
Abstract
Ewing’s sarcoma family of tumors (EFTs) is a group of rare and aggressive tumors. Data on EFTs in patients (pts) ≥ 50 years are limited and these pts are often not eligible for clinical trials. Some, but not all, studies have reported inferior outcome for older pts with EFTs. We conducted an IRB-approved retrospective analysis among centers of the French Sarcoma Group on pts diagnosed with EFTs at age ≥50 between 2000 and 2012. Clinical features, treatment modality and outcomes were analyzed. Seventy-seven pts were identified, including 36 females (46.8%) and the median age at diagnosis was 56 years (range: 50–86). The primary tumor was located in soft tissue in 59 pts (76.6%). Fifty-six pts (72.7%) had localized disease, among them 49 (87.5%) received chemotherapy in addition to local therapy. Their estimated 3-yr OS and event-free survival (EFS) rates were respectively 73.3% and 62.2%. Recurrence occurred in 43 pts. The estimated 3-yr OS rate was 37% in pts with metastatic disease at presentation. EFTs in pts ≥50 years are more likely to originate from soft tissue and their outcomes appear to be worse than that of younger pts treated with modern protocols.
Similar content being viewed by others

Introduction
The Ewing’s sarcoma family of tumor (EFTs) is a group of aggressive tumors affecting most often children, adolescent and young adult1,with a median age of 15 years at diagnosis. The estimated annual incidence of Ewing sarcoma is 3/1.000.000 in France and in the USA2,3. In 85–90% of EFTs cases, the tumor is characterized by a translocation involving the EWSR1 (EWS RNA-Binding Protein 1) gene on chromosome 22, and an ETS (E26 transformation-specific)-family gene such as FLI-1 or ERG1. The primary site of disease is the bone in 80% of cases and about one fourth of patients present with metastasis at initial diagnosis1.
The current standard of care for patients with EFTs includes intensive induction chemotherapy followed by local therapy (surgery, radiotherapy or a combination of both) followed by adjuvant chemotherapy4. Recent studies have shown the benefit of four or five drug regimens and interval time compression of chemotherapy, suggesting a beneficial effect of dose-intensity in this chemo-sensitive disease5,6,7. However, most of these studies have shown that the benefit of more intensive chemotherapy regimens is limited to patients with localized disease8.
Although rare, EFTs in older subjects represents a clinical challenge because many of these patients will not be suitable candidates for intensive induction chemotherapy. Therefore, the optimal management of these patients remains to be defined. Limited data are available on age-specific prognostic and treatment, with discordant conclusions. A recent report for the SEER database has shown an inferior survival for EFTs patients older than 40 years9, while three other reports suggest that adherence to chemotherapy protocol is similar in patients less than 40 and patients older than 4010,11,12. Authors of these reports therefore argue that older patients should be managed with similar chemotherapy protocols. These studies have in most cases focused on patient treated in the 1990’s, before the advent of 5 drug regimens. Furthermore, some have compared the outcome of patients less than 40 to that of patients aged 40 to 50 years11,12.
The EURO EWING 99 study has served as the guide for the management of patients with EFTs in the French Sarcoma Group (FSG) from 2000 to 2012, however only patients 50 years or younger were eligible. The objective of the present study was to describe the management and outcome of older patients and compare them to that of younger patients10.
Results
Patients and tumors’ characteristics
Seventy-seven patients were enrolled, including 36 females (46.8%) and their characteristics are described in Table 1. Briefly, the median age at diagnosis was 56 years (range: 50–86) and performance status was ≤1 for 57 patients (74%). The primary tumor was located in soft tissue in 59 pts (76.6%) and in bone in 18 (23%). Sites of extra-osseous tumors included: gluteal muscle, retroperitoneum, rhinopharynx, pleura, cervical muscles. Median tumor size was 6.8 cm and tumor size was >8 cm in 25 patients (40%). Fifty-six patients (73%) had localized disease whereas 21 (27%) presented with metastases at diagnosis and among them, 11(52%) had lung-only metastases. There were no statistical differences between stage at diagnosis for patients <65 and ≥65 years (p = 0.84).
Molecular analysis was available for 53 patients (69%): EWSR1-FLI1 fusion was found in 26 patients, FUS-ERG in one patient and rearrangement of the EWSR1 gene not otherwise specified (NOS) in 22 patients (Table 1).
Treatment modality
Thirty-seven patients (48%) were initially managed outside of a FSG institution. Their survival was not statistically different from those initially treated in FSG group. Six of these patients were managed with surgery alone and one with radiotherapy alone.
