
Abstract
Malaria, caused by parasites of the genus Plasmodium, leads to over half a million deaths per year, 90% of which are caused by Plasmodium falciparum. P. vivax usually causes milder forms of malaria; however, P. vivax can remain dormant in the livers of infected patients for weeks or years before re-emerging in a new bout of the disease. The only drugs available that target all stages of the parasite can lead to severe side effects in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency; hence, there is an urgent need to develop new drugs active against blood and liver stages of the parasite. Different groups have demonstrated that triclosan, a common antibacterial agent, targets the Plasmodium liver enzyme enoyl reductase. Here, we provide 4 independent lines of evidence demonstrating that triclosan specifically targets both wild-type and pyrimethamine-resistant P. falciparum and P. vivax dihydrofolate reductases, classic targets for the blood stage of the parasite. This makes triclosan an exciting candidate for further development as a dual specificity antimalarial, which could target both liver and blood stages of the parasite.
Similar content being viewed by others

Introduction
Over half a million deaths per year result from malaria, caused by parasites of the genus Plasmodium. Approximately 90% of these deaths (generally of children in Africa) are caused by Plasmodium falciparum, the parasite responsible for the most severe forms of the disease. However, Plasmodium vivax is the malaria parasite with the largest global distribution, threatening 2.5 billion people - mainly in South America and Asia1. Although P. vivax usually causes milder forms of malaria, it is still a killer2. Moreover, P. vivax can remain dormant in the livers of infected patients for weeks or years before re-emerging in a new bout of the disease3.
There is an urgent need to develop new anti-malarial drugs as resistance has arisen against all existing drugs4,5,6, and there is no immediate prospect of an effective vaccine7. Primaquine is the only drug in use that targets all stages of the parasite. However, it greatly increases the risk of hemolysis in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency, as does the promising new drug, tafenoquine1. G6PD deficiency is prevalent in areas where malaria is endemic since it offers some protection against infection8.
Triclosan is a simple antimicrobial agent that has been used for more than 40 years, and which is safe enough to be incorporated into consumer products from toothpastes to toys9,10. In 2001, it was reported that triclosan inhibits the in vitro propagation of P. falciparum11. Since then, triclosan and its analogs have been repeatedly shown to inhibit the growth of blood-stage P. falciparum in culture12,13,14,15,16,17,18,19. It was assumed that the mechanism of action of triclosan against Plasmodium was the same as that against bacteria - inhibition of enoyl reductase in the FAS-II11 fatty-acid synthesis pathway. Indeed, phenotypical, biochemical, structural evidence, and in silico simulations demonstrated that triclosan is a strong inhibitor of the P. falciparum enoyl reductase (PfENR)13,20,21. However Yu et al.20 showed convincingly that the FAS-II pathway is not important for the propagation of P. falciparum in erythrocytes and so inhibition of PfENR cannot explain the effect of triclosan on blood-stage parasites.
Efforts to optimize the compound for use as an antimalarial showed no correlation between PfENR inhibition in vitro and triclosan’s activity against the living parasite21,22. Since it has been demonstrated that PfENR is important for the liver stage of the parasite, but not for the erythrocyte stage22,23, action against a different target must be responsible for triclosan’s inhibition of the growth of blood-stage P. falciparum.
We have developed an automated yeast-based assay for use in high-throughput screens for compounds that are selectively active against target enzymes from parasites24,25. Our assay is based on replacing yeast genes essential for growth, with coding sequences specifying orthologous proteins from either parasites or humans, making the strains dependent on the activity of the parasite or the human enzyme in order to grow25. These strains are then labeled with different fluorescent proteins and pooled to allow monitoring of the growth, in real time, of each strain in competition with the others.
We carried out growth competition experiments between 3 yeast strains dependent on a DHFR enzyme from P. falciparum, P. vivax, or the human DHFR (these three strains were differentiated from one another by each expressing either the fluorescent proteins mCherry, Venus, or Sapphire26,27). These competitions took place in single wells of microtiter trays, enabling a Robot Scientist to screen thousands of different compounds and identify candidates with no general cytotoxicity, but which inhibited the parasite target without affecting its human counterpart24,28.
