
Abstract
The global prevalence of hepatitis C Virus (HCV) is approximately 3% and one-fifth of all HCV carriers live in the Middle East, where Egypt has the highest global incidence of HCV infection. Quantitative structure-activity relationship (QSAR) models were used in many applications for predicting the potential effects of chemicals on human health and environment. The adaptive neuro-fuzzy inference system (ANFIS) is one of the most popular regression methods for building a nonlinear QSAR model. However, the quality of ANFIS is influenced by the size of the descriptors, so descriptor selection methods have been proposed, although these methods are affected by slow convergence and high time complexity. To avoid these limitations, the antlion optimizer was used to select relevant descriptors, before constructing a nonlinear QSAR model based on the PIC50 and these descriptors using ANFIS. In our experiments, 1029 compounds were used, which comprised 579 HCVNS5B inhibitors (PIC50 < ~14) and 450 non-HCVNS5B inhibitors (PIC50 > ~14). The experimental results showed that the proposed QSAR model obtained acceptable accuracy according to different measures, where \({{\boldsymbol{R}}}^{{\boldsymbol{2}}}\) was 0.952 and 0.923 for the training and testing sets, respectively, using cross-validation, while \({{\boldsymbol{R}}}_{{\boldsymbol{L}}{\boldsymbol{O}}{\boldsymbol{O}}}^{{\boldsymbol{2}}}\) was 0.8822 using leave-one-out (LOO).
Similar content being viewed by others


Introduction
Hepatitis C virus (HCV) is a member of the Flaviviridae family and it comprises six major genotypes, with a huge number of subtypes in each genotype1. The HCV genotype distributed throughout the world include genotype 1 (Japan, Europe, and North America), genotype 2 (Japan and North America), genotype 3 (Indian subcontinent), genotype 4 (North Africa and the Middle East), genotype 5 (South Africa), and genotype 6 (South East Asia)2,3.
The global prevalence of HCV is about 3%4,5 and one-fifth of all HCV carriers live in the Middle East2. About 20% of Egyptians are estimated to have HCV infections with almost half a million infections per year6,7. Thus, Egypt has the highest global incidence of HCV infection8,9, which varies from 9% to 50% in some rural areas due to the specific modes of infection5. The mechanism of infection has been fully elucidated, but viral entry and replication are not completely understood7. HCV possesses different enzymes and it has been suggested that HCV polymerase is the main enzyme involved in the viral replication process10.
Similar to other DNA polymerases, the architecture of HCV NS5b polymerase resembles a right hand with “thumb”, “palm” and “fingers” domains. The palm domain catalyzes the phosphoryl transfer reaction, whereas the fingers domain participates in interactions with the incoming nucleoside triphosphate as well as the template base with which it is paired10. HCV NS5b is an interesting target for antiviral therapy with limited side effects and it has been the subject of extensive trials to design nucleoside and non-nucleoside inhibitors11.
In recent years, the quantitative structure-activity relationship)QSAR(model has attracted much attention in pharmaceutical research because it can produce high-quality leads in the early stages of drug discovery12. In addition, QSAR reduces the costs of experiments and the failure rate when identifying lead compounds. Hansch et al. were the first to apply QSAR more than 50 years ago13, but Cros actually developed the QSAR concept in the toxicology field in 1863 by establishing a relationship between the toxicity of alcohols and their water solubility14. Thus, QSAR was developed in physical chemistry, before being applied to data sets containing many thousands of compounds with varied molecular structures, while it also evolved from modest regression approaches to wide-ranging statistical and machine learning methods. The QSAR technique has been used extensively in academic research, industry, and governmental institutions throughout the world. QSAR methods are employed to guide lead optimization approaches15. QSAR models can be applied broadly to evaluate the potential effects of chemicals, materials, and nanomaterials on human health and the environment.
In general, QSAR is used to determine the relationship between the chemical structural features (molecular descriptors) of compounds and their biological activity by using mathematical equations. For example, Hansch et al. proposed a relationship between lipophilicity and biological potency based on a study by Veldstra16. Fujita et al. used quantum-chemical calculations to measure the differences in activity regulation in growing plants17, where the experimental value of the octanol–water property (logP) was measured. Other studies have employed computational approaches to determine different effects of substituents on potency18,19.
Dimensionality can be used to define the type of QSAR used according to the chemical structure dimension. one-dimensional (1D)-QSAR describes the activity based on the total molecular properties such as logp and constitutional properties (no. of atoms of oxygen, nitrogen, etc.). 2D-QSAR associates the activity with the chemical structure such as a pharmacophore.3D-QSAR relates the activity to the interaction fields of molecules. 4D-QSAR is represented by using a group of 3D-ligand conformations, 5D-QSAR considers diverse induced-fit models of 4D-complexes, and 6D-QSAR includes various solvation models20.
QSAR models can be calculated using the following two groups of methods21. The first group comprises linear methods such as linear regression, partial least-squares, multiple linear regression, and principal component analysis (PCA). However, these methods are affected by various limitations, e.g., the interactions between the dependent and independent variables cannot be interpreted easily due to nonlinear relationships. In contrast to linear methods, the second group comprising nonlinear QSAR methods can determine the nonlinear mappings based on physicochemical and biological descriptors of the molecules22,23 while they also avoid some drawbacks of the linear QSAR methods. The nonlinear QSAR methods include artificial neural networks (ANNs), k-nearest neighbors, Bayesian neural nets20, fuzzy mappings24 and the adaptive neuro-fuzzy inference system (ANFIS)25.
ANNs have been applied to determine activity levels26, but this method is sensitive to the parameters and it can be trapped by local optima. Therefore, other methods may be used such as support vector machines (SVMs)27 and ANFIS28. ANFIS is a combination of ANN and fuzzy logic systems, which exploits the advantages of both (i.e., good reasoning using fuzzy logic systems and the simplicity of ANN), and it obtains good results for regression problems (especially the QSAR problem)29.
However, ANFIS has some limitations because it is influenced by the approach used to learn its parameters. In addition, a large number of descriptors may affect the performance of ANFIS (or any other QSAR model). Therefore, selecting the best descriptors is very important for reducing the computational cost and eliminating irrelevant descriptors that might reduce the accuracy of ANFIS. Several methods can be used to select the optimal subset of descriptors, including a method25 comprising two stages where the first employed a genetic algorithm (GA) to select suitable descriptors for the inhibitory activity of cathepsin K, before ANFIS was then used to predict the bioactivity values for cathepsin K. PCA has also been used to determine the largest eigenvectors representing the best descriptors, where these descriptors were then used as inputs for ANFIS and SVM to predict biologically active catechol structures21. However, most descriptor selection approaches, such as GA, are affected by limitations such as slow convergence and time complexity, while PCA and other dimension reduction methods change the original data set. Therefore, the antlion optimizer (ALO) was proposed to solve this problem30. ALO is a recently developed swarm intelligence technique, which emulates the natural interactions between antlions and ants. The ALO algorithm has several advantages such as small number of parameters, free gradient and good ability to balance between the exploration and exploitation30. Therefore, it has been applied in several applications such as, Esha Gupta et al. have proposed method based on ALO algorithm to determine the optimal parameters of primary governor loop of thermal generators, also it used to solve the process planning and scheduling functions in the manufacturing process31, and to find the optimal sizing and location of renewable distributed generations32. In addition, it has been used in several power system problem for example, load forecasting33, economic power dispatch34, and load frequency control35.
According to the previous literature, in this study, we developed an accurate, simple, reliable, and less computationally expensive technique for calculating bioactivity values by combining the ALO algorithm with ANFIS. The proposed model, called ALO-ANFIS, comprises two phases. In the first phase, the ALO algorithm was used to determine relevant descriptors. In the second phase, the ANFIS method was used in QSAR for modeling the relationships between the bioactivities of 1029 HCVNS5B compounds and their structural descriptors (those selected by ALO). In order to evaluate the performance of the selected descriptors, the data set was split into training and testing sets, where the training set comprised 772 compounds for refining the model and the testing set comprised 257 appropriately selected chemicals for testing the model. The accuracy of the ALO-ANFIS model was assessed using leave-one-out (LOO), Y-randomization, and external validation techniques.
The remainder of this paper is organized as follows. In Section 2, we introduce the methods and provide a brief discussion of the data set and their chemical descriptors, as well as the approach used for splitting the data and the basic concepts employed in ANFIS and ALO. In Section 3, we explain the proposed QSAR model. The experimental results are presented in Section 4. In Section 5, we give our conclusions and suggestions for future research.
Methods
Data sets and chemical descriptors
Chemical descriptors define the construction and function of designated chemicals. Many descriptors are now easy to calculate due to advances in computer technology36. These descriptors are classified according to several types such as compositional descriptors, topological descriptors, quantum descriptors, electronic parameters, and geometric parameters. Descriptors may be integers, such as the molecular weight, which describes the entire compound, or substitutions such as a steric effect constant, which refers to a precise fragment or group. Integral descriptors are used only for specific compounds, whereas substitutional descriptors can be used for several compounds, e.g., trichloromethane and trichloroacetic acid have equal steric effect constant values for chlorine37.
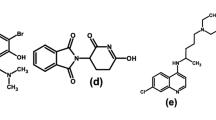
In the current work, Six HCV inhibitors (Fig. 1) (PDB ID: 3HHK, 3SKA, 2BRK, 4DRU, 2GIR and 3PHE) with their derivatives which were collected from literature38,39,40,41,42,43 and gathered in the current dataset44. Their structures in smile format, PIC50 and their literature are represented in the supplementary information.