Among 56 patients with localized disease, 49 (88%) received chemotherapy in addition to local therapy and among these, chemotherapy was considered intensive in 34 patients (61%) (Table 2). Most of the patients who received intensive chemotherapy were younger than 65 years (p = 0.025). The median number of cycles given was six (range 0–17). Median cumulative administrated dose was 341 mg/m2 for doxorubicin and 49.000 mg/m2 for ifosfamide. Timing of chemotherapy (neoadjuvant versus adjuvant) was not associated with significant survival difference. Local therapy was surgery in 20 patients, surgery and radiotherapy in 28 patients and radiotherapy alone in five patients. Median radiation dose was 50.3 Gy (range 12–66). Twenty-nine patients (55%) with localized disease were treated with surgery first.
All patients with metastatic disease (n = 21) at presentation received chemotherapy, except three who had bulky primary tumors. One patient had surgery, while another had local radiotherapy but both died rapidly after local treatment. A third patient died of myocardial infarction three weeks after diagnosis, without receiving any specific treatment. Among patients receiving chemotherapy for metastatic disease, chemotherapy was considered intensive for 11 patients. Eight patients (38%) with metastatic disease had surgery of their primary tumors.
Survival
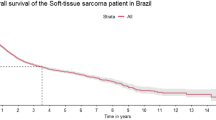
Median follow-up was 71 months and median overall survival (OS) was 92.8 months for the whole cohort, 128 months for patients with localized disease and 23 months for patients with metastatic disease. There was no significant survival difference between centers. For patients with localized disease, the estimated 3-year OS and event free survival (EFS) rates were respectively 73.3% (95% CI: 59.1; 83.3) and 62.2% (95% CI: 47.7; 73.8). There was no difference in OS or EFS between patients receiving intensive chemotherapy vs standard dose or no perioperative chemotherapy (Fig. 1). For patients with metastatic disease at presentation, the estimated 3-yr OS rate was 37% (95% CI: 15.7; 58.7).

Event free survival of patients with localized disease according to chemotherapy regimen (intensive/non intensive/no). Kaplan Meier plots for event free survival, calculated from date of diagnosis to first event (progression, death) or date of last patient contact or date of death (of any cause). Log Rank: 0.8168 Chemotherapy regimens which contained high doses of anthracycline and high dose alkylating agents were considered intensive.
In univariate analysis only initially high LDH level were marginally associated with worse overall survival in patients with localized disease. Other prognosis factor established in pediatric population such as age, tumor size, tumor location were tested but were not significantly associated with OS (Table 3).
Management of recurrent disease
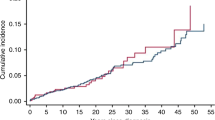
Forty-three patients (55%) experienced recurrence, survival data was available for 39 patients with a median time to recurrence of 18.2 months (range: 2–134). Recurrences were most often metastatic only (58.1%), followed by local only (23.2%). The site of recurrence was lung for 16 patients (37.2%) and bone for nine patients (20.9%). Median post-recurrence survival was 11.2 months (Fig. 2). Most patients (n = 32/43) received second line chemotherapy for recurrent disease with various chemotherapy regimens including cisplatine + etoposide; doxorubicin + cyclophosphamide; temozolomide + irinotecan and vinorelbine + cyclophosphamide.

Post-recurrence survival. Kaplan Meier plots for post-recurrence survival in patients (localized + metastatic) who first achieved remission (n = 39), calculated from time of recurrence to death.
Discussion
There are few published studies on adult patients with EFTs and there results are conflicting. Some studies reported similar outcomes for adult and pediatric patients with EFTs. However, median age in these studies was relatively low, for instance: 21.5 years in Fizazi study13 and 27.1 years in Ahmed study14. Three studies reported similar outcomes for patients over 40 years old with EFTs10,11,12. Other studies reported shorter survival for adult patients15,16,17,18,19. Recent retrospective analyses of large cohorts of patients with EFTs have shown older age to be significantly associated with poorer prognosis20,21,22,23. A recent analysis from the SEER database has shown an inferior survival for EFTs patients older than 40 years9. One of the main questions to address is whether this difference in survival is due to differences in management or intrinsic differences in tumor and/or host biology. Recently published data in patients with synovial sarcoma suggests that there are biological differences between tumors in younger vs older patients24 but similar data are not available for EFTs.
