3 and CUL3 occur in 80% of Gordon’s syndrome patients [13].
WNK1 mutations are intronic deletions that result in increased expression of L-WNK1 [39]. The expression of L-WNK1 is normally low in the distal tubule. However, this mutation causes increased expression in the distal tubule [39]. As a result, the WNK-SPAK/OSR1-NCC pathway is overactivated, and NaCl reabsorption is increased, leading to hypertension.
Mutations in WNK4 are exonic missense mutations that occur in the C-terminal domain next to the second coiled-coiled motif and a unique amino acid sequence called the acid motif [34, 37]. Mutations in the acidic motif of WNK4 markedly impair its binding to KLHL3 [34]. The acid motif, which is present in all WNK kinases, is the binding site for KLHL3 [9]. Thus, the disruption of this binding site hinders the ability of WNK4 to be ubiquitinated by the CUL3-KLHL3 E3 ubiquitin ligase complex. The resulting lack of WNK4 degradation leads to an accumulation of WNK4, which, in turn, causes NCC phosphorylation and activation. Interestingly, a number of polymorphisms in both WNK1 [40,41,42] and WNK4 [43] have been identified that are not associated with Gordon’s syndrome but are still affiliated with an increased risk of hypertension, which provides a stronger justification for potentially targeting the WNK-SPAK/OSR1-NCC pathway as a treatment option for other types of hypertension.
The mutations in KLHL3 that cause Gordon’s syndrome are predominantly missense mutations [13, 14]. These mutations mainly occur within two locations. The first location is the C terminal domain of KLHL3, which is responsible for binding substrate proteins (i.e., WNKs). The second location is the N terminus BTB domain of KLHL3, which is the binding site for the Cullin 3-Ring E3 ubiquitin ligase complex [13, 14]. Thus, a mutation in either of these locations results in insufficient WNK kinase degradation: The C terminal domain mutation impairs the ability of KLHL3 to bind WNKs, thereby preventing the ubiquitination of WNKs. The N terminal domain mutation impairs the ability of KLHL3 to bind the Cullin 3-Ring E3 ubiquitin ligase complex, thus preventing the initial formation of the CUL3-KLHL3 ubiquitin ligase complex.
Mutations in CUL3 give rise to the most severe phenotype of Gordon’s syndrome. Compared to mutations in WNK1, WNK4, and KLHL3, mutations in CUL3 have been shown to cause significantly more severe metabolic acidosis and hyperkalemia [13]. Additionally, the onset of hypertension occurs before the age of 18 in 94% of patients. Therefore, the development of a treatment that targets CUL3 would be particularly significant [13]. Mutations in CUL3 that cause Gordon’s syndrome are located at sites involved in the splicing of exon 9. As a consequence, exon 9 is skipped during transcription, leading to a 57-amino-acid deletion in the CUL3 protein (CUL3-Δ9) [13, 44]. The expression of CUL3-Δ9 results in the malfunction of the CUL3-KLHL3 ubiquitin ligase complex, thus impairing the degradation of WNKs [34]. There are a number of mechanisms by which this has been proposed to occur. One suggestion is that there is a reduced availability of wild-type CUL3 because it becomes dimerized with CUL3-Δ9 in an unstable structure that cannot ubiquitinate substrate proteins [45]. Another suggestion is that the CUL3 mutation is a gain-of-function mutation: CUL3 can be activated through a process termed neddylation, which involves the covalent attachment of Nedd8. This process is increased in the mutant CUL3-Δ9 [46]. The increased activation of CUL3-Δ9 allows it to ubiquitinate off-target substrates. KLHL3 has been suggested to be ubiquitinated by CUL3-Δ9 as a result of this gain-of-function mutation [47]. This would lead to the degradation of KLHL3 and the prevention of CUL3-KLHL3 ubiquitin ligase complex formation [47]. It has also been suggested that this gain-of-function mutation allows the autoubiquitination of CUL3-Δ9. This has been demonstrated in vivo with CUL3-Δ9 transgenic mice that exhibited decreased levels of CUL3-Δ9 expression and normal KLHL3 expression [48]. Wild-type CUL3 is also expressed in the vasculature, where the expression of CUL3-Δ9 in smooth muscle interferes with the expression and function of wild-type CUL3, consequently affecting blood pressure [49,50,51].
Current pharmacological treatments for Gordon’s syndrome
Hypertension and metabolic abnormalities in Gordon’s syndrome can be controlled pharmacologically using thiazide diuretics [52,53,54]. Thiazide diuretics can acutely reduce hypertension by decreasing NaCl reabsorption from the distal tubule, which is achieved through the inhibition of NCC [55, 56], which decreases NaCl reabsorption. The mechanism of action of chronic thiazide diuretic use is not fully understood [57].
There are a number of limitations to using thiazide diuretics as a treatment option for hypertension. Diuretics are ineffective as a monotherapy in a portion of hypertensive patients. Furthermore, a study by Morsing and colleagues found adaptations of the distal convoluted tubule of rats after chronic infusion of thiazide diuretics: The membrane density of NCC was increased in response to chronic thiazide use [57]. However, despite the increase in NCC membrane expression, the reabsorption of NaCl was reduced [57]. Thiazide diuretics also have a range of side effects, including hyperglycemia, dyslipidemia, and an increased risk of developing type 2 diabetes [58, 59]. For these reasons, research into other treatment options for hypertension, including Gordon’s syndrome, is becoming increasingly important.