HCV inhibitors.
In this study, 1029 compounds were used to build the QSAR model, which comprised 579 HCV NS5B inhibitors, (PIC50 < ~14) and 450 non-HCVNS5B inhibitors (PIC50 > ~14)45. These compounds were collected from previous studies and extracted from the BindingDB and CHEMBL databases10,46. The chemical descriptors were calculated using the DataWarrior package47. Thus, DataWarrior was used to calculate the properties of compound such as the drug-likeness, atom counts, and functional groups, where these descriptors represented 29 properties, as shown in Table 1.
Data splitting
In order to evaluate the performance of the proposed model, the training and testing sets were determined using the cross-validation method. In the cross-validation, the data set was split into a number of classes, before selecting one class as the testing set, whereas the other classes were used to construct the training set. We performed a 10-fold cross validation, with nine classes representing the training set and one representing the testing set, where this process was performed 10 times and the average accuracy was calculated based on all the runs.
Adaptive Neuro-Fuzzy Inference System
The ANFIS method combines fuzzy logic and ANN48,49. In recent years, ANFIS has attracted much attention because of its many applications to renewable energy50 and wind prediction51.
In general, the Takagi–Sugeno inference method is one of most popular methods used in ANFIS48. This method generates a nonlinear mapping of fuzzy rules from the input space to the output space by using a number of fuzzy IF–THEN rules. The definition of the first-order Sugeno type is given by Eq. (1)45:
where Q1, Q2 and P1, P1, P2 are the member functions (MFs) for x and y, respectively, and l i , m i , and n i (i = 1, 2) are the associated parameters of the output functions.
The structure of the ANFIS model comprises five layers (Fig. 2), where each layer represents part of the fuzzy inference system (FIS). In particular, the first layer represents the fuzzification process, the second layer is responsible for computing the inference rules and their firing strengths, the third layer is the normalization layer, the adoption layer in FIS is represented by the fourth layer, and the defuzzification output is represented by the last layer. The details of each layer are given in the following. In the structure of ANFIS, the FIS parameters are encoded as weights between layers in the ANN, which allows optimization methods (used in ANN) to be used to determine the optimal FIS parameters.