We tried to address this issued in the present study, and we analyzed treatment modalities and outcome of older patients with EFTs. Several similarities in disease presentation with younger patients were noted such as a slight male predominance25, a predominance of non-bulky tumors (<8 cm) and localized disease at presentation (≈75%). The most common sites of metastasis and recurrence were the lung and the bone, which is consistent with EFTs in younger patients.
As previously reported, the majority of patients (77%) had soft tissue primary tumors, which is very different from EFTs in younger patients who present in the majority of cases with bone primaries3,10,11,12. Older series emphasized the need to treat extraosseous EFTs with EFTs protocol26, while recent studies suggest that extraosseous origin may be a favorable prognosis factor, at least in the pediatric population27,28. Our data suggest that the outcome of older patients with EFTs may be worse than in younger patients. Indeed, the 3-year OS rate in our study was 73.3% in patients with localized disease while the 3-year OS rate was 85.7% for patients with localized disease treated with VIDE as induction chemotherapy, and VAC or VAI as consolidation treatment in the EURO-EWING99 trial. Similarly, the Children Oncology Group reported a 5-year OS rate of 83% for patients with localized EFTs treated with compressed VDC-IE protocol and 77% for patients with standard interval7. These differences should, however, be interpreted with caution as our analysis is retrospective and included unselected patients as opposed to selected clinical trials patients described in the EURO-EWING99 and COG AEWS0031 studies. Nevertheless, we observed high recurrence rate (55%) in our cohort compared to pediatric series (recurrence rate 30%29), associated with a short post-relapse survival (median 9.5 months). The median time to recurrence of 18 months was however comparable to those reported in series of younger patients29,30.
For patients with metastatic disease at diagnosis, prognosis appears similar to that of the pediatric population with an estimated 3-year OS rate of 37%. However, outcomes for patients with metastatic tumors are heterogeneous, depending on metastatic sites (lung-only versus other)31,32, which therefore limits comparisons.
We observed significant differences with regards to treatment modalities: many patients in this study did not receive preoperative chemotherapy. In most cases, these patients were initially managed in non-expert centers. Atypical presentation (age, soft tissue involvement) may have led to misdiagnosis in first intention and consequently affect therapeutic strategy.
The worse prognosis of adult patients in previous studies has been attributed to less intensive treatment9,17. However, we did not observe any survival differences between patients receiving intensive chemotherapy versus non-intensive in our study. Interpretation of these results is however, limited by the small size of our study. Because many studies conducted in EFTs have overall shown an improvement in outcome with more intensive chemotherapy regimens33,34,35,36, we can only hypothesize that the inferior outcome may be related to treatment toxicity and difficulty to complete optimal treatment. We were not able to collect toxicity data in our retrospective study. The feasibility of intensive induction chemotherapy in older patients, with higher comorbidity remains uncertain. Subgroup analysis of the EE99 trial revealed no increase in toxicity in adult patients (19–50 years) who were not shown to have more dose modifications or delays than younger patients37. Other studies reported significantly higher toxicity and lower mean dose intensity of treatment for older patients compared to pediatric patients10,17.
Differences in clinical presentation and outcomes may support the hypothesis of biologic/genomic differences of EFTs occurring in adult than in children, as recently shown in synovial sarcoma38. EFTs are characterized by pathognomonic EWSR1 gene translocation with a member of the ETS transcription factor family and genomic studies have shown that the number of additional mutations or genomic alterations increased with age39,40, which suggests increasing genomic instability with age. The clinical relevance of these additional mutations is uncertain.
Clinical presentation and outcomes of patients diagnosed with EFTs at age over 50 years are different from pediatric population. The differences in clinical presentation underline possible differences in tumor biology. However, differences in outcome may also result of the differences in management, thus the optimal treatment strategy for older adults diagnosed with Ewing sarcoma remains to be defined. Prospective trials are needed to clarify the optimal treatment strategy for patients with EFTs diagnosed after 50 years.
Methods
Patients
The study protocol was approved by the relevant regulatory and ethics committee (CCTIRS, CNIL) as well as by the French Sarcoma Group (FSG). In addition, all methods were performed in accordance with the relevant guidelines and regulations. Patients were identified at each participating site using the following criteria: histologically confirmed diagnosis of Ewing sarcoma (by an expert pathologist of the FSG) at age 50 years or older at diagnosis and treated between 2000 and 2012. Seventy-seven patients treated at eight institutions were identified. Data on clinical features, treatment modality and outcomes were extracted from individual patient files. Due to the heterogeneity of chemotherapy regimens, those were classified into two groups: intensive regimens which contained high doses of anthracycline (≥60 mg/m2 per cycle) and high dose alkylating agents (MAI, VIDE, CADO, IVA-IVAD) versus non-intensive which comprised single agent chemotherapy, regimens without anthracycline, as well as “Memphis-like” regimens.