This review examines whether the WNK-SPAK/OSR1-NCC pathway could be an effective target for the treatment of Gordon’s syndrome. By altering the activity of various components in this pathway, NCC activation can be prevented, thereby reducing blood pressure. Research has focused on developing molecules that inhibit either the kinase activity or the binding activity of WNKs, SPAK/OSR1, or MO25. Therefore, this review discusses the ability of the currently reported molecules to reduce blood pressure in Gordon’s syndrome and possibly in other forms of hypertension [60].
WNK kinase inhibitors
Due to their integral role in the WNK-SPAK/OSR1-NCC pathway, WNK kinases have been targeted for the treatment of hypertension in Gordon’s syndrome. Although homozygous WNK1-knockout mice have been shown to die during embryonic development [61,62,63], only one of these three independent studies investigating heterozygous WNK1-knockout mice reported mild hypotension [61]. Homozygous WNK4-knockout mice show mild Gitelman-like symptoms, including hypotension and reduced phosphorylation of NCC [64,65,66]. This suggests that targeting specific WNK kinases may be beneficial in reducing blood pressure through the reduced activation of NCC. Research has focused on developing molecules that can bind to WNK kinases and inhibit their function, thereby preventing their participation in the WNK-SPAK/OSR1-NCC pathway. These molecules can be broadly divided into two groups: orthosteric (ATP competitive) and allosteric (ATP noncompetitive) inhibitors. The binding properties of some key WNK inhibitory molecules are presented in Table 1.
Orthosteric WNK kinase inhibitors
The currently reported orthosteric WNK kinase inhibitors take advantage of a unique structural irregularity found in the kinase domain of the WNK kinases. WNK kinases are characterized by the abnormal placement of a catalytic lysine residue (Lys233) in their binding site for ATP [67]. Since this variation is specific to WNK kinases, a high level of selectivity can be achieved for WNK kinase inhibition over other kinases, which do not share this structural configuration. Thus, highly selective inhibitory molecules that exploit the catalytic lysine residue have been generated for WNK kinases.
A high-throughput screen conducted by Yamada et al. led to the generation of a potent WNK kinase inhibitor called WNK463 [68]. The X-ray co-crystal structure of this molecule in complex with the kinase-dead mutant of WNK1 (S382A) revealed its unique binding method, which arises due to the abnormal placement of the catalytic lysine residue [68]. This provides excellent binding selectivity for the WNK kinases over other kinases. This selectivity for WNK was demonstrated when a high concentration of WNK463 (10 μM) showed >50% inhibition against only two kinases out of the 442 that were tested [68]. This suggests that WNK463 will incur fewer unwanted side effects, as fewer off-target kinases will be inhibited. In vitro, WNK463 inhibited the kinase activity of each of the four WNK kinases (Table 1) [68]. Additionally, WNK463 was shown to prevent the WNK-mediated phosphorylation of OSR1, both in a biochemical assay and in human embryonic kidney 923 (HEK293) cells [68]. Oral dosing of WNK463 (1, 3, or 10 mg/kg) in spontaneously hypertensive rats led to a dose-dependent reduction in blood pressure and an increase in urine output with an increase in urinary Na+ and K+ excretion [68]. In addition, in transgenic mice that overexpress human L-WNK1, oral dosing of WNK463 led to reduced blood pressure and a dose-dependent decrease in SPAK and OSR1 phosphorylation [68]. These results show that the physiological effects of WNK463 indeed resulted from WNK inhibition.
An issue with such ATP competitive inhibitors is that they must compete with ATP for their binding site on WNKs. This means that a molecule with high potency is required to overcome competition from physiological ATP levels. In addition, since the ATP binding site is highly conserved among the WNK kinases [69], achieving selectivity among them has proven difficult [68]. This lack of selective inhibition among WNKs is problematic, as WNK kinases show various levels of activity in physiological processes other than renal ion homeostasis [27, 70]. It should be noted that the development of WNK463 has ceased due to issues in preclinical safety, elicited ataxia, and breathing difficulties at 1–10 mg/kg doses [71]. This may be due to this lack of selectivity for the WNK family members, resulting in off-target effects. For these reasons, molecules that inhibit WNK allosterically have gained traction; they can target less conserved regions of the WNK kinases, possibly allowing them to exhibit selectivity between the WNK family members. Additionally, they do not need to compete with ATP for their binding site on WNKs.
Allosteric WNK kinase inhibitors
Another high-throughput screen was conducted by Yamada et al. to identify allosteric WNK kinase inhibitors [72]. A total of 1.2 million compounds were screened at a high concentration (50 μM) so that any weakly binding compounds would still be detected. Initially, the screen identified 8257 compounds, but further prioritization and modification led to the development of compound 2. The co-crystallization of compound 2 with L-WNK1 revealed a new allosteric binding pocket that is exclusive to the WNK kinases, thus demonstrating the selectivity for WNK kinases that can be achieved through an allosteric binding site [72]. This selectivity was validated when a high concentration of compound 2 (10 μM) showed >50% inhibition only in L-WNK1 against a panel of 61 other kinases [72]. In addition, no phosphorylation of OSR1 occurred when exposed to a high concentration of compound 2 (10 μM), again showing selectivity for WNKs [72]. This high level of selectivity for WNKs is vital for the development of a kinase inhibitor, given the broad range of physiological processes that other kinases are involved in. In vitro, compound 2 inhibited each of the WNK kinases with similar potency (Table 1) [72]. To test the effects of this inhibition on electrolyte homeostasis, an NKCC1 assay was developed that measured rubidium flux in place of K+ uptake [72]. Compound 2 inhibited rubidium uptake in a dose-dependent manner (IC50 = 0.24 μM). Despite the positive in vitro results of compound 2, in vivo studies are not warranted due to the inadequate pharmacokinetic profile of the compound [72].
