
Abstract
Familial hypercholesterolemia (FH) is an autosomal dominant disorder associated with premature cardiovascular disease (CVD). Mutations in the LDLR, APOB, and PCSK9 genes are known to cause FH. In this study, we analysed the genetic spectrum of the disease in subjects from the Iranian population with a clinical diagnosis of FH. Samples were collected from 16 children and family members from five different cities of Iran. Probands were screened for mutations in the LDLR, APOB, and PCSK9 genes using next generation sequencing, with results confirmed by Sanger sequencing. The likely pathology of identified variants was examined using in silico tools. Of the probands, 14 had a clinical diagnosis of homozygous FH and two of heterozygous FH. No mutations were found in either APOB or PCSK9, but nine probands were homozygous for seven different LDLR mutations, with p.(Trp577Arg) occurring in three and p.Val806Glyfs*11 occurring in two patients. Two mutations were novel: p.(Leu479Gln) and p.(Glu668*). Seven probands with a clinical diagnosis of FH were mutation negative. This pilot study, integrating clinical and molecular-based techniques, begins to elucidate the FH heterogeneity and the mutation spectrum in the Iranian population. Such information is important for future disease management and cost savings.
Similar content being viewed by others


Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant disorder resulting in elevated plasma low-density lipoprotein cholesterol (LDL-C). Heterozygous FH (HeFH) is common, with an estimated prevalence of at least 1/5001 in most European populations, but being higher in some populations such as the Danes2, the Afrikaners in South Africa3, French Canadians4 and the Dutch5, with recent estimates in the UK suggesting around 1/2706,7. Plasma LDL-C in HeFH is two-to-three-fold higher than normal. Individuals with this form of FH are more likely to develop premature coronary heart disease (CHD) in the second or third decade of their lives1. The second form is homozygous FH (HoFH), which has an estimated prevalence of 1 in 1 million in most populations, however, some populations have reported higher prevalence such as 1:300,000 in the Netherlands5. Individuals with HoFH have six-to-eight-fold higher levels of LDL-C plasma than normal and develop CHD in the early stages of their lives, often dying before the age of 201. FH is caused by a mutation in one of three genes: the low-density lipoprotein cholesterol receptor (LDLR), Apolipoprotein B gene (APOB), or a gain-of-function mutation in the gene for proprotein convertase subtilisin/kexin type-9 (PCSK9). Autosomal recessive hypercholesterolemia (ARH), a rare inheritance of FH, has been reported8 and occurs when individual inherits two pathogenic variants in the low-density lipoprotein adaptor protein 1 (LDLAP1) gene. In the UK, the vast majority (93%) of identified mutations are found in the LDLR gene, a further 5% in APOB, and about 2% in the PCSK9 gene9,10.
FH can be diagnosed using well-established clinical criteria. The Simon Broome Register Criteria (UK)11 and The Dutch Lipid Clinic Network (DLCNC)12 (Europe) use total cholesterol (TC) and LDL-C levels, presence of tendon xanthoma (TX), a family history of hypercholesterolemia and premature CHD in a first and/or second degree relative. The most common cholesterol-lowering drugs are statins, which are used as the primary intervention method to prevent premature CHD. However, statin efficacy ranges from a 30% to 60% reduction in LDL-C, which depends on statin potency, type and dose, and mutation type13. Identifying and treating FH patients in childhood or early adulthood is important to prevent or reduce morbidity and mortality from premature CHD14.
Although the FH molecular basis and prevalence have been demonstrated in many populations, the FH prevalence and spectrum are unknown in Iran. In 2015, clinical practice guidelines for the diagnosis and treatment of HoFH in the Middle East region was published, which showed several factors such as consanguineous marriages, treatment accessibility and cascade screening limitations that were not found in Western countries15. Previous Iranian FH Genetic studies have used limited genetic screening methods based on PCR technology16,17,18,19,20. Fard-Esfahani et al. in 2005, who examined 30 patients with clinical possible FH, identified one novel mutation in exon 4 of the LDLR p.(Gly445Cy) using the PCR-single-strand conformation polymorphism (PCR-SSCP) method18, while another study used PCR-Restriction Fragment Length Polymorphism (PCR-RFLP) and PCR-SSCP methods, where no mutations were reported either in the promoter or coding region of LDLR or in exon 26 of APOB 16,17. A recent study examined 80 patients with a clinical diagnosis of FH, and after testing for the two common APOB mutations and screened the LDLR gene only for exons 3, 4, 9 and 10 only, identified two mutations in exons 3 and 420. We believe that our study, using Next Generation Sequencing for the whole of LDLR/APOB/PCSK9, is the first to carry out a thorough genetic screen in Iranian FH patients.”