Results and Discussion
Dihydrofolate reductase (DHFR) is an enzyme that catalyzes the NADPH-dependent reduction of dihydrofolate to tetrahydrofolate. This reaction is essential for the de novo synthesis of purines and certain amino acids, making DHFR essential for rapid growth, and is the target for the action of the important antimalarial drugs pyrimethamine and proguanil. Employing our yeast-based high-throughput screening approach24, we screened the Johns Hopkins Library of FDA-approved compounds against yeast strains expressing human DHFR (HsDHFR), P. falciparum DHFR (PfDHFR), P. vivax DHFR (PvDHFR) as well as the pyrimethamine-resistant P. vivax and P. falciparum DHFRs (PvRdhfr and PfRdhfr)28. The automated screen identified triclosan (Eve ID 21658, JHU-10450) as a specific inhibitor of P. vivax and P. falciparum DHFRs, including the pyrimethamine-resistant variants of the two enzymes. The Robot Scientist Eve prepared titration experiments with 1 to 20 μM of triclosan and successfully confirmed the hits. We prepared serial dilutions of yeast cultures expressing the human or Plasmodium DHFRs and spotted those onto agar plates containing 10 μM of triclosan, further confirming the specificity of the drug for the parasite target (Fig. 1).

Triclosan specifically inhibits Plasmodium DHFRs in yeast. Yeast strains expressing either Homo sapiens DHFR (yHsDHFR), Plasmodium falciparum wild-type DHFR (yPfDHFR), pyrimethamine-resistant P. falciparum DHFR (yPfRdhfr), or P. vivax DHFR were cultivated overnight and dilutions 5, 25, 125, 625 and spotted onto YNB-glucose-agar plates containing 0 or 10 μM irgasan (triclosan).
We expressed the human and P. vivax DHFRs in E. coli, and purified the proteins using methotrexate affinity columns. We then performed in vitro enzyme assays and observed that the P. vivax DHFR is >20× more sensitive to triclosan (IC50 of 775 ± 384 nM) than the human enzyme (IC50 of 17.2 ± 4.9 μM) (Fig. 2).

Triclosan inhibits Plasmodium DHFR in vitro. (A) DHFR catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF), coupled with the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH). This reaction was followed for HsDHFR and PvDHFR in the absence (blue) or presence (red) of 100 µM Triclosan at 340 nm. (B) Initial rates of reaction were determined and used to derive IC50 values, with the standard deviation calculated from multiple kinetic runs (n = 3). To deal with variations in enzyme activity in different preparations, kinetic data were normalized as relative activities and fitted to % activity = % activitymax/(1 + [triclosan]/IC50). Reaction conditions: [triclosan] = 0–200 μM, [HsDHFR] = 30.5 nM, [PvDHFR] = 18.5 nM, [Tris/HCl] = 0.1 M, pH 7.4, [DHF] = [NADPH] = 100 μM, 21 °C.
To further validate DHFR as a target of triclosan in Plasmodium, we transfected blood-stage P. falciparum NF54 with a plasmid expressing the human DHFR enzyme (HsDHFR). If the blood-stage target of triclosan is indeed the Plasmodium DHFR, expression of HsDHFR (which our yeast and in vitro assays showed to be resistant to triclosan) should protect the parasite against the compound.
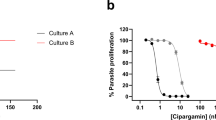
We found that our transfected Plasmodium strain was more tolerant to exposure to triclosan than the wild type (Fig. 3), consistent with DHFR being the compound’s target when the parasite is within the erythrocyte. The difference in the rate of progression of the parasitemia in wild-type and transfected strains was compared at a range of triclosan concentrations and found to be statistically significant (SEM). It should be noted that, in contrast to the in vitro enzyme inhibition kinetics, those for growth inhibition are complex. This is likely to be due to the fact that our parasite lines have been transfected with episomal constructs bearing the coding sequence for human DHFR. The resulting range of plasmid copy numbers would result in differences in the levels of human DHFR between individual Plasmodium cells in the population29. The consequence is that a considerable proportion of cells do not express human DHFR at levels that are sufficiently high to confer drug resistance.