Further optimization and refinement of three molecules (compounds 1–3) identified by Yamada et al. [72] were conducted to develop molecules that could be tested in vivo [73]. Various aspects of these molecules were interchanged to identify compounds that had improved pharmacokinetic profiles. This led to the development of 9 additional molecules (compounds 4–12) [73]. In a panel of 440 kinases, compound 11 showed >35% inhibition in only four kinases other than L-WNK1, showing the high level of selectivity that can be achieved when utilizing a binding site other than the highly conserved ATP binding site [73]. Compound 11 showed allosteric inhibition of the WNK kinases, with L-WNK1 and WNK3 being potently inhibited and WNK2 being moderately inhibited compared to WNK4 (Table 1). In HEK293 cells, compound 11 showed a dose-dependent inhibition of OSR1 phosphorylation, suggesting its potential to impede the WNK-SPAK/OSR1-NCC pathway [73]. In vivo testing of compound 11 in rats revealed a reasonable pharmacokinetic profile, with low oral bioavailability and moderate clearance [73]. Therefore, compound 11 was modified, leading to the development of compound 12. This molecule showed an improved pharmacokinetic profile, with lower clearance and a twofold increase in oral bioavailability [73]. Oral dosing of compound 12 (10, 30, or 100 mg/kg) led to reductions in systolic blood pressure in a dose-dependent manner [73]. Additionally, in spontaneously hypertensive rats, ascending oral doses of compound 12 on successive days led to a dose-dependent increase in urine production with increased Na+ and K+ excretion [73]. The good pharmacokinetic profile of compound 12 and its efficacy in vivo make it a promising candidate for further development as an antihypertensive drug.
In another study conducted by Pinkas et al., a fluorescence-based thermal shift assay was used to screen 860 kinase inhibitors in an attempt to identify inhibitors of WNKs [74]. The kinase inhibitor PP121 was the top hit identified in the screen. This inhibitor was originally discovered by Apsel et al. [75]. PP121 strongly inhibited L-WNK1 and WNK3 (Table 1), indicating its potential as a treatment for Gordon’s syndrome [74]. A selectivity profile for PP121 was conducted against a panel of 135 serine/threonine kinases, the same family of kinases to which WNK belongs [75]. At 1 μM, PP121 inhibited only 19 kinases >50%. This suggests that PP121 may not incur many off-target effects in vivo, since this high concentration of PP121 showed low levels of inhibition in the same family of kinases as WNK. In addition, another selectivity profile was conducted that included a wider variety of kinases [74]. At 1 μM, PP121 showed >75% inhibition in 29 out of 144 kinases, including L-WNK1. This moderate level of selectivity suggests that further modification of PP121 will be necessary in order to limit off-target effects in a wider range of kinases. The family of kinases that were most affected in this selectivity profile were the tyrosine kinases, suggesting that PP121 may have therapeutic potential as a tyrosine kinase inhibitor. Accordingly, a number of studies have investigated the inhibitory effects of PP121 on various tyrosine kinases [75,76,77]. The overall moderate level of selectivity for WNK in these selectivity assays suggests that further modification of PP121 as a WNK inhibitor will be necessary to limit off-target effects.
One of the major issues in the development of WNK inhibitors is the lack of inhibitor molecule specificity among the WNK kinases. For orthosteric WNK inhibitors, this appears to be a limiting factor that cannot be overcome, since the ATP binding site of WNK kinases is highly conserved [69]. However, recent chemoinformatic analysis has revealed that structural specificities among the WNK isoforms do exist, particularly in the allosteric binding sites [78]. The exploitation of these structural differences may be an effective strategy for the development of specific WNK kinase inhibitors and certainly merits further investigation. Compound 12 [73] may be a strong candidate for further development in this manner.
SPAK and OSR1 inhibitors
SPAK and OSR1, the kinases downstream of WNKs in the WNK-SPAK/OSR1 pathway, have become attractive targets for inhibition. Heterozygous SPAK-knockout mice display reduced blood pressure, while homozygous SPAK-knockout mice exhibit significant electrolyte abnormalities in addition to hypotension [79, 80]. Heterozygous OSR1-knockout mice also exhibit hypotension and are associated with reduced phosphorylation of SPAK [79]. This information suggests that the inhibition of SPAK or OSR1 may be beneficial in reducing blood pressure in Gordon’s syndrome. The main features of some key SPAK and OSR1 inhibitory molecules are presented in Table 2.
A high-throughput screen and a drug repositioning strategy conducted by Kikuchi et al. identified two allosteric SPAK inhibitors: Stock 1S-14279 and closantel [81]. Stock 1S-14279 was identified in a novel ELISA-based screen of >20,000 small-molecule compounds, while closantel was screened from a library of 840 existing drugs and had, therefore, already passed several stages of clinical development. Both compounds share a similar chemical structure, and both inhibited SPAK independently of ATP concentration, suggesting that they are allosteric inhibitors (Table 2) [81]. The specificity of these compounds was tested in a panel of 48 kinases [81]. At a concentration of 10 μM, closantel inhibited 6 kinases >50%, and Stock 1S-14279 inhibited 2 kinases >50%, suggesting that these molecules have good selectivity for SPAK. In mouse renal distal tubule–derived (mpkDCT) cells, both compounds inhibited NCC phosphorylation in a dose-dependent manner [81]. In vivo testing in mice revealed that single intraperitoneal injections of Stock 1S-14279 or closantel resulted in a significant decrease in NCC abundance and phosphorylation after 30 min [81]. However, by 120 min, these effects had reversed. Thus, chronic administration of these compounds was tested. Repeated injections of Stock 1S-14279 were lethal in mice, while closantel had no significant effects on blood pressure or electrolyte levels in the urine. Overall, these tests showed promising results in vitro; however, further pharmacological modification may yield better in vivo effects.