ANFIS layers.
The first layer receives the input data (x and z) at node i and then computes the membership values (A i and B i ) of the MFs as the output of each node, as given by Eq. (2):
where μ A and μ B are the MFs defined as:
where ρ i and σ i are the data mean and standard deviation, respectively, which represents the premise parameters set (note that Eq. (3) is the generalized Gaussian MF)48.
The second layer computes the firing strength of a rule (ω i ) by each node as follows.
The third layer computes the normalization of the firing strength \(({\bar{w}}_{i})\) as:
The fourth layer nodes compute the output using the following equation:
where l i , m i , and n i are the consequent parameters for node i.
The final layer computes the output of the whole model where this layer comprises a single node:
The ANFIS model employs two sets of adjustable parameters: the premise and consequent parameters, which need to be updated throughout the learning phase until the optimal global values are achieved based on the desired response.
Antlion Optimizer
The Antlion Optimizer (ALO) algorithm starts by generating two random populations of antlions and ants, before assessing each position in these two populations using a fitness function, and the optimal ant position is then found30. In general, the ants update their solution based on the antlions, which are selected by random walk or according to an elite individual. Each ant’s position is updated based on the antlion selected by random walk, where the random walks are normalized according to the following equation30:
where \({a}_{i},{b}_{i},{c}_{i}^{t},\) and \({d}_{i}^{t}\) represent the minimum and maximum of the random walk, and the minimum and maximum of the i-th variable in the t-th iteration, respectively.
The antlion traps affect the random walks of ants, so the ants move within a hyper-sphere (defined by c and d) around a selected antlion \(Antlio{n}_{j}^{t}\). This process is emulated by the ALO algorithm as follows30:
where dt and ct are the maximum and minimum of all the variables, respectively.
The next process is defined as trap building where the roulette wheel method is used to select \(Antlio{n}_{j}^{t}\) based on a fitness function. Thus, a fitter antlion has a higher likelihood of catching the ants.
After building traps, the antlions shoot sands outward from the pit center when ants are in the trap. This behavior is emulated by making ct and dt (defining the radius of the hyper-sphere of ant random walks) decrease with respect to the current iteration t as:
where w is a constant for adjusting the exploitation performance and its value is given as: (1) w = 2 when t > 0.1 T, (2) w = 3 when t > 0.5 T, (3) w = 4 when t > 0.75 T, (4) w = 5 when t > 0.9 T, and (5) w = 6 when t > 0.95 T.
When the ant prey reaches the bottom of the pit, the antlion catches it and takes it into the sand to begin eating. The mathematical definition of this process is given as:
where \(An{t}_{i}^{t}\) is the position of the i-th ant in the t-th iteration. This equation considers that antlions catch prey if they are fitter than other antlions. The position of the antlion is updated to the latest position of the hunted ant to enhance its chance of catching new prey.
In addition to selecting an antlion by random walk, it can be determined using an elite individual strategy, where the best position is used to represent the elite individual. The movement of each ant around a selected antlion may be selected simultaneously by the roulette wheel method and the elite individual strategy, which is formulated as30:
where \({R}_{A}^{t}\) and \({R}_{E}^{t}\) are the random walk around the antlion selected by the roulette wheel and the best position selected by the elite individual strategy, respectively.
Proposed QSAR model
In this section, we explain the proposed QSAR model for predicting the activity of HCV NS5B inhibitors and non-inhibitors. This approach is called the ALO-ANFIS QSAR model and it comprises two phases, where the first is the descriptor selection phase and the second is the PIC50 prediction phase, as given by Algorithm 1. The details of each phase are explained in the following subsections, where the first step in the proposed algorithm is dividing the HCV NS5B data set into training and testing sets using suitable method (such as 10-fold cross-validation method).
Feature selection phase
In this phase, the ALO algorithm is used to select the most relevant features from the training set as follows.
The ALO algorithm starts by generating two populations of ants and antlions, which each solution is converted into binary vector (representing the selected features) using the following equation52:
where ε ∈ [0, 1] is a threshold and x ij represents the jth feature of x i . For example, if x i = 01100, then the second and third features are selected.
The fitness function f i is calculated for each solution as:
where α ∈ [0, 1] is a random number, and |x| and D are the number of selected descriptors and the total number of features, respectively. The \({\hat{Y}}_{i}\) and Y i represent the predict and the actual value of PIC50, respectively, for the training set. Also, N_S is the total number of samples in the PIC50. The fitness function considers the root mean squared error (RMSE) and the number of selected descriptors with the aim of minimizing both.
Based on the best fitness function, the elite individual is selected as the best position for the antlion, and each ant (from the population of ants) updates its position based on the antlion selected using the roulette wheel method. Next, c and d defining the radius of the hyper-sphere are updated, before updating the position of each ant based on the random walk around the selected antlion and the elite individual. The positions of the ants are evaluated and their fitness values are compared with those of the antlions, before the antlions replace their position with those of the corresponding fittest ant. These steps are repeated until the maximum number of iterations is reached or when the difference between the two fitness function becomes smaller than a threshold.
Regression phase
In the regression phase, the reduced training set (after select the features from the first phase) is used as the input for the ANFIS model where it is used to learn the ANFIS parameters. After the ANFIS training stage has finished, the testing set (with the same selected features) is used as the input and the ANFIS output is computed. The performance of the output is compared with the actual PIC50 value, where the RMSE and other measures are computed.
Algorithm 1: ALO-ANFIS QSAR model.
-
1.
Input: Data set for QSAR D, and the target yIC50
-
2.
Output: Predicted values and performance measures.
-
3.
Define: Number of solutions N in the population of ants and antlions, Iter max as the maximum number of iterations, and c, d defining the radius of the hyper-sphere
-
4.
Divided the dataset D into training (D train ) and testing (D test ) sets using 10-fold CV method.
Feature Selection Phase:
-
5.
X = ALO(Iter max , N, c, d, D train )
Regression Phase
-
6.
Create new training set based on the selected features D newtrain = D train (:,X)
-
7.
Learn the ANFIS parameters based on the training set to obtain the (trained model = ANFIS(D newtrain )).
-
8.
Create new testing set based on the selected features D newtest = D test (:, X)
-
9.
Apply the trained model to the testing set to compute the predicted value of PIC50 (\({\hat{y}}_{IC50}={\rm{Trained}}\,{\rm{model}}({D}_{newtest})\))
-
10.
Evaluate the performance of the predicted output \({\hat{y}}_{IC50}.\)
Algorithm 2: ALO algorithm (Iter max , N,c, d, D).
-
1.
Generate a random two populations of ants and antlions.
-
2.
For i = 1: N
-
3.
Evaluate the fitness fAL i of antlions i using Eq. (15).