Human DHFR protects Plasmodium from triclosan. P. falciparum NF54 strain was transfected with a plasmid expressing human dihydrofolate reductase, and treated with either triclosan or pyrimethamine for 48 h. Relative parasitemias were calculated following JC1 staining and flow cytometry analysis. Error bars indicate SEM.
Finally, triclosan was docked into the X-ray structures of human and Plasmodium DHFRs (Fig. 4). Although triclosan displayed slightly altered binding poses in the different parasitic DHFRs, the inhibitor showed the same interactions with all four DHFRs, indicating comparable affinity (binding free energies in the range −7.24 and −7.56 kcal/mol). In contrast, triclosan was not interacting with the HsDHFR through hydrogen bonding, resulting in reduced affinity (∆G = −6.57 kcal/mol) and supporting the contention that triclosan discriminates between the Plasmodium and human enzymes.

Differential binding of triclosan to Plasmodium and human DHFRs. Predicted binding poses of triclosan (in purple) bound to the active site of DHFRs. Wild-type P.vivax DHFR (PDB ID: 2BL9), double mutant P. vivax DHFR (PDB ID: 2BLC), P. falciparum DHFR (PDB ID: 4DPD), quadruple mutant P. falciparum DHFR (PDB ID: 4DP3), and human DHFR (PDB ID: 3NTZ). NADPH is colored in blue; hydrogen bonds are represented by black dashed lines.
DHFR inhibitors are routinely used as prophylactic drugs and are given to over a million children during the malaria season. However, DHFR inhibitors are no longer a standard treatment for the disease because of the evolution of drug-resistant variants of Plasmodium. Extensive efforts to discover a DHFR-targeted antimalarial that is effective against pyrimethamine-resistant strains have yet to produce a clinically approved entity, thus a novel Triclosan-derived compound with activity against drug-resistant forms of DHFR is of high potential value.
The presence of triclosan in consumer products in concentrations 2500-fold above the present IC5030, suggests that it can be administered in quantity and short-term toxicity effects can be disregarded, notwithstanding possible long-term hazards31. This tolerance is important, as delivering effective concentrations in vivo may be challenging since data on the effect of administering triclosan to mice infected with P.berghei are contradictory11,18, with sub-cutaneous delivery being more effective than oral administration18. It may be that further engineering of triclosan by synthetic chemistry is necessary to improve delivery and half-life, but with regard to affinity triclosan is a viable non-toxic lead with a mechanism based on hitherto undiscovered selectivity. A number of triclosan analogs have already been synthesized16,21, demonstrating the ample opportunity for its improvement for use as an antimalarial.
In this work, we have provided four different lines of evidence (screens using recombinant yeast strains, enzyme assays, growth assays with blood-stage Plasmodium, and in silico simulations of drug-enzyme interactions) demonstrating that triclosan specifically targets the DHFR enzyme of both P. vivax and P. falciparum, and that it is capable of selectively inhibiting both the wild-type and pyrimethamine-resistant enzymes compared to human DHFR. Our conclusion that triclosan targets Plasmodium DHFR also explains much of the inconsistency in the literature regarding the efficacy of triclosan22: HsDHFR is commonly used as a Plasmodium selective marker as it provides pyrimethamine resistance, and controls for this confounding factor are not always performed.
Given the earlier evidence that triclosan inhibits ENR, it may be that triclosan is an example of “polypharmacology”32, poised for multitarget therapies. The totality of data on this compound suggests that it is capable of inhibiting blood-stage Plasmodium, via its action against DHFR, and the liver stage of the parasite, via the inhibition of a key enzyme in fatty-acid synthesis20. Interestingly, polyphenol compounds such as epigallocathechin gallate (EGCG), also inhibit fatty acid synthesis and have antiplasmodial activity33, and have demonstrated antifolate activity34. Given the recent success in developing drug candidates with multi-stage activity35, it is realistic to envision the development of a new dual-action drug, based on triclosan, that is effective against both the blood and liver stages of wild-type and pyrimethamine-resistant Plasmodium species. The fact that triclosan has two distinct enzyme targets in different domains of metabolism may militate against the development of resistance. All of this engenders the hope that our discovery of a second enzyme target for triclosan may prove to be the first step in the development of a novel class of antimalarials.