AlAmri et al. recently reported that a highly conserved secondary pocket found in the CCT domain of SPAK and OSR1 may be the binding site for Stock1S-14279 and closantel [82]. Therefore, further testing was conducted in silico to identify other inhibitory molecules that bind to this secondary pocket [82]. The screening of 1200 US Food and Drug Administration (FDA)-approved compounds was performed with the intension of finding molecules that bind to the secondary pocket in OSR1. Rafoxanide, a molecule structurally similar to closantel, was one of the best molecules identified in the screen. In vitro, it was shown to inhibit constitutively active forms of OSR1 (OSR1 T185E) and SPAK (SPAK T233E) (Table 2) [82]. In the presence of MO25, a SPAK/OSR1 activator, rafoxanide was still able to inhibit OSR1 with a similar potency (Table 2) [82]. Additionally, in HEK293 cells, the titration of rafoxanide (1–50 μM) caused dose-dependent inhibition of NKCC1 phosphorylation [82]. This suggests that SPAK and OSR1, the upstream kinases of NKCC1, were inhibited. These findings advocate the importance of the CCT domain of SPAK and OSR1 for their kinase activity and highlight the secondary pocket as a good target for inhibitory molecules. In vivo testing and compound modification will be important next steps in the development of clinically available SPAK and OSR1 inhibitors.
Given the encouraging results from these in silico screening methods, AlAmri et al. recently conducted a high-throughput screen of 1200 FDA-approved drugs with the hope of identifying novel SPAK and OSR1 inhibitors [83]. The drugs were screened at 20 μM and led to the identification of 7 compounds that inhibited OSR1 T185E in a dose-dependent manner. Of these, verteporfin emerged as the most potent OSR1 T185E inhibitor (Table 2). In addition, verteporfin was equally potent in its inhibition of SPAK T233E (Table 2). An in vitro kinase assay revealed that the inhibition of OSR1 by verteporfin was not significantly affected by ATP concentration, suggesting that it binds in an ATP-independent manner [83]. In silico docking of verteporfin to OSR1 suggested that binding occurs in an allosteric site adjacent to the kinase domain, similar to the allosteric WNK kinase inhibitors mentioned above [83]. To test the selectivity of verteporfin for SPAK and OSR1, a screen of 140 kinases was conducted at 1 μM [83]. Other than SPAK and OSR1, eight kinases were inhibited ≥70%. This may be problematic for the future development of verteporfin as an antihypertensive drug, as the inhibition of these off-target kinases may lead to undesired side effects. The inhibitory effects of verteporfin were then tested in HEK293 cells [83]. Verteporfin dose-dependently inhibited NKCC1 phosphorylation, suggesting the inhibition of SPAK or OSR1. Importantly, this inhibition did not occur in the WNK phosphorylation site on SPAK and OSR1. This indicates that verteporfin inhibits only active SPAK and OSR1 rather than inhibiting the activation of SPAK and OSR1 by WNKs. In addition, verteporfin did not affect the ability of MO25 to activate OSR1 in vitro [83]. Although animal testing was not conducted in this study [83], there have been reports of hypotension in animals being treated with verteporfin, suggesting that the inhibition of SPAK and OSR1 can reduce blood pressure [84].
Although these findings are promising, there are a number of limitations to targeting SPAK and OSR1. NCC and NKCC1 become activated when phosphorylated by SPAK and OSR1, whereas KCCs are inhibited when phosphorylated by SPAK and OSR1 [85]. The neuronal excitability of GABAergic neurons is partly mediated by Cl− efflux through KCCs [86]. Thus, the inhibition of SPAK/OSR1 may prevent the phosphorylation and inhibition of these transporters, leading to undesirable effects in the central nervous system. This effect has been observed with the inhibition of WNK3, which is an upstream kinase of SPAK and OSR1 [87], suggesting that a similar phenotype may occur with SPAK and OSR1 inhibition. Another potential limitation is that the kidneys employ adaptive mechanisms to counteract physiological change. Although SPAK-null mice display hypotension, compensatory mechanisms for NaCl reabsorption have been observed in SPAK-knockout mice [88]. These adaptations may explain why the hypotension observed is relatively mild in comparison to the predicted effects of the loss of NCC function. Such mechanisms include distal nephron modeling and the activation of the paracrine signaling system to activate salt transport pathways in the distal nephron and ultimately enhance NaCl reabsorption.
Prevention of SPAK and OSR1 protein–protein interactions
A number of molecules have been identified that target protein–protein interactions between SPAK/OSR1 and other components of the WNK-SPAK/OSR1-NCC pathway. Some of these molecules are involved in the inhibition of SPAK/OSR1 binding to WNK, while others prevent MO25 from binding to SPAK/OSR1. By preventing these interactions, the activity of the WNK-SPAK/OSR1-NCC pathway can be reduced, thereby leading to a decrease in blood pressure. The main features of some key molecules that inhibit SPAK and OSR1 binding are presented in Table 3.