HoFH may have a higher prevalence in Iran because consanguineous marriages are more common in the Persian culture (35–54%)21. Historically, a patient was clinically diagnosed with HoFH, if he/she had untreated LDL-C > 13 mmol/L (500 mg/dL) or treated LDL-C > 8 mmol/L (300 mg/dL), and the presence of TXs before the age of 10 years, or the patient’s parents were diagnosed with HeFH22. However, the range of LDL-C levels reported in HoFH patients is broad and can overlap with ranges found in other types of FH15,22, which is another challenge for identification. Two cases of Iranian children with HoFH have reported severe septum and complex CHD outcomes23,24. Therefore, it has become essential to identify FH patients in Iran, and find diagnostic strategies and appropriate treatment in order to reduce morbidity and mortality due to CHD. This study provides a diagnostic guideline of FH in Iran including clinical criteria, cascade screening by using the next generation sequencing (NGS)-based method followed by Sanger sequencing, in addition to mutation pathogenesis analyses.
Results
Patient characteristics and sample selection
Clinical and lipid parameter details of the 57 recruited participants are shown in Supplementary Table S2. We selected 16 probands from unrelated families. Fourteen probands had been diagnosed with clinical HoFH, had LDL-C > 13 mmol/L or treated LDL-C > 8 mmol/L, TX, and a positive family history of hyperlipidaemia or MI. Two probands (FH9-P1 and FH10-P2) were suspected of having FH, and as shown in Supplementary Table 2, although their treated cholesterol levels did not exceed the FH cut-off (LDL-C = 3.3 and 4.9 mmol/l, respectively), but they reported a strong family history of hyperlipidaemia and/or MI. The baseline characteristics of the16 selected cases are shown in Table 1.
Mutation Spectrum
Out of the 16 patients examined, a FH-causing variant was found in nine patients (56%), while no mutation was detected in seven individuals (44%), five clinically diagnosed with FH and two suspected of FH. All identified mutations were found in LDLR. As shown in Table 2, although TC and LDL-C levels were higher in the mutation positive group compared to the mutation negative group, in this small sample these differences were not statistically significant (TC p = 0.10 and LDL-C p = 0.09).
In eight patients with a clinical diagnosis of HoFH, we identified six different LDLR mutations (Table 3, Fig. 1). The DNA mutations were reported in five different exons of LDLR (4, 10, 11, 12, and17). Five of these identified mutations have been reported in the UCL-FH mutation database. Three nonsense mutations were identified, c.389 C > G, p. (Ser130*) in exon 4, and c.1599 G > A, p.(Trp533*) in exon 11 and p.(Cys668*). Also, we found missense mutation, c.1729T > C p.(Trp577Arg) in exon 12. Three patients carried the LDLR mutation p.(Trp577Arg), and one carried p.(Cys667Trp). The fifth mutation was a frame-shift mutation in exon 17, c.2146dupG p.(Val806Glyfs*11), where an insertion of G changed the amino acid at position 806 from Valine to Glycine and led to a shift in the reading frame and the creation of a stop codon downstream.

Diagram of LDLR gene showing the mutations identified in this study. Seven mutations were identified in this study: two novel mutations are indicated in red. Exons are shown as vertical boxes and introns as the lines connecting them.
Three patients (FH 4-P, FH 12-P, and FH 16-P) were clinically diagnosed with HoFH, however, no mutation was found in any of the FH genes. Also, no mutation was found in the subjects with suspected FH.
Novel LDLR variants
The functionality of the missense mutations was assessed by in-silico mutation prediction tools, including PolyPhen2, SIFT, and Mutation Taster, and were predicted to be functional. As the nonsense mutations and insertion frame-shift mutation led to peptide truncation, they were also predicted to be functional. We found two novel mutations in LDLR in two HoFH probands, which are not reported in the UCL-FH mutation database, one in exon 10 c.1436 T > A, p.(Leu479Gln) and another in exon 14 c.2001_2002delinsGT p.(Glu668*). The mutation heritability of the mutations was confirmed by investigating the proband’s family members. The homozygous p.(Leu479Gln) variant was found in a 10year-old boy who had high levels of TC (16.5 mmol/l) and LDL-C (15 mmol/l). Co-segregation analyses showed that this mutation is an FH-causing mutation and was present in all members of the family including both parents and a sibling (Fig. 2). They were all heterozygous for this mutation; both parents had raised cholesterol levels [Father (FH14-F): TC = 9.8 mmol/l, LDL-C = 6.6 mmol/l; Mother (FH14-M): TC = 8.5 mmol/l, LDL-C = 6.8 mmol/l], but not the sibling.