Methods
Yeast strains and fluorescent plasmids
All yeast strains and marker plasmids used in this work are described in Bilsland et al.24,25. Briefly: plasmids, bearing genes for the expression of fluorescent proteins, were constructed by replacing the coding region of yEmRFP from yEpGAP-Cherry with Venus or Sapphire, and replacing the URA3 marker with LEU2. These three fluorescent proteins have very distinct excitation and emission wavelengths: mCherry (λex 580 nm; λem 612 nm), Sapphire (λex 405 nm; λem 510 nm) and Venus (λex 500 nm; λem 540 nm).
A strain expressing the drug-resistant P. vivax DHFR (yPvRdhfr) was constructed by making the following site-directed mutations in the coding sequence for the P. vivax enzyme: S58R, S117N and I173L. This plasmid was transformed into dfr1Δ/DFR1 a yeast strain with the BY4743 genetic background. The strain was sporulated and MATα haploids were selected and used in drug screens. The drug-resistant yPfRdhfr strain, which is a triple mutant for residues N51I, C59R, and S108N, was generated in a similar manner.
Library-screening assays and hit confirmation using the Robot Scientist Eve
Automated screens were performed as described in Williams et al.28. Starter cultures of individual strains, labeled with different fluorescent proteins, were grown to stationary phase in YNB-glucose (0.67% yeast nitrogen base without amino acids, 2% ammonium sulphate and 2% glucose) with the appropriate auxotrophic supplements. An aliquot (1 mL) of each pre-culture was inoculated into 100 mL of fresh medium. Pools of three strains were incubated for 4 h at 30 °C, with shaking, to ensure exponential growth. Doxycycline (Sigma-Aldrich) (5 μg/mL) was then added to the culture to reduce expression of the gene for the heterologous enzymes24. The culture was attached to a Thermo Combi multidrop within the Robot Scientist Eve work cell24. The culture was stirred continuously and maintained at 23 °C during assay plate set-up.
High-throughput drug screens were performed by the Robot Scientist Eve using the mixed cultures described above and the ~1,600 FDA- and foreign- approved drugs from the Johns Hopkins University Clinical Compound Library. Strains were grown in competition in the presence of a library compound, and the relative growth rates used to estimate the activity of the drug against the parasite target. For hit confirmation assays, the Robot Scientist Eve prepared plates with eight replicates of eight different compounds, at six different concentrations (0, 1, 2.5, 5, 10, 20 μM), and 64 negative control wells.
Recombinant protein expression and purification
Synthetic genes encoding versions of the Homo sapiens and P. vivax dihydrofolate reductases25, tagged with maltose-binding protein (MBP) and polyhistidine (HIS) sequences, were cloned into the BamHI/NsiI sites of pMAT10 (Keily Littlefield and Darerca Owen, personal communication). Escherichia coli BL21 (DE3) transformants, harboring each of the plasmids, were grown in LB medium and recombinant protein expression was induced with 0.4 mM isopropyl-1-β-D-thio-galactopyranoside (IPTG) for 16 hours at 15 °C. Cultures were centrifuged at 4000 rpm for 10 min, the supernatant discarded, and cell pellets resuspended in 1/50th of the culture volume in 20 mM Tris-HCl, pH 7.4, 300 mM NaCl and Roche Complete EDTA-free Protease Inhibitor tablets, DNase I (RNase-free; Thermo-Scientific 2 U.mL−1), 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (Sigma). Cell suspensions were passed three times through an EmulsiFlex-C5 to facilitate cell lysis.