Inhibition of SPAK/OSR1 binding to WNK
A screen of 16,902 compounds conducted by Mori et al. led to the identification of 10 potential SPAK binding inhibitor compounds [89]. Two of these compounds, Stock 1S-50699 and Stock 2S-26016, showed the greatest inhibition of SPAK binding to WNK4 out of the compounds that were screened (Table 3). Both compounds were shown to bind to the CCT domain of SPAK, thereby preventing interactions with WNK [89]. Stock 1S-50699 exhibited good drug properties, such as slow binding and dissociation, compared to Stock 2S-26016, suggesting that it may have greater potential to be developed into an antihypertensive drug [89]. To test the specificity of these compounds to SPAK, two negative controls were conducted [89]. First, at high concentrations (200 μM), neither molecule inhibited mitogen-activated protein kinase, a closely related kinase that is not involved in the WNK-SPAK-NCC pathway. Second, a noninhibitory analog of Stock 2S-26016 had no effect on the phosphorylation of SPAK and NCC, showing that the inhibitory activity of Stock 2S-26016 was not due to nonspecific effects. The dosing of both compounds in mpkDCT cells (25, 50, 100, and 200 μM) inhibited the phosphorylation of SPAK and NCC in a dose-dependent manner [89]. These in vitro and in vivo tests are encouraging; therefore, further testing in animal models will be beneficial for their development.
In a recent study conducted by Zhang et al., a novel SPAK binding inhibitor named ZT-1a was developed [71]. Pharmacophores from closantel, rafoxanide, and STOCK1S-14279 were combined to create this specific SPAK inhibitor; from closantel, the 2-(4-amino-2-chloro-5-methylphenyl)-2-(4-chlorophenyl)acetonitrile moiety was used, and from STOCK1S-14279 and rafoxanide, the chloro-substituted 2-hydroxybenzoic acid was used. To test the selectivity of ZT-1a for SPAK, a selectivity profile was conducted at 10 μM ZT-1a [71]. Inhibition ≥50% was achieved in 6 out of the 140 kinases that were tested, suggesting a high level of selectivity for SPAK. In vitro, ZT-1a reduced the kinase activity of SPAK in a dose-dependent manner [71]. Interestingly, the potency of this inhibition was only marginally reduced in the presence of the SPAK activator MO25 (Table 3). ZT-1a was shown to inhibit SPAK independently of ATP concentration, suggesting an allosteric binding site [71]. A SPAK antibody pull-down assay using HEK293 cell lysates was conducted to determine the inhibitory action of ZT-1a [71]. Coimmunoprecipitation of SPAK with L-WNK1 was reduced significantly in a dose-dependent manner, suggesting that ZT-1a inhibits the binding of SPAK and WNKs. In vivo testing in mice revealed that although there was no mortality in naive mice treated with ZT-1a and closantel, all ischemic stroke mice died within 2 days, while the ZT-1a-treated mice showed prolonged median survival. Thirty minutes after intraperitoneal injection of ZT-1a, the phosphorylation of NCC (pThr46/50/55/60) was significantly reduced in a dose-dependent manner. Similar effects were seen in other cation-chloride cotransporters, suggesting a potential role for ZT-1a in other areas of physiology, such as neuronal signaling [71]. This could be a limitation, as ZT-1a may interfere with other SPAK regulatory activities, notably the SPAK regulation of GABA signaling via KCC2. Unlike NCC, KCC phosphorylation inactivates the cotransporter. However, the elevated selectivity for SPAK, achieved by combining the functional moieties of closantel, rafoxanide, and STOCK1S-14279, coupled with the prolonged median survival observed in mice could make ZT-1a a promising therapy option for Gordon’s syndrome. Although the results from mouse studies demonstrate effective inhibition of NCC, further in vivo testing will be required to assess the effects of ZT-1a on blood pressure.
Inhibition of MO25 binding to SPAK/OSR1
To prevent the MO25-dependent activation of SPAK and OSR1, research into binding inhibitors of MO25 has been conducted. This approach would inhibit only MO25-mediated activation of SPAK/OSR1; therefore, basal kinase activity would not be affected. This mild approach may in fact be beneficial, given the strong phenotypic effects of SPAK/OSR1-knockout mice [79, 80]. A study by Kadri et al. developed a fluorescent polarization assay to screen a library of 4000 compounds [90]. The assay measured the binding of MO25 to a peptide that is derived from SPAK/OSR1 and contains two highly conserved tryptophan residues. The alteration of the tryptophan residues prevented MO25 binding, thus confirming the importance of these residues in SPAK/OSR1 and MO25 interactions [90]. This assay may be useful for further identification of MO25 binding inhibitors. The screen led to the identification of HK01. The selectivity of HK01 for MO25 was demonstrated when a high concentration of HK01 (500 μM) did not inhibit the protein kinase MST3, which belongs to the same family of kinases as SPAK and OSR1 [90]. In vitro, HK01 reduced MO25-mediated activation of OSR1 T185E in a dose-dependent manner [90]. In addition, HK01 was shown to inhibit MO25 from binding the SPAK/OSR1-derived peptide, suggesting that it would also inhibit SPAK/OSR1 (Table 3) [90]. This was confirmed in vitro using HEK293 cells, which exhibited a decrease in SPAK/OSR1-mediated phosphorylation of NKCC1 as the HK01 dose increased [90]. Overall, this study has revealed a novel method of WNK-SPAK/OSR1-NCC pathway inhibition, demonstrated by reduced SPAK/OSR1 activity that was caused by the inhibition of MO25. This indirect inhibition of SPAK/OSR1 could form a promising new class of antihypertensive drugs for Gordon’s syndrome and other forms of hypertension.
CUL3 and KLHL3 stabilization
Although no CUL3- or KLHL3-stabilizing molecules have been reported, they have been suggested as a potential treatment option for Gordon’s syndrome. Stabilizing CUL3 or KLHL3 could result in improved degradation of WNK kinases, thereby inhibiting the WNK-SPAK/OSR1-NCC pathway and lowering blood pressure in Gordon’s syndrome. An issue with targeting CUL3 and KLHL3 is that developing molecules that can stabilize or enhance their function would be more challenging than developing the WNK, SPAK, OSR1, and MO25 inhibitory molecules discussed above. For instance, Gordon’s syndrome subtype PHA2D is caused by mutations in two locations in the KLHL3 protein [13, 14]; this means that, to treat this subtype of Gordon’s syndrome, molecules would need to be developed that can counteract either of these mutations. In comparison, inhibitory molecules need to prevent only the activity of their target; they do not need to restore function. Although patients with Gordon’s syndrome are effectively treated with hydrochlorothiazide, research to identify compounds that could reduce blood pressure will benefit patients with hypertension in the general population.