-
4.
End
-
5.
t = 1 //initial value of the current iteration
-
6.
Do
-
7.
Determine the best solution (Elite) by selecting the best fAL best .
-
8.
For i = 1: N
-
9.
Choose the antlion using the roulette wheel method (antlions RW ).
-
10.
Update the radius of the hyper-sphere, c and d, using Eqs. (9)–(11).
-
11.
Use Eq. (8) to perform a random walk around antlions RW with the roulette wheel method \(\,{R}_{A}^{t}\).
-
12.
Use Eq. (8) to perform a random walk around the elite individual \({R}_{E}^{t}\).
-
13.
Use Eq. (13) to update the position of ant i .
-
14.
Evaluate the fitness fant i of ant i using Eq. (15).
-
15.
If fant i ≤ fAL i
-
16.
antlions i = ant i (using Eq. (12))
-
17.
End IF
-
18.
End For
-
19.
t = t + 1
-
20.
Until (t < Iter max )
-
21.
Return elite X
Algorithm 3: ANFIS(D tain ).
-
1.
Normalize the training set (D tain ) to be D norm .
-
2.
Construct the ANFIS network with Gaussian MF.
-
3.
DO
-
4.
Compute the membership values for each node in the first layer
-
5.
Computes the firing strength of the rule for each node in the second layer
-
6.
Compute the normalized firing strength in the third layer.
-
7.
Compute the output for each node in the fourth layer using Eq. (6).
-
8.
Compute the output \(\hat{y}\) using Eq. (7).
-
9.
If the difference between the \(\hat{y}\) and y is smaller than ε,
-
10.
Break
-
11.
Else
-
12.
Update the parameters of the ANFIS model using the backpropagation method.
-
13.
End
-
14.
t = t + 1
-
15.
Until the maximum number of iterations is reached.
-
16.
Return the trained model
The complexity of the proposed QSAR model
The computational complexity of the proposed ALO-ANFIS is depended on some elements (1) the population size (N). (2) Maximum number of iterations (Iter max ), (3) the number of features (N f ), (4) sample size of the dataset (N s ). (5) The number of cluster in ANFIS model (N C ), number of selected features (N sf ), (6) the sorting algorithm (we used the Quicksort).
Where the complexity of Quicksort in best case is O(NlogN) and in the worst case is O(N2). Therefore, the proposed ALO-ANFIS model has complexity
Experiments and Results
QSAR Model
The experiments were implemented in Matlab and run in a 64-bit Windows environment. The parameters comprised a population size N = 25, maximum number of iterations = 100, and the stopping condition was reaching the maximum number of iterations. We evaluated the performance of the proposed model by changing the maximum number of iterations, but we found that when the maximum number of iterations exceeded 100, the increase in performance became very small as the computational time increased. In addition, when the maximum number of iterations was less than 100, the computational time was reduced but the performance of the proposed model was greatly degraded. The same effects were obtained when the size of the population was varied.
Molecular docking
After filtration the 1029 chemical structures to reach 140 structures, molecular docking process was carried out to validate the QSAR technique, supplementary information.
The protein crystal structure, 3HHK, were cleaned and treated as described in literature53; the crystal structure was used because of its high resolution 1.7 Å. Chimera was used for crystal structure preparation54, only one chain of 3HHK protein was used, all ligands or any solvent molecules were discarded, and polar hydrogens were added by MGLTools.
The MTiOpenScreen is a virtual screening server, which uses Autodock Vina as a docking tool55 to investigate the highly potent and selective molecular compounds as mentioned before56. When using a big data set reaches to 1000 structures, a certain criteria will be applied to ensure a good absorption/penetration of the drugs. this criteria includes Lipenski rule of five (no more than 5 hydrogen bonds, no more than 10 hydrogen bond acceptors, the molecular weight less than or equal to 500 daltons and an octanol-water partition coefficient not be greater than 5) and other two criteria the rotatable bonds less than 10 and the polar surface area will be less than 140 Å57.
Molegro molecular view58 was used to extract the docked inhibitors from their receptors for the graphic representation.
Performance measures
Three groups of measures were used to evaluate the performance of the proposed model. The first group comprised statistical measures for evaluating the performance by comparing the predicted output obtained from the proposed model and the actual value. The second group used a set of criteria to determine whether the proposed model obtained suitable predictions. The third group determined the applicable domain for the proposed model. Definitions of each of these measures are given in the following.
Statistical measures
-
(a)
The mean squared error (MSE) measures the difference between the predicted value and the actual values as:
$$MSE=\frac{1}{{N}_{\mbox{--}}S}\sum _{i=1}^{{N}_{\mbox{--}}S}{({\hat{Y}}_{i}-{Y}_{i})}^{2}.$$(17) -
(b)
RMSE represents the data dispersion around zero deviation, which is defined by:
$$RMSE=\sqrt{\frac{1}{{N}_{\mbox{--}}S}\sum _{i=1}^{{N}_{\mbox{--}}S}{({\hat{Y}}_{i}-{Y}_{i})}^{2}}.$$(18) -
(c)
Coefficient of determination (R2) measures the goodness of fit between the results obtained by a method and the actual data. If the value of R2 is closer to 1, the correlation between the experimental and predicted values is better. R2 is defined by:
Predictive criteria
In addition, we used the criteria proposed by Tropsha et al.59 for determining whether a model is predictive (when they are satisfied) or not (when they are not satisfied), as follows:
where R2represents the regression correlation coefficient of between Y and \(\hat{Y}\) (the actual values of PIC50 and their predicted values, respectively) in both sets (training and testing), \({R}_{O}^{2}\) (\({R}_{O}^{\text{'}2})\,\,\)represent the regression correlation coefficients between \(\hat{Y}\) and Y (Yversus \(\hat{Y}\)) through the origin, and K and K'represent the slopes of the regression lines through the origin (for more information about these measures, please refer to Tropsha et al.59.
Moreover, in another study60, R2 to was modified to determine the difference between \({R}_{O}^{2}\) and \({R}_{O}^{\text{'}2}\) as:
where the model is considered to have good external predictability if the value of \({R}_{m}^{2} > 0.5\).
Applicable domain
Tropsha et al.59 used the Williams plot to measure the applicable domain (AD) for the QSAR model, where this type of graph plots leverage values against the standardized residuals. The leverage value h i for the elements of the independent variable is computed as:
where Xis the trained model constructed from the training set and x i is the principal component vector of the element considered.
The warning leverage value (h*) is computed by 3K + 1/N_S, where K represents the total number of independent variables. The probability of accordance between the predicted and experimental values is high for the elements (molecules) in the training set when h < h*. In addition, h > h* indicates that this element will enhance the QSAR model when the element is present in the training set. However, if this element is in the testing set, this indicates that the predicted value is unreliable. If the residual of this element is low, then this element cannot be considered as an outlier. Therefore, in order to determine the applicable domain of the QSAR model, the standardized residual and the leverage must be considered at the same time.
Molecular docking evaluation
To validate the molecular docking of the current data set structures, the ligand 7ZZ was re-docked to HCVNS5b or 3HHK as mentioned before, the RMSD between the experimental and docked 7ZZ structure was less than 2 angstrom, see Fig. 3.