Cell lysates were centrifuged at 20,000 rcf for 50 min at 8 °C. MBP_HIS-tagged proteins were purified in HIS-Select® Nickel Affinity Gel (Sigma-Aldrich) gravity-flow columns. MBP_HIS-tagged DHFRs were dialyzed into Thrombin Cleavage Buffer (20 mM Tris-HCl, pH 8.4, 2.5 mM CaCl2 and 150 mM NaCl) and incubated for 5 hours at 20 °C with 24 mM lactose and 5 U/mL bovine thrombin (GE Healthcare). Cleavages were terminated by raising the NaCl concentration of the solution to 0.5 M and binding to 1 mL HiTrap Benzamidine FF columns (GE Healthcare) pre-equilibrated with High Salt Thrombin Buffer (20 mM Tris-HCl, pH 8.4, 2.5 mM CaCl2 and 500 mM NaCl). Cleaved samples were dialyzed into Methotrexate Equilibration Buffer (50 mM Tris- HCl, pH 8.0, 2 mM EDTA, 0.5 M NaCl, 2 mM DTT and 10% v/v glycerol) and applied to columns containing 1.25 mL methotrexate-agarose beads (Sigma). The columns were washed with 20 mL of Methotrexate Equilibration Buffer then eluted in 1.5 mL fractions using Methotrexate Elution Buffer (Methotrexate Equilibration Buffer + 4 mM DHF).
We attempted to express and purify P. falciparum DHFR and pyrimethamine-resistant P.vivax DHFR under multiple conditions, but were unable to recover stable proteins for enzyme assays.
Enzyme Assays
DHFR catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF), coupled to the oxidation of nicotinamide adenine dinucleotide phosphate (NADPH). This reaction was followed in vitro by absorbance at λabs = 340 nm by spectrophotometry (SpectraMax 190, Molecular Devices) in UV-transparent 96-well plates (Costar, Corning) at 21 °C. NADPH and DHF absorb at 340 nm with a combined extinction coefficient of ε340nM = 12,300 M−1 cm−1, which takes into consideration the oxidation of NADPH and the reduction of DHF.
H. sapiens DHFR (HsDHFR) and P. vivax DHFR (PvDHFR) were thawed and stored at 4 °C for use in the enzyme assay. DHF and NADPH stocks were made to 1 mM in assay buffer (0.1 M Tris, pH 7.4) and stored at −20 °C until use. Triclosan (Irgasan; Sigma-Aldrich) (stored as a 1 mM stock in 10% (v/v) DMSO in 0.1 M Tris, pH 7.4) was incubated with HsDHFR or PvDHFR for 10 minutes in assay buffer (0.1 M Tris, pH 7.4) before addition of 100 μM DHF and 100 μM NADPH. Reaction conditions: [triclosan] = 0 or 100 µM, [HsDHFR] = 30.5 nM, [PvDHFR] = 18.5 nM, [DHF] = [NADPH] = 100 µM.
Initial rates of reaction at various concentrations of triclosan were monitored by spectrophotometry (SpectraMax 190, Molecular Devices) and data were analyzed using Excel (Microsoft). Initial rates were determined by following the linear absorbance decrease and used to derive IC50 values. Reaction turnover was assayed in UV-transparent 96-well plates (Costar, Corning) with a total volume of 100 μL. DHF and NADPH stocks were made up to 1 mM in assay buffer (0.1 M Tris, pH 7.4) and stored at −20 °C until use.
To deal with variations in enzyme activity in different preparations, kinetic data were normalized as relative activities and fitted to the following equation:
assuming competitive inhibition and using the Cheng-Prusoff equation36. Errors were calculated from multiple kinetic runs. The kinetics of PvDHFR carried a larger error than HsDHFR because PvDHFR was prone to loss of activity during purification and storage. Duplicates were performed and IC50 values were obtained by fitting the data shown in Fig. 1C to the above equation (MatLab software, Mathworks – curves were fitted by Fabrice Gielen, Cambridge).