Although mutations in CUL3 cause the most severe form of Gordon’s syndrome [13], CUL3 is probably not the most suitable target for blood pressure reduction. Both the activation and the inhibition of CUL3 can lead to increased WNK kinase signaling: overactivation might lead to off-target or self-ubiquitylation, as demonstrated by CUL3-∆9 [47], while the inhibition of CUL3 would prevent the degradation of WNK kinases, again leading to increased activation of the WNK-SPAK/OSR1-NCC pathway. An alternative treatment option for PHA2E (the Gordon’s syndrome subtype that gives rise to CUL3-∆9) [13, 44] may be to inhibit the process of neddylation, which is responsible for CUL3 activation [45].
Conclusion
The inhibition of various components of the WNK-SPAK/OSR1-NCC pathway has been demonstrated to be an effective target for reducing blood pressure in Gordon’s syndrome and potentially other forms of hypertension. A number of molecular inhibitors have been developed; however, it is clear that further modification and in vivo testing are required before clinical trials can begin. One of the main issues with thiazide diuretic usage as a treatment for Gordon’s syndrome is that chronic usage can lead to adaptation in the nephron. It remains to be seen whether similar effects will occur with chronic usage of the newly developed WNK pathway inhibitors discussed in this review.
References
World Health Organisation. A global brief on hypertension: silent killer, global public health crisis. Geneva: World Health Organisation; 2013. p. 40. https://www.who.int/cardiovascular_diseases/publications/global_brief_hypertension/en/.
World Health Organisation. Global status report on noncommunicable diseases 2010. Geneva: World Health Organisation; 2011. p. 176. https://www.who.int/nmh/publications/ncd_report2010/en/
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–23.
World Health Organization. Global health estimates 2016: deaths by cause, age, sex, by country and by region, 2000–2016. Geneva: World Health Organization; 2018. https://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html
Simonetti GD, Mohaupt MG, Bianchetti MG. Monogenic forms of hypertension. Eur J Pediatr. 2012;171:1433–9.
Bergaya S, Vidal-Petiot E, Jeunemaitre X, Hadchouel J. Pathogenesis of pseudohypoaldosteronism type 2 by WNK1 mutations. Curr Opin Nephrol Hypertens. 2012;21:39–45.
Hall ME, Hall JE. Pathogenesis of hypertension. In: Bakris GL, Sorrentino MJ, editors. Hypertension: a companion to Braunwald’s heart disease, 3rd ed. Philadelphia. Elsevier; 2018. pp. 33–51.
Mansfield TA, Simon DB, Farfel Z, Bia M, Tucci JR, Lebel M, et al. Multilocus linkage of familial hyperkalaemia and hypertension, pseudohypoaldosteronism type II, to chromosomes 1q31-42 and 17p11-q21. Nat Genet. 1997;16:202–5.
Disse-Nicodeme S, Achard JM, Desitter I, Houot AM, Fournier A, Corvol P, et al. A new locus on chromosome 12p13.3 for pseudohypoaldosteronism type II, an autosomal dominant form of hypertension. Am J Hum Genet. 2000;67:302–10.
Delaloy C, Lu J, Houot AM, Disse-Nicodeme S, Gasc JM, Corvol P, et al. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol. 2003;23:9208–21.
Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391:17–24.
Chavez-Canales M, Zhang C, Soukaseum C, Moreno E, Pacheco-Alvarez D, Vidal-Petiot E, et al. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 2014;64:1047–53.
Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102.
Louis-Dit-Picard H, Barc J, Trujillano D, Miserey-Lenkei S, Bouatia-Naji N, Pylypenko O, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44:456–60. S451–53
Sohara E, Uchida S. Kelch-like 3/Cullin 3 ubiquitin ligase complex and WNK signaling in salt-sensitive hypertension and electrolyte disorder. Nephrol Dial Transpl. 2016;31:1417–24.
Sherwood L. Human physiology: from cells to systems. 9th ed. Boston, MA, USA: Cengage Learning; 2016. p. 1. Volume (various pagings).
Moes AD, van der Lubbe N, Zietse R, Loffing J, Hoorn EJ. The sodium chloride cotransporter SLC12A3: new roles in sodium, potassium, and blood pressure regulation. Pflug Arch. 2014;466:107–18.
Gamba G. The thiazide-sensitive Na+-Cl− cotransporter: molecular biology, functional properties, and regulation by WNKs. Am J Physiol Ren Physiol. 2009;297:F838–48.
Arroyo JP, Kahle KT, Gamba G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Asp Med. 2013;34:288–98.
Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–6.
Yang CL, Angell J, Mitchell R, Ellison DH. WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J Clin Invest. 2003;111:1039–45.
Kahle KT, Macgregor GG, Wilson FH, Van Hoek AN, Brown D, Ardito T, et al. Paracellular Cl− permeability is regulated by WNK4 kinase: insight into normal physiology and hypertension. Proc Natl Acad Sci USA. 2004;101:14877–82.
de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, et al. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem J. 2014;458:559–73.
Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, et al. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem. 2005;280:42685–93.
Gagnon KB, Delpire E. Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol Rev. 2012;92:1577–617.
Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem. 2002;277:50812–9.
Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK kinase signaling in ion homeostasis and human disease. Cell Metab. 2017;25:285–99.
Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci. 2008;121:3293–304.
Dhiani BA, Mehellou Y. The Cul4-DDB1-WDR3/WDR6 complex binds SPAK and OSR1 kinases in a phosphorylation-dependent manner. Chembiochem. 2020;21:638–43.
Filippi BM, de los Heros P, Mehellou Y, Navratilova I, Gourlay R, Deak M, et al. MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J. 2011;30:1730–41.
Alessi DR, Zhang J, Khanna A, Hochdorfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal. 2014;7:re3.
Zhang J, Siew K, Macartney T, O’Shaughnessy KM, Alessi DR. Critical role of the SPAK protein kinase CCT domain in controlling blood pressure. Hum Mol Genet. 2015;24:4545–58.
Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic S, et al. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med. 2010;2:63–75.
Ohta A, Schumacher FR, Mehellou Y, Johnson C, Knebel A, Macartney TJ, et al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: disease-causing mutations in KLHL3 and WNK4 disrupt interaction. Biochem J. 2013;451:111–22.
Ferdaus MZ, McCormick JA. The CUL3/KLHL3-WNK-SPAK/OSR1 pathway as a target for antihypertensive therapy. Am J Physiol Ren Physiol. 2016;310:F1389–96.
Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82.
Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013;3:858–68.
Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol. 2016;78:367–89.
Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, et al. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA. 2013;110:14366–71.
Tobin MD, Raleigh SM, Newhouse S, Braund P, Bodycote C, Ogleby J, et al. Association of WNK1 gene polymorphisms and haplotypes with ambulatory blood pressure in the general population. Circulation. 2005;112:3423–9.
Putku M, Kepp K, Org E, Sober S, Comas D, Viigimaa M, et al. Novel polymorphic AluYb8 insertion in the WNK1 gene is associated with blood pressure variation in Europeans. Hum Mutat. 2011;32:806–14.
Newhouse S, Farrall M, Wallace C, Hoti M, Burke B, Howard P, et al. Polymorphisms in the WNK1 gene are associated with blood pressure variation and urinary potassium excretion. PLoS One. 2009;4:e5003.
Guo XG, Ding J, Xu H, Xuan TM, Jin WQ, Yin X, et al. Comprehensive assessment of the association of WNK4 polymorphisms with hypertension: evidence from a meta-analysis. Sci Rep. 2014;4:6507.
Ferdaus MZ, McCormick JA. Mechanisms and controversies in mutant Cul3-mediated familial hyperkalemic hypertension. Am J Physiol Ren Physiol. 2018;314:F915–20.
Ibeawuchi SR, Agbor LN, Quelle FW, Sigmund CD. Hypertension-causing mutations in Cullin3 protein impair RhoA protein ubiquitination and augment the association with substrate adaptors. J Biol Chem. 2015;290:19208–17.
Picard HLD, Latreche S, Thurairajasingam N, Auzan C, Fiquet B, Frayssinet R, et al. Cullin-3 mutations leading to skipping of exon 9 are responsible for severe cases of familial hyperkalaemic hypertension. J Hypertens. 2015;33:E79.
McCormick JA, Yang CL, Zhang C, Davidge B, Blankenstein KI, Terker AS, et al. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J Clin Invest. 2014;124:4723–36.
Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7:1285–306.
Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, et al. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARgamma and RhoA/Rho-kinase. Cell Metab. 2012;16:462–72.
Agbor LN, Ibeawuchi SC, Hu C, Wu J, Davis DR, Keen HL, et al. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight. 2016;1:e91015.
Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7:1285–306.
Farfel Z, Iaina A, Rosenthal T, Waks U, Shibolet S, Gafni J. Familial hyperpotassemia and hypertension accompanied by normal plasma aldosterone levels: possible hereditary cell membrane defect. Arch Intern Med. 1978;138:1828–32.
Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. 2002;87:3248–54.
Gordon R, Klemm S, Tunny T, Stowasser M. Gordon’s syndrome: a sodium-volume dependent form of hypertension with a genetic basis. In: Laragh J, Brenner B, editors. Hypertension: pathophysiology, diagnosis, and management. 2nd ed. New York: Raven Press; 1995. p. 2111–23.
Ellison DH, Velazquez H, Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol. 1987;253:F546–54.
Duarte JD, Cooper-DeHoff RM. Mechanisms for blood pressure lowering and metabolic effects of thiazide and thiazide-like diuretics. Expert Rev Cardiovasc Ther. 2010;8:793–802.
Morsing P, Velazquez H, Wright FS, Ellison DH. Adaptation of distal convoluted tubule of rats. II. Eff chronic thiazide Infus Am J Physiol. 1991;261:F137–43.
Sica DA. Diuretic-related side effects: development and treatment. J Clin Hypertens (Greenwich). 2004;6:532–40.
Maitland-van der Zee AH, Turner ST, Schwartz GL, Chapman AB, Klungel OH, Boerwinkle E. Demographic, environmental, and genetic predictors of metabolic side effects of hydrochlorothiazide treatment in hypertensive subjects. Am J Hypertens. 2005;18:1077–83.
Tobin MD, Tomaszewski M, Braund PS, Hajat C, Raleigh SM, Palmer TM, et al. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension. 2008;51:1658–64.
Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, BeltrandelRio H, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci USA. 2003;100:14109–14.
Susa K, Kita S, Iwamoto T, Yang SS, Lin SH, Ohta A, et al. Effect of heterozygous deletion of WNK1 on the WNK-OSR1/ SPAK-NCC/NKCC1/NKCC2 signal cascade in the kidney and blood vessels. Clin Exp Nephrol. 2012;16:530–8.