The structures of experimental (aqua) and docked structure (orange) to 3HHK receptor.
In Fig. 3, compound 7ZZ colored in different colors to differentiate between experimental and docked structure. Amino acid residues appear as thin sticks while ligand atoms are represented as bold sticks. Hydrophilic residues have a red color, while hydrophobic residues have blue color. Atoms of residues are colored according to the hydropathy index proposed by Kyte and Doolittle in 198261, the blue dashed line represents the hydrogen bond.
Results and discussion
The HCVNS5B QSAR model was built using the proposed ALO-ANFIS approach. The computed Pearson’s correlation coefficients between the selected descriptors (molecular flexibility (var1), molecular complexity (var2), non-C/H atoms (var3), electronegative atoms (var4), stereo centers (var5), rotatable bonds (var6), rings (var7), aromatic atoms (var8), and symmetric atoms (var9)) are shown in Fig. 4, which indicates there were positive and negative correlations between various descriptors, but the values were not high so no further descriptors were removed.

Correlation matrix.
Figures 5–6 show the predicted values and the experimental values for the training set and testing set, respectively, which indicate that the predicted PIC50 values agreed well with the experimental values (prediction percentage error was less than 5%). The model did not exhibit proportional and systematic error because the distribution of the residuals to both sides of zero was random. In addition, Fig. 7 shows the regression plot for the training set, testing set, and all the actual PIC50 values.

Training set results obtained by the proposed QSAR model (the output in the legend refers to the actual training set), (A) the predicted versus the actual, (B) the MSE and RMSE values, (C) the histogram of the Error.

Testing set results obtained by the proposed QSAR model (the output in the legend refer to the actual testing set), (A) the predicted versus the actual, (B) the MSE and RMSE values, (C) the histogram of the Error.

The Correlation results for the experimental PIC50 values versus the values predicted by the ALO-ANFIS model.
Table 2 shows the results obtained for the proposed ALO-ANFIS model according to the set of measures, which indicates that the RMSEand MSE values were 0.1104 and 0.012, respectively. The value of \({R}_{var}^{2}\,\)was higher than 0.6, but all of the results indicate that the goodness of fit for the proposed model was satisfactory. However, several QSAR models obtained good fits (such as ANN with a sufficient number of layers and neurons) but these models were not predictive.
Therefore, several criteria must be satisfied in order to determine whether a model is predictive, as mentioned in the previous section. These criteria are divided into internal and external validation measures59. To assess the internal measures, the model was evaluated using only the available data (training) and no other external data (testing set) (e.g., the Y-randomization test). In contrast to the internal measures, the external measures depended on the testing data that had not been used already.
To test the predictability of the proposed model, we divided the data into a training set (823) and testing set (206) (which were selected randomly and they had not been used for training the model). The statistical measures obtained for the ALO-ANFIS model using the training and testing sets are given in Table 2. The RMSE of the proposed model was small for the testing set (~0.588), but after comparing this value with those reported previously25,26, we considered that this value was high, possibly because the testing set was larger than those used in previous studies. In addition, the R2 of values for the training and testing sets were 0.952, and 0.923, respectively, which are greater than 0.5.
Moreover, the results in Table 2 satisfy the criteria mentioned above59, where the \({R}^{2}\) value obtained for the testing set was greater than 0.6, and the \({R}^{2}\) value for the training set (or Q2) was greater than 0.5. In addition, the value of R2 was very close to \({R}_{0}^{2}\) (and \({R^{\prime} }_{0}^{2}),\) where the values of \({R}^{2}-{R}_{0}^{2}/{R}^{2}\) and \({R}^{2}-{R^{\prime} }_{0}^{2}/{R}^{2}\) were smaller than 0.11. Thus, based on the values of the \({R}_{m}^{2}\) parameter (the values were greater than 0.5 for both the training and testing sets), the ALO-ANFIS QSAR model was considered to be a predictive model.
We also obtained evidence that the ALO-ANFIS QSAR model is robust and predictive based on the chance correlation results produced using the LOO test and the Y-randomization test. The results of the LOO cross-validation for the training set (in Table 2) as \({R}_{LOO}^{2}\,\,\)and RMSE LOO were 0.8822 and 0.4440, respectively, so it is reasonable to use the ALO-ANFIS model in QSAR. In the Y-randomization test, we constructed a number of models (set to 10) by randomly permuting the data in the original model, and the expected values of R2 and Q2 for these constructed models were smaller than those for the original ALO-ANFIS QSAR model, as shown in Table 3.
The applicable domain for the proposed ALO-ANFIS QSAR model was determined using the Williams graph shown in Fig. 8, where the leverage value and standardized residuals are plotted. Figure 8 shows that one molecule has a h value higher than h* = 0.5, which indicates that this molecule represents an outlier for the structure; therefore, the predicted values for this molecule should be considered as being extrapolated using the ALO-ANFIS QSAR model.

Williams plot for the ALO-ANFIS model with h* = 0.5.
Comparison with other models
The performance of the proposed QSAR model is compared with other two models namely, PSO-ANFIS and GA-ANFIS (see the supplementary file for more information about PSO and GA) as given in Table 4. From this table it can be concluded that the PSO-ANFIS model is better than GA-ANFIS model in terms of R2 and RMSE for both training and testing sets. However, the proposed QSAR model is still better than the other two models except the R2 for training set the PSO-ANFIS is better. In addition, the ALO-ANFIS selects smaller number of descriptors than the other models with less times computational.
Moreover, (Figs 9–12) show the actual PIC50 value versus the predicted values by using PSO-ANFIS and GA-ANFIS, respectively, for training set (testing set). From these figures we can observed that the PSO-ANFIS is closed to the actual values than the GA-ANFIS, especially, in the testing set this goodness of fit is appeared.

Training set results obtained by the PSO-ANFIS QSAR model (the output in the legend refer to the actual testing set), (A) the predicted versus the actual, (B) the MSE and RMSE values, (C) the histogram of the Error.

Training set results obtained by the GA-ANFIS QSAR model (the output in the legend refer to the actual testing set), (A) the predicted versus the actual, (B) the MSE and RMSE values, (C) the histogram of the Error.