Enzyme molarity calculations were based on the following sequences:
HsDHFR: GSMVGSLNCIVAVSQNMGIGKNGDLPWPPLRNEFRYFQRMTTTSSVEGKQN LVIMGKKTWFSIPEKNRPLKGRINLVLSRELKEPPQGAHFLSRSLDDALKLTE QPELANKVDMVWIVGGSSVYKEAMNHPGHLKLFVTRIMQDFESDTFFPEIDL EKYKLLPEYPGVLSDVQEEKGIKYKFEVYEKND.
PvDHFR:
GSMEDLSDVFDIYAICACCKVAPTSEGTKNEPFSPRTFRGLGNKGTLPWKCNS VDMKYFSSVTTYVDESKYEKLKWKRERYLRMEASQGGGDNTSGGDNTHGG DNADKLQNVVVMGRSSWESIPKQYKPLPNRINVVLSKTLTKEDVKEKVFIIDS IDDLLLLLKKLKYYKCFIIGGAQVYRECLSRNLIKQIYFTRINGAYPCDVFFPEF DESQFRVTSVSEVYNSKGTTLDFLVYSKVGG.
Culture of wild-type and transgenic P. falciparum
P. falciparum strain NF54 was transfected with a pDC10 derivate37 and cultivated as described previously38. Wild-type and transgenic parasites were cultivated in the presence of increasing concentrations of triclosan (0–2.6 µM) or pyrimethamine (0–35 nM). Drug treatment of cultures was started at 0.5–1% parasitemia (trophozoite stage) and run for 48 h. Parasitemias were quantified using the SYBR method39 and resulting fluorescence data were analysed and plotted using Excel. The experiments were done twice in biological triplicates.
Docking
The starting coordinates of the human DHFR (PDB ID: 3NTZ), P. vivax DHFR (PDB ID: 2BL9), P. falciparum DHFR (PDB ID: 4DPD), P. vivax double mutant DHFR (PDB ID: 2BLA), and P. falciparum quadruple mutant DHFR (PDB ID: 4DP3) were collected from the Protein Data Bank (www.rcsb.org)40,41,42 and employed in Glide (Small-Molecule Drug Discovery Suite 2014-2: Glide, version 6.3, Schrödinger, LLC, New York, NY, 2014) docking calculations.
For Glide calculations, all the crystallographic structures were first imported to Maestro (Schrödinger Release 2014-2: Maestro, version 9.8, Schrödinger, LLC, New York, NY, 2014). With the exception of NADPH, all co-crystallized ligands and water molecules were identified and removed from the structures and the enzymes were treated using the Protein Preparation Wizard (Schrödinger Release 2014-2: Schrödinger Suite 2014-2 Protein Preparation Wizard; Epik version 2.8, Schrödinger, LLC, New York, NY, 2014) provided by Maestro. All the enzymatic structures were also checked for missing atoms, bonds, and contacts. The structure of triclosan was first constructed using Maestro’s building tool and energetically minimized for processing using LigPrep, which allowed the acquisition of a valid low-energy 3D structure for this inhibitor with correct protonation state in the pH range of 7 ± 2. Molecular Docking was initiated by generating a Grid file using Receptor Grid Generation tool of Glide. Grid files containing receptor and binding site information required for molecular docking were prepared using the default options of the Receptor Grid Generation tool with the grid box being centered at the coordinates average of the co-crystallized ligand for each crystallographic structure. To test the reliability of the docking procedure, all the co-crystallized ligands were first docked into the corresponding crystallographic structure using the Extra Precision (XP) Glide algorithm. Subsequently, triclosan was also successfully docked in each active site and the lowest energy conformation provided by Glide Score was chosen.
Ethics statement
Human blood and plasma were obtained from the local blood bank and ethical clearance for using this blood for this research was granted by the Ethics Committee of the Institute of Biomedical Sciences at the University of São Paulo (No. 842/2016).
References
Wells, T. N., van Huijsduijnen, R. H. & Van Voorhis, W. C. Malaria medicines: a glass half full? Nature reviews. Drug discovery 14, 424–442 (2015).