Bergaya S, Faure S, Baudrie V, Rio M, Escoubet B, Bonnin P, et al. WNK1 regulates vasoconstriction and blood pressure response to alpha 1-adrenergic stimulation in mice. Hypertension. 2011;58:439–45.
Wang Y, O’Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, et al. From the Cover: whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci USA. 2009;106:226–31.
Takahashi D, Mori T, Nomura N, Khan MZ, Araki Y, Zeniya M, et al. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci Rep. 2014;34:e00107.
Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, et al. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA. 2012;109:7929–34.
Min X, Lee BH, Cobb MH, Goldsmith EJ. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure. 2004;12:1303–11.
Yamada K, Park HM, Rigel DF, DiPetrillo K, Whalen EJ, Anisowicz A, et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat Chem Biol. 2016;12:896–8.
Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907.
Rodan AR, Jenny A. WNK kinases in development and disease. Curr Top Dev Biol. 2017;123:1–47.
Zhang J, Bhuiyan MIH, Zhang T, Karimy JK, Wu Z, Fiesler VM, et al. Modulation of brain cation-Cl− cotransport via the SPAK kinase inhibitor ZT-1a. Nat Commun. 2020;11:78.
Yamada K, Zhang JH, Xie X, Reinhardt J, Xie AQ, LaSala D, et al. Discovery and characterization of allosteric WNK kinase inhibitors. ACS Chem Biol. 2016;11:3338–46.
Yamada K, Levell J, Yoon T, Kohls D, Yowe D, Rigel DF, et al. Optimization of allosteric with-no-lysine (WNK) kinase inhibitors and efficacy in rodent hypertension models. J Med Chem. 2017;60:7099–107.
Pinkas D, Bufton J, Bartual S, Chen Z, Daubner G, Schumacher F, et al. Human with no lysine kinase 3 (WNK3); a target enabling package. https://doi.org/10.5281/zenodo.1219718. 2017.
Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–9.
Dunna NR, Kandula V, Girdhar A, Pudutha A, Hussain T, Bandaru S, et al. High affinity pharmacological profiling of dual inhibitors targeting RET and VEGFR2 in inhibition of kinase and angiogeneis events in medullary thyroid carcinoma. Asian Pac J Cancer Prev. 2015;16:7089–95.
Zhou Z, Chen X, Chen X, Qin A, Mao Y, Pang Y, et al. PP121 suppresses RANKL-Induced osteoclast formation in vitro and LPS-Induced bone resorption in vivo. Exp Cell Res. 2020;388:111857.
Kuenemann MA, Fourches D. Cheminformatics analysis of dynamic WNK-inhibitor interactions. Mol Inf. 2018;37:e1700138.
Lin SH, Yu IS, Jiang ST, Lin SW, Chu P, Chen A, et al. Impaired phosphorylation of Na+-K+-2Cl− cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci USA. 2011;108:17538–43.
Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, et al. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21:1868–77.
Kikuchi E, Mori T, Zeniya M, Isobe K, Ishigami-Yuasa M, Fujii S, et al. Discovery of novel spak inhibitors that block WNK kinase signaling to cation chloride transporters. J Am Soc Nephrol. 2015;26:1525–36.
AlAmri MA, Kadri H, Alderwick LJ, Simpkins NS, Mehellou Y. Rafoxanide and closantel inhibit SPAK and OSR1 kinases by binding to a highly conserved allosteric site on their C-terminal domains. ChemMedChem. 2017;12:639–45.
AlAmri MA, Kadri H, Alderwick LJ, Jeeves M, Mehellou Y. The photosensitising clinical agent verteporfin is an inhibitor of SPAK and OSR1 kinases. Chembiochem. 2018;19:2072–80.
Charisis SK, Naoumidi II, Ginis HS, Detorakis ET, Tsilimbaris MK. Contact transcleral ciliary body photodynamic therapy with verteporfin in pigmented rabbits: effect of repeated treatments. Photochem Photobio. 2010;86:194–9.
de Los Heros P, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, et al. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl− co-transporters. Biochem J. 2014;458:559–73.
Stein V, Nicoll RA. GABA generates excitement. Neuron. 2003;37:375–8.
Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, et al. WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci USA. 2005;102:16783–8.
Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, et al. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest. 2015;125:2136–50.
Mori T, Kikuchi E, Watanabe Y, Fujii S, Ishigami-Yuasa M, Kagechika H, et al. Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem J. 2013;455:339–45.
Kadri H, Alamri MA, Navratilova IH, Alderwick LJ, Simpkins NS, Mehellou Y. Towards the development of small-molecule MO25 binders as potential indirect SPAK/OSR1 kinase inhibitors. Chembiochem. 2017;18:460–5.
Acknowledgements
This study was in part supported by the University of Exeter Medical School start-up fund (JZ) and the National Natural Science Foundation of China 81970238 (JZ).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Brown, A., Meor Azlan, N.F., Wu, Z. et al. WNK-SPAK/OSR1-NCC kinase signaling pathway as a novel target for the treatment of salt-sensitive hypertension. Acta Pharmacol Sin 42, 508–517 (2021). https://doi.org/10.1038/s41401-020-0474-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0474-7
Keywords
This article is cited by
-
Hybrid parameters for fluid identification using an enhanced quantum neural network in a tight reservoir
Scientific Reports (2024)
-
An update regarding the role of WNK kinases in cancer
Cell Death & Disease (2022)
-
S-wave velocity inversion and prediction using a deep hybrid neural network
Science China Earth Sciences (2022)
-
SPAK-p38 MAPK signal pathway modulates claudin-18 and barrier function of alveolar epithelium after hyperoxic exposure
BMC Pulmonary Medicine (2021)