Testing set results obtained by the PSO-ANFIS QSAR model (the output in the legend refer to the actual testing set), (A) the predicted versus the actual, (B) the MSE and RMSE values, (C) The histogram of the Error.

Testing set results obtained by the GA-ANFIS model (the output in the legend refer to the actual testing set), (A) the predicted versus the actual, (B) the MSE and RMSE values, (C) The histogram of the Error.
From all the pervious results we can conclude that the proposed ALO-ANFIS model is better than the other two models in terms of performance measures (R2, RMSE Time(s), No. Descriptors).
From the previous results we concluded that the 9 features that selected by the proposed QSAR model have the largest influencing the HCVNS5B inhibitors. However, the proposed model cannot write its output as a regression equation to describe the relation between the 9 descriptors and the PIC50, so, it is difficult to interpret the obtained results. However, the harmony between these descriptors create potent HCV inhibitors as the following:
Molecular flexibility: The word flexibility is depending on the chemical graph and refers to the molecular mass, branches, rings and heteroatoms. The importance of molecular flexibility in chemistry and biology are many. As in receptor-ligand interactions, the flexible molecules near pharmacophores may led to reaction inhibition and in chemical reactions, it may led to intermolecular interactions and consequently physical changes62.
Molecular complexity: It represents the sum of bond connectivity’s of molecular structures. It is a simple tool to design a synthetic pathway to a specific molecule. The relation between molecular complexity and biological activity is mentioned before63, it is noticed that more simple molecular structure will produce more potent drug64.
Hydrogen bonding: Is a weak bond which formed between hydrogen atom and an electronegative atom like Oxygen, nitrogen and sulfur. There are two types of Hydrogen bonds, if it is formed between molecules, it will be named intermolecular or within a molecule and it will be called intramolecular65.
Number of rotatable bonds (nrotb): It represents the change in conformational entropy of a molecule66. It also express molecular flexibility and oral bioavailability of drugs67. Searle and Williams revealed that each rotatable bond introduces about 1.2–1.6 kcal/mol in changing of binding free energy, assuming complete loss of rotational freedom68.
Rings and Aromatic Rings: Debnath et al.69, noticed that increasing hydrophobicity is in linear relation with mutagenic effect.
Symmetric atoms: It represents 3D property and it describes atom distribution with respect to some invariant reference frames70.
Finally, from the previous discussion it can be observed that the proposed ALO-ANFIS QSAR model provides an efficiency and effectiveness. Since it can selecting an optimal subset of descriptors that increase the regression accuracy. This promising results are achieved due to the ALO algorithm has good ability to balance between the exploration and exploitation during the search process about the optimal solution. Also, due to the good properties of the ANFIS model to solve the QSAR regression problem, since it combines ANN and fuzzy logic system.
Molecular docking and QSAR
In Fig. 13, Amino acids were colored according to hydropathy while ligand according to atom type in electrostatic interactions in A and C view. Also ligand was colored according to hydrophobicity in B and D image segments. Hydrogen bond was represented in blue dashed line. The size of compound in C is more accommodate to protein binding site than A, this explain A(the lowest hit) with binding energy = −5.1 kcal/mol and C (the best hit) with binding energy = −13.5 kcal/mol.

The lowest and best hit in the current docking set were shown in hit (A–D) respectively.
In the current study, the highly potent and selective molecular compounds56 were depend on several descriptors like molecular flexibility (var1), molecular complexity (var2), non-C/H atoms (var3), electronegative atoms (var4), stereo centers (var5), rotatable bonds (var6), rings (var7), aromatic atoms (var8) symmetric atoms (var9), so a curve will be made between these descriptors versus binding energy and PIC50 as in Fig. 14. From this figure, it can be observed that the value of the PIC50 is increased with increase the value of the following descriptors (var2, var3, var4, var5, and var7), however, with the other descriptors its value is decreased, with small value, by increasing their value. In addition, the value of binding energy is decreased with decreasing the value of var2, var5, and var7 descriptors, and increased with other descriptors.