Melo, G. C. et al. Expression levels of pvcrt-o and pvmdr-1 are associated with chloroquine resistance and severe Plasmodium vivax malaria in patients of the Brazilian Amazon. PLoS One 9, e105922 (2014).
Mikolajczak, S. A. et al. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17, 526–535 (2015).
Miotto, O. et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nature genetics 47, 226–234 (2015).
Fairhurst, R.M. Understanding artemisinin-resistant malaria: what a difference a year makes. Current opinion in infectious diseases (2015).
Chugh, M. et al. Identification and deconvolution of cross-resistance signals from antimalarial compounds using multidrug-resistant Plasmodium falciparum strains. Antimicrob Agents Chemother 59, 1110–1118 (2015).
Dunachie, S., Hill, A. V. & Fletcher, H. A. Profiling the host response to malaria vaccination and malaria challenge. Vaccine (2015).
Howes, R. E. et al. Spatial distribution of G6PD deficiency variants across malaria-endemic regions. Malar J 12, 418 (2013).
Bhargava, H. N. & Leonard, P. A. Triclosan: applications and safety. Am J Infect Control 24, 209–218 (1996).
Han, J., Qiu, W., Campbell, E. C., White, J. C. & Xing, B. Nylon Bristles and Elastomers Retain Centigram Levels of Triclosan and Other Chemicals from Toothpastes: Accumulation and Uncontrolled Release. Environ Sci Technol 51, 12264–12273 (2017).
Surolia, N. & Surolia, A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat Med 7, 167–173 (2001).
Singh, A. P., Surolia, N. & Surolia, A. Triclosan inhibit the growth of the late liver-stage of Plasmodium. IUBMB Life 61, 923–928 (2009).
Perozzo, R. et al. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J Biol Chem 277, 13106–13114 (2002).
Nicola, G. et al. Discovery of novel inhibitors targeting enoyl-acyl carrier protein reductase in Plasmodium falciparum by structure-based virtual screening. Biochem Biophys Res Commun 358, 686–691 (2007).
McLeod, R. et al. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of apicomplexan Fab I. Int J Parasitol 31, 109–113 (2001).
Freundlich, J. S. et al. Synthesis and biological activity of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 2: 2′-substituted triclosan derivatives. Bioorg Med Chem Lett 16, 2163–2169 (2006).
Frecer, V., Megnassan, E. & Miertus, S. Design and in silico screening of combinatorial library of antimalarial analogs of triclosan inhibiting Plasmodium falciparum enoyl-acyl carrier protein reductase. Eur J Med Chem 44, 3009–3019 (2009).
Baschong, W. et al. Triclosan is minimally effective in rodent malaria models. Nat Med 17, 33–34; author reply 34–35 (2011).
Anderson, J. W. et al. Novel diaryl ureas with efficacy in a mouse model of malaria. Bioorg Med Chem Lett 23, 1022–1025 (2013).
Freundlich, J. S. et al. X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. J Biol Chem 282, 25436–25444 (2007).
Kumar, S. P., George, L. B., Jasrai, Y. T. & Pandya, H. A. Prioritization of active antimalarials using structural interaction profile of Plasmodium falciparum enoyl-acyl carrier protein reductase (PfENR)-triclosan derivatives. SAR QSAR Environ Res 26, 61–77 (2015).
Yu, M. et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 4, 567–578 (2008).
Vaughan, A. M. et al. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol 11, 506–520 (2009).
Bilsland, E. et al. Yeast-based automated high-throughput screens to identify anti-parasitic lead compounds. Open Biol 3, 120158 (2013).
Bilsland, E. et al. Functional expression of parasite drug targets and their human orthologs in yeast. PLoS Negl Trop Dis 5, e1320 (2011).
Keppler-Ross, S., Noffz, C. & Dean, N. A new purple fluorescent color marker for genetic studies in Saccharomyces cerevisiae and Candida albicans. Genetics 179, 705–710 (2008).
Sheff, M. A. & Thorn, K. S. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21, 661–670 (2004).
Williams, K. et al. Cheaper faster drug development validated by the repositioning of drugs against neglected tropical diseases. J R Soc Interface 12, 20141289 (2015).