Statistical analysis
Overall survival (OS) was calculated from the time of diagnosis to date of death or date of last patient contact. Event free survival (EFS) was calculated from date of diagnosis to first event (progression, death) or date of last patient contact or date of death (of any cause). OS- and EFS-rates were estimated according to time using the Kaplan–Meier method. Median follow-up was calculated using a reverse Kaplan–Meier estimate. Only univariate analysis was performed due to the small number of patients in each group. Univariate (Cox proportional hazards regression model) analyses were used to examine the predictive value of significant factors. Associations between different clinico-pathological parameters were estimated by the chi-square test. Statistical analyses were carried out using the SAS software (version 9.4). A p value ≤ 0.05 was considered significant.
References
Lawlor, E. R. & Sorensen, P. H. Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit Rev Oncog 20, 155–171 (2015).
Balamuth, N. J. & Womer, R. B. Ewing’s sarcoma. Lancet Oncol. 11, 184–192 (2010).
Esiashvili, N., Goodman, M. & Marcus, R. B. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J. Pediatr. Hematol. Oncol. 30, 425–430 (2008).
Gaspar, N. et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 33, 3036–3046 (2015).
Mascarenhas, L. et al. Pilot Study of Adding Vincristine, Topotecan, and Cyclophosphamide to Interval-Compressed Chemotherapy in Newly Diagnosed Patients With Localized Ewing Sarcoma: A Report From the Children’s Oncology Group. Pediatr Blood Cancer 63, 493–498 (2016).
Le Deley, M.-C. et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J. Clin. Oncol. 32, 2440–2448 (2014).
Womer, R. B. et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. J. Clin. Oncol. 30, 4148–4154 (2012).
Grier, H. E. et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 348, 694–701 (2003).
Karski, E. E., Matthay, K. K., Neuhaus, J. M., Goldsby, R. E. & Dubois, S. G. Characteristics and outcomes of patients with Ewing sarcoma over 40 years of age at diagnosis. Cancer Epidemiol 37, 29–33 (2013).
Bacci, G. et al. Adjuvant and neoadjuvant chemotherapy for Ewing sarcoma family tumors in patients aged between 40 and 60: report of 35 cases and comparison of results with 586 younger patients treated with the same protocols in the same years. Cancer 109, 780–786 (2007).
Kaidar-Person, O., Haim, N. & Bar-Sela, G. Treatment of adult patients with Ewing’s sarcoma: compliance with chemotherapy protocols & toxicity. Med. Oncol. 28(Suppl 1), S685–689 (2011).
Pieper, S. et al. Ewing’s tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-E.W.I.N.G. 99. Onkologie 31, 657–663 (2008).
Fizazi, K. et al. Ewing’s family of tumors in adults: multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J. Clin. Oncol. 16, 3736–3743 (1998).
Ahmed, S. K., Robinson, S. I., Okuno, S. H., Rose, P. S. & Laack, N. N. I. Adult ewing sarcoma: survival and local control outcomes in 102 patients with localized disease. Sarcoma 2013, 681425 (2013).
Siegel, R. D., Ryan, L. M. & Antman, K. H. Adults with Ewing’s sarcoma. An analysis of 16 patients at the Dana-Farber Cancer Institute. Am. J. Clin. Oncol. 11, 614–617 (1988).
Baldini, E. H. et al. Adults with Ewing’s sarcoma/primitive neuroectodermal tumor: adverse effect of older age and primary extraosseous disease on outcome. Ann. Surg. 230, 79–86 (1999).
Bar-Sela, G., Peer, A., Rothschild, S. & Haim, N. Treatment of patients aged over 50 years with non-osseous Ewing’s sarcoma family tumors: five cases and review of literature. Tumori 94, 809–812 (2008).
Gupta, A. A. et al. Clinical outcome of children and adults with localized Ewing sarcoma: impact of chemotherapy dose and timing of local therapy. Cancer 116, 3189–3194 (2010).
Diaz-Beveridge, R. et al. Multimodality Treatment of Pediatric and Adult Patients With Ewing Sarcoma: A Single-institution Experience. J. Pediatr. Hematol. Oncol. 37, e278–284 (2015).
Cotterill, S. J. et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 18, 3108–3114 (2000).