The correlation between the PIC50, Bending energy and the 9 descriptors.
Conclusion
HCV NS5B is an interesting target for antiviral therapy with limited side effects and it has been the subject of extensive trials to develop nucleoside and non-nucleoside inhibitors. In this study, we used a series of HCV NS5B inhibitors to build a QSAR model. The chemical descriptors were calculated using the DataWarrior package. However, any QSAR model is usually influenced by the number of descriptors and the regression method employed. Therefore, in this study, we developed a new QSAR model for assessing inhibitors and non-inhibitors of HCV NS5B. The proposed model comprised two stages, where we used the ALO algorithm to determine the most relevant descriptors related to the PIC50 values in the first stage, and ANFIS was then used to determine the nonlinear relationships between the selected descriptors and the PIC50 values in the second stage. The results obtained by the proposed model indicate that it is an acceptable approach for predicting the activity of drugs as HCV NS5B inhibitors.
According to the promising results obtained using the proposed model, we will apply this method to other complex problems in drug design in future research, as well as other applications such as wind speed prediction by making suitable improvements.
References
Simmonds, P. et al. Evolutionary analysis of variants of hepatitis C virus found in South-East Asia: comparison with classifications based upon sequence similarity. Journal of General Virology. 77, 3013–3024 (1996).
Ramia, S. & Eid-Fares, J. Distribution of hepatitis C virus genotypes in the Middle East. International Journal of Infectious Diseases. 10, 272–277 (2006).
Watson, J. P., Al-Mardini, H., Awadh, S., Ukabam, S. & Record, C. O. Hepatitis C virus genotypes in a cohort of Middle Eastern patients. Annals of Saudi medicine. 19, 410–412 (1999).
Fallahian, F. & Najafi, A. Epidemiology of hepatitis C in the Middle East. Saudi Journal of Kidney Diseases and Transplantation. 22, 31 (2011).
Craxì, A. et al. EASL clinical practice guidelines: management of hepatitis C. J Hepatol. 55, 245–64 (2011).
Yahia, M. Global health: a uniquely Egyptian epidemic. Nature. 474, S12–S3 (2011).
El-Zanaty, F. & Way, A. Egypt Demographic and Health Survey 2008 Egyptian: Ministry of Health. Cairo: El-Zanaty and Associates and Macro International. 421 (2009).
Mohamed, M. K. et al. Intrafamilial transmission of hepatitis C in Egypt. Hepatology. 42, 683–687 (2005).
Alter, M. J. Epidemiology of hepatitis C virus infection. World Journal of gastroenterology. 13, 24–36 (2007).
Das, D. et al. Recent advances in drug discovery of benzothiadiazine and related analogs as HCV NS5B polymerase inhibitors. Bioorganic & medicinal chemistry. 19, 4690–4703 (2011).
Tomei, L., Altamura, S., Paonessa, G., De Francesco, R. & Migliaccio, G. Review HCV Antiviral Resistance: The Impact of in vitro Studies on the Development of Antiviral Agents Targeting the Viral NS5B Polymerase. Antiviral chemistry and chemotherapy. 16, 225–245 (2005).
Myint, K. Z. & Xie, X.-Q. Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. Int J Mol Sci. 11(10), 3846–3866 (2010).
Hansch, C., Maloney, P. P., Fujita, T. & Muir, R. M. Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients (1962).
Cros, A. Action de l’alcool amylique sur l’organisme (1863).
Cramer, R. D. The inevitable QSAR renaissance. Journal of computer-aided molecular design. 26, 35–8 (2012).
Veldstra, H. The Relation of Chemical Structure to Bio-Logical Activity in Growth Substances. Annual Review of Plant Physiology. 4, 151–98 (1953).
Fujita, T., Imai, S., Koshimizu, K., Mitsui, T. & Kato, J. Plant Growth Activities of 5-and 8-Halogeno-dihydro-and-tetrahydro-1-naphthoic Acids (1959).
Hansch, C. et al. The correlation of biological activity of plant growth regulators and chloromycetin derivatives with Hammett constants and partition coefficients. Journal of the American Chemical Society. 85, 2817–2824 (1963).
Hansch, C. & Fujita, T. p-σ-π Analysis. A method for the correlation of biological activity and chemical structure. Journal of the American Chemical Society. 86, 1616–1626 (1964).
Verma, J., Khedkar, V. M. & Coutinho, E. C. 3D-QSAR in drug design–a review. Current topics in medicinal chemistry. 10, 95–115 (2010).
Khoshneviszadeh, M. & Sakhteman, A. Exploring quantitative structure–activity relationship (QSAR) models for some biologically active catechol structures using PC-LS-SVM and PC-ANFIS. Appl. Biol. Chem. 59(3), 433–441 (2016).
Dastorani, M. T., Moghadamnia, A., Piri, J. & Rico-Ramirez, M. A. Application of ANN and ANFIS models for reconstructing missing flow data. Env. Monit Assess. 166, 421–434 (2010).
Mizuta, K. et al. The dopamine D1 receptor is expressed and facilitates relaxation in airway smooth muscle. Respir Res. 14, 89 (2013).
Buyukbingol, E., Sisman, A., Akyildiz, M. & Alparslan, F. N. Adaptive neuro-fuzzy inference system (ANFIS): a new approach to predictive modeling in QSAR applications: a study of neuro-fuzzy modeling of PCP-based NMDA receptor antagonists. Bioorg Med Chem. 15, 4265–4282 (2007).
Shahlaei, M., Madadkar-Sobhani, A., Saghaie, L. & Fassihi, A. Application of an expert system based on Genetic Algorithm-Adaptive Neuro-Fuzzy Inference System (GA-ANFIS) in QSAR of cathepsin K inhibitors. Expert Syst. Appl. 39(6), 6182–6191 (2012).
Kompany-Zareh, M. & Akhlaghi, Y. Correlation weighted successive projections algorithm as a novel method for variable selection in QSAR studies: investigation of anti-HIV activity of HEPT derivatives. J. Chemom. 21, 239–250 (2007).
Gharaghani, S. G. & Fatemi, M. H. A novel QSAR model for prediction of apoptosis-inducing activity of 4-aryl-4-H-chromenes based on support vector machine. Bioorg. Med. Chem. 15, 7746–7754 (2007).
Goodarzi, M. & Freitas, M. MIA-QSAR coupled to principal component analysis-adaptive neuro-fuzzy inference. Eur. J. Med. Chem. 45, 1352–1358 (2010).
Du, H., Du, X. Y. H., Wang, J., Hu, Z. & Liu, M. Prediction of relative sensitivity of the olfactory and nasal trigeminal chemosensory systems for a series of the volatile organic compounds based on local lazy regression method. Sensors Actuators B Chem. 138, 55–63 (2009).
Mirjalili, S. Ant Lion Optimizer. Adv. Eng. Softw. 83, 80–98 (2015).
Petrović, M. et al. The Ant Lion Optimization Algorithm for Integrated Process Planning and Scheduling. Applied Mechanics and Materials. 834, 187–192 (2016).
Ali, E. S., Abd Elazim, S. M. & Abdelaziz, A. Y. Ant Lion Optimization Algorithm for optimal location and sizing of renewable distributed generations. Renewable Energy. 101, 1311–1324 (2017).
Zhao, H. & Guo, S. An optimized grey model for annual power load forecasting. Energy. 107, 272–286 (2016).
Nischal, M. M. & Mehta, S. Optimal load dispatch using ant lion optimization. Int. J Eng Res Appl. 5(8) (2015).
Satheeshkumar, R. & Shivakumar, R. Ant lion optimization approach for load frequency control of multi-area interconnected power systems. Circuits Syst. 7, 2357–2383 (2016).