Witmer, K. et al. Analysis of subtelomeric virulence gene families in Plasmodium falciparum by comparative transcriptional profiling. Molec Microbiol 84, 243–259 (2012).
Jenkins, S., Addy, M. & Newcombe, R. Toothpastes containing 0.3% and 0.5% triclosan. I. Effects on 4-day plaque regrowth. Am J Dent 2(Spec No), 211–214 (1989).
Srinivasan, S. et al. Plaque triclosan concentration and antimicrobial efficacy of a new calcium carbonate toothpaste with 0.3% triclosan compared to a marketed 0.3% triclosan toothpaste. J Clin Dent 24, 68–72 (2013).
Hopkins, A. L. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 4, 682–690 (2008).
Tasdemir, D. et al. Inhibition of Plasmodium falciparum fatty acid biosynthesis: evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. J Med Chem 49, 3345–3353 (2006).
Navarro-Peran, E. et al. The antifolate activity of tea catechins. Cancer Res 65, 2059–2064 (2005).
Baragana, B. et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522, 315–+ (2015).
Cheng, Y. & Prusoff, W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical pharmacology 22, 3099–3108 (1973).
Fidock, D. A., Nomura, T. & Wellems, T. E. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Molecular pharmacology 54, 1140–1147 (1998).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
Smilkstein, M., Sriwilaijaroen, N., Kelly, J. X., Wilairat, P. & Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48, 1803–1806 (2004).
Kongsaeree, P. et al. Crystal structure of dihydrofolate reductase from Plasmodium vivax: pyrimethamine displacement linked with mutation-induced resistance. Proc Natl Acad Sci USA 102, 13046–13051 (2005).
Yuthavong, Y. et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc Natl Acad Sci USA 109, 16823–16828 (2012).
Zhang, X. et al. Design, synthesis, biological evaluation and X-ray crystal structure of novel classical 6,5,6-tricyclic benzo[4,5]thieno[2,3-d]pyrimidines as dual thymidylate synthase and dihydrofolate reductase inhibitors. Bioorg Med Chem 19, 3585–3594 (2011).
Acknowledgements
This work was supported by: the UK Biotechnology and Biological Sciences Research Council (BB/F008228/1) and a contract from the European Commission under the FP7 Collaborative Programme, UNICELLSYS, both to S.G.O. and R.D.K.; the Bill and Melinda Gates foundation (Op1087646 to EB and SGO), São Paulo Research Foundation - FAPESP (2012/23306-5 to WLF, EFGC and GW and 2015/19103-0 and 2015/03553-6 to EB), the ERC (208813 to FH). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We would like to thank Prof Fabio T. M. Costa for his critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
E.B. performed the plate based yeast-based assays, cloned and contributed with the DHFR purification for in vitro assays; L.V. and F.H. contributed with the in vitro enzyme assays; K.W. and V.J. performed the yeast-based HTP screens; J.F. contributed with the DHFR purification; M.P.C. and M.G. performed the docking experiments; W.L.F., E.F.G.C. and G.W. contributed with the validation in transgenic Plasmodium; E.B., R.D.K. and S.G.O. designed the study and co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bilsland, E., van Vliet, L., Williams, K. et al. Plasmodium dihydrofolate reductase is a second enzyme target for the antimalarial action of triclosan. Sci Rep 8, 1038 (2018). https://doi.org/10.1038/s41598-018-19549-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19549-x
This article is cited by
-
To quest new targets of Plasmodium parasite and their potential inhibitors to combat antimalarial drug resistance
Journal of Parasitic Diseases (2024)
-
Geo-environmental factors and the effectiveness of mulberry leaf extract in managing malaria
Scientific Reports (2023)
-
Biochemical and Structural Insights Concerning Triclosan Resistance in a Novel YX7K Type Enoyl-Acyl Carrier Protein Reductase from Soil Metagenome
Scientific Reports (2019)
-
High-performance medicine: the convergence of human and artificial intelligence
Nature Medicine (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