Duchman, K. R., Gao, Y. & Miller, B. J. Prognostic factors for survival in patients with Ewing’s sarcoma using the surveillance, epidemiology, and end results (SEER) program database. Cancer Epidemiol 39, 189–195 (2015).
Karski, E. E. et al. Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr Blood Cancer 63, 47–53 (2016).
Lee, J., Hoang, B. H., Ziogas, A. & Zell, J. A. Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer 116, 1964–1973 (2010).
Lagarde, P. et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J. Clin. Oncol. 31, 608–615 (2013).
Ludwig, J. A. Ewing sarcoma: historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr Opin Oncol 20, 412–418 (2008).
Applebaum, M. A. et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 117, 3027–3032 (2011).
Castex, M.-P. et al. Extraosseous localized ewing tumors: improved outcome with anthracyclines–the French society of pediatric oncology and international society of pediatric oncology. J. Clin. Oncol. 25, 1176–1182 (2007).
Cash, T. et al. Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children’s Oncology Group. Pediatr Blood Cancer. https://doi.org/10.1002/pbc.26096 (2016).
Barker, L. M., Pendergrass, T. W., Sanders, J. E. & Hawkins, D. S. Survival after recurrence of Ewing’s sarcoma family of tumors. J. Clin. Oncol. 23, 4354–4362 (2005).
Ferrari, S. et al. Post-relapse survival in patients with Ewing sarcoma. Pediatr Blood Cancer 62, 994–999 (2015).
Ladenstein, R. et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J. Clin. Oncol. 28, 3284–3291 (2010).
Dirksen, U., Le Deley, M. C., Brennan, B. & Judson, I. R. Efficacy of busulfan-melphalan high dose chemotherapy consolidation (BuMel) compared to conventional chemotherapy combined with lung irradiation in ewing sarcoma (ES) with primary lung metastases: Results of EURO-EWING 99-R2pulm randomized trial (EE99R2pul). J. Clin. Oncol. 34, (suppl, abstr 11000).
Paulussen, M. et al. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment–cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J. Clin. Oncol. 26, 4385–4393 (2008).
Loschi, S. et al. Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant. 50, 1083–1088 (2015).
Jahnukainen, K., Kallio, P., Koivusalo, A. & Saarinen-Pihkala, U. M. High-dose Thiotepa as Consolidation Therapy With Autologous Hematopoietic Stem Cell Transplantation for High-risk Ewing Family Tumors: Single-institution Experience. J. Pediatr. Hematol. Oncol. 37, 536–542 (2015).
Whelan, J., Le Deley, M. C., Dirksen, U., Judson, I. R. & Hawkins, D. S. Efficacy of busulfan-melphalan high dose chemotherapy consolidation (BuMel) in localized high-risk Ewing sarcoma (ES): Results of EURO-EWING 99-R2 randomized trial (EE99R2Loc). J. Clin. Oncol. 34, (suppl, abstr 11001).
Juergens, C. et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 47, 22–29 (2006).
Vlenterie, M. et al. Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br. J. Cancer 113, 1602–1606 (2015).
Sand, L. G. L., Szuhai, K. & Hogendoorn, P. C. W. Sequencing Overview of Ewing Sarcoma: A Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int J Mol Sci 16, 16176–16215 (2015).
Tirode, F. et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4, 1342–1353 (2014).
Author information
Authors and Affiliations
Contributions
Conceptualization: P.R., P.C. Methodology: P.R., P.C. Validation: P.R., P.C. Formal analysis: P.R., P.C., A.L.C. Investigation: P.R., P.C., A.I., V.L., N.P., O.M., C.C., F.B., E.B., L.C., D.L., T.R., S.D., P.M., D.R., J.Y.B. Resources: P.R., P.C., A.I., V.L., N.P., O.M., C.C., F.B., E.B., L.C., D.L., T.R., S.D., P.M., D.R., J.Y.B. Writing – review and editing: P.R., P.C., A.I., V.L., N.P., A.L.C., O.M., C.C., F.B., E.B., L.C., D.L., T.R., S.D., P.M., D.R., J.Y.B.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rochefort, P., Italiano, A., Laurence, V. et al. A Retrospective Multicentric Study of Ewing Sarcoma Family of Tumors in Patients Older Than 50: Management and Outcome. Sci Rep 7, 17917 (2017). https://doi.org/10.1038/s41598-017-17733-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17733-z
This article is cited by
-
Outcome of multidisciplinary treatment of peripheral primitive neuroectodermal tumor
Scientific Reports (2020)
-
Ewing sarcoma
Nature Reviews Disease Primers (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