Emmert-Streib, F., Dehmer, M., Varmuza, K. & Bonchev, D. Statistical modelling of molecular descriptors in QSAR/QSPR: John Wiley & Sons (2012).
Chen, B., Zhang, T., Bond, T. & Gan, Y. Development of quantitative structure activity relationship (QSAR) model for disinfection byproduct (DBP) research: A review of methods and resources. Journal of hazardous materials. 299, 260–79 (2015).
Cummings, M. D. et al. Structure-based macrocyclization yields hepatitis C virus NS5B inhibitors with improved binding affinities and pharmacokinetic properties. Angew Chemie—Int Ed. 51, 4637–4640 (2012).
Shaw, A. N. et al. Substituted benzothiadizine inhibitors of Hepatitis C virus polymerase. Bioorg Med Chem Lett. Elsevier Ltd. 19, 4350–4353 (2009).
Anilkumar, G. N. et al. I. Novel HCV NS5B polymerase inhibitors: Discovery of indole 2-carboxylic acids with C3-heterocycles. Bioorganic Med Chem Lett. Elsevier Ltd. 21, 5336–5341 (2011).
Di Marco, S. et al. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J Biol Chem. 280, 29765–29770 (2005).
Le Pogam, S. et al. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J Virol. 80, 6146–6154 (2006).
Kumar, D. V et al. Quinolones as HCV NS5B polymerase inhibitors. Bioorg Med Chem Lett. Elsevier Ltd. 21, 82–87 (2011).
Wei, Y. et al. Discovery of novel hepatitis C virus NS5B polymerase inhibitors by combining random forest, multiple e-pharmacophore modeling and docking. PloS one. 11(2), e0148181 (2016).
Lü, W. & Xue, Y. Prediction of hepatitis c virus non-structural proteins 5B polymerase inhibitors using machine learning methods. Acta Physico-Chimica Sinica. 27, 1407–1416 (2011).
May, M. M. et al. Discovery of substituted N-phenylbenzenesulphonamides as a novel class of non-nucleoside hepatitis C virus polymerase inhibitors. Antiviral research. 95, 182–91 (2012).
Sander, T., Freyss, J., von Korff, M. & Rufener, C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. Journal of chemical information and modeling. 55, 460–73 (2015).
Sugeno, M. & Kang, G. T. Structure identification of fuzzy method. Fuzzy Sets Syst. 28(1), 15–33 (1988).
Jang, J.-S. R. System, ANFIS: adaptive-network-based fuzzy inference. Trans. Syst. Man Cybern. 23(3), 665–685 (1993).
Suganthi, L., Iniyan, S. & Samuel, A. A. Applications of fuzzy logic in renewable energy systems – A review. Renew. Sustain. Energy Rev. 48, 585–607 (2015).
Lin, W. M., Cheng, F. S. & Hong, C. M. Fuzzy neural network output maximization control for sensor less wind energy conversion system. Energy. 35, 592–601 (2010).
Abd El Aziz, M. E. & Hassanien, A. E. Modified cuckoo search algorithm with rough sets for feature selection, Neural Comput. Applic. (2016).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Pettersen, E. F. et al. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem. 25, 1605–1612 (2004).
Labbé, C. M., Rey, J., Lagorce, D. et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Research. 43 (Web Server issue): W448–W454 (2015).
Mejdrová, I. et al. Rational design of novel highly potent and selective phosphatidylinositol 4-kinase IIIβ (PI4KB) inhibitors as broad-spectrum antiviral agents and tools for chemical biology. Journal of medicinal chemistry 60(1), 100–118 (2016).
Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45(12), 2615–2623 (2002).
Thomsen, R. & Christensen, M. H. MolDock: A New Technique for High-Accuracy Molecular Docking. J Med Chem. 49, 3315–3321 (2006).
Tropsha, A., Gramatica, P. & Gombar, V. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR & Combinatorial Science 22, 69–77 (2003).
Roy, P. P. & Roy, K. On some aspects of variable selection for partial least squares regression models. QSAR & Combinatorial Science. 27, 302–313 (2008).
Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 157, 105–132 (1982).
Kier, L. B. An Index of Molecular Flexibility from Kappa Shape Attributes. Quantitative Structure-Activity Relationships. 8, 221–224 (1989).
Schuffenhauer, A., Brown, N., Selzer, P., Ertl, P. & Jacoby, E. Relationships between Molecular Complexity, Biological Activity, and Structural Diversity. Journal of Chemical Information and Modeling. 46, 525–535 (2006).
Hann, M. M., Leach, A. R. & Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. Journal of chemical information and computer sciences. 41, 856–864 (2001).
Roy, K, Kar, S. & Das, R. N. Understanding the basics of QSAR for applications in pharmaceutical sciences and risk assessment. Academic press. (2015).
Kubinyi, H., Folkers, G. & Martin, Y. C. eds 3D QSAR in Drug Design: Volume 2: Ligand-Protein Interactions and Molecular Similarity. 2. Springer Science & Business Media (1998).
Zlatkov, A. B., Peikov, P. T., Momekov, G. C., Pencheva, I. & Tsvetkova, B. Synthesis, Stability and Computational Study of some Ester Derivatives of Theophylline-7-acetic Acid with Antiproliferative Activity. Der Pharma Chemica. 2, 197 (2010).
Searle, M. S. & Williams, D. H. The cost of conformational order: Entropy changes in molecular associations. J Am Chem Soc. 114, 10690–10697 (1992).
Debnath, A. K. et al. A QSAR investigation of the role of hydrophobicity in regulating mutagenicity in the Ames test: 1. Mutagenicity of aromatic and heteroaromatic amines in Salmonella typhimurium TA98 and TA100. Environmental and molecular mutagenesis 19(1), 37–52 (1992).
Mamy, L., Patureau, D. & Barriuso, E. et al. Prediction of the Fate of Organic Compounds in the Environment From Their Molecular Properties: A Review. Critical Reviews in Environmental Science and Technology. 45(12), 1277–1377 (2015).
Acknowledgements
This work was partially supported by National Key Research & Development Program of China (No. 2016YFD0101903), National Natural Science Foundation of China (No. 61702386, 61672398), Major Technical Innovation Program of Hubei Province (No. 2017AAA122), Key Natural Science Foundation of Hubei Province of China (No. 2017CFA012) and Applied Foundamental Research of Wuhan (No. 20160101010004).
Author information
Authors and Affiliations
Contributions
All authors contributed equally to this study, Yasmine S. Moemen collected and prepared the data, Mohamed Abd Elaziz and Aboul Ella Hassanien developed the algorithm used in this study, Shengwu Xiong and the other three authors wrote the main manuscript and discussed the results.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elaziz, M.A., Moemen, Y.S., Hassanien, A.E. et al. Quantitative Structure-Activity Relationship Model for HCVNS5B inhibitors based on an Antlion Optimizer-Adaptive Neuro-Fuzzy Inference System. Sci Rep 8, 1506 (2018). https://doi.org/10.1038/s41598-017-19122-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-19122-y
This article is cited by
-
Soft computing techniques for biomedical data analysis: open issues and challenges
Artificial Intelligence Review (2023)
-
Enhanced Harris hawks optimization with genetic operators for selection chemical descriptors and compounds activities
Neural Computing and Applications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