Co-segregation analysis of family with LDLR novel mutation p(.Leu479Gln). (A) NGS sequencing reads alignment using Integrated Genomics Viewer (http://software.broadinstitute.org/software/igv/) for proband FH14-P reports a variant at exon 10 of the LDLR gene [c.1436 T > A, p(.Leu479Gln)], the calling rate A allele of the variant is 95% which indicates the proband is a homozygote for the variant. (B) Co-segregation analysis of the FH14 family shows the 3 other family members are heterozygotes of the variant. (C) Sanger sequencing shows a base change between homozygote and heterozygote inherited modes.
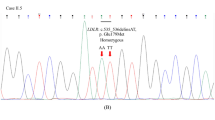
The second novel mutation, p.(Glu668*),was found in a 4 year-old girl who had high levels of TC (15.0 mmol/l) and LDL-C (13.6 mmol/l), but no family history of either hyperlipidaemia or MI. It was also found that the proband was homozygous for another mutation that changes amino acid 667, p.(Cys667Trp) (Fig. 3), but due to the presence of the sequence change creating the nonsense mutation at position 668, we designate the pathogenic mutation in this subject as p.(Glu668*). Unfortunately, family members of the proband were not available for segregation analysis.

LDLR novel mutation p.(Glu668*) sequencing data. (A) NGS sequencing reads alignment using Integrated Genomics Viewer (http://software.broadinstitute.org/software/igv/) for proband FH17-P reports two base change c.2001_2002delinsGT at exon 14 of the LDLR gene which affecting two amino acid [p.(Cys667Trp) and p.(Glu668*)], the calling rate of minor alleles of the variants are ≥94% which indicate the proband is a homozygote of the variants. (B) Sanger sequencing shows a base change between wild type and mutant homozygote inherited mode.
Discussion
This study has identified a number of previously reported as well as novel mutations among patients with a clinical diagnosis of HoFH and HeFH attending various Iranian clinics. Amongst the 14 child probands clinically diagnosed with HoFH, mutations were found in nine patients (57%). No mutations were found in two additional child patients included because of having a strong family history of hyperlipidaemia and/or MI. Different mutations were identified using the targeted NGS method and were confirmed by Sanger sequencing, all in LDLR, where five had been previously reported as FH-causing mutations, but were not previously reported in the Iranian population20, and two, p.(Leu479Gln) and p.(Glu668*) that were first reported in this study.
This study has found two mutations occurring in more than one of the patients examined. The mutation p.(Trp577Arg), which was seen in three patients with HoFH. This was previously reported as very common in the Turkish population, explaining 21.4% of the predicted defective LDLR alleles25,26. For populations that are geographically close, genetic and geographic distances are often highly correlated27. Since there is close geographical and cultural links between Turkey and Iran it it very likely that they will share some common founder mutations. The three probands were from Esfarayen (FH1-P, FH2-P) and Guchan (FH7-P) which are in the north-east of Iran on the border with Turkmenistan, and 74% of the Turkmenistan population are Turkmen (Turkish background).
The second mutation found in more than one patient in the study was a frame-shift mutation p.(Val806Glyfs*11) in exon 17, which was found in two patients with HoFH. This mutation has been reported in different populations in the USA28, South Sweden29, Czech30, Netherlands31 and Japan32. There are two possible explanations for the presence of the same mutation in different populations. First, there may be an ancestral mutation occurring in an individual living somewhere in the European continent, which has then spread throughout the world due to human migration. The FDB mutation p.(R3527Q), for example, is an ancestral mutation found in western Europe 6000–7000 years ago, and distributed to the East and West via human migration in the past 2000–3000 years33. This possibility could be tested by determining if the mutation occurs on the same SNP haplotype, but data is not available to carry this out.
The second possible explanation is that the mutation may be located in a CpG (5′—C—phosphate—G—3′) mutation hotspot34. Cytosine in the CpG dinucleotide carries a methyl group which is added via methyltransferase to form 5-methylcytosine, which causes DNA methylation and affects gene transcription via preventing transcription factors and other proteins from binding35. However, by looking for the sequence around the point mutation and the CpG island map of LDLR gene, the insertion of guanine (G) nucleotide (c.2416_2417insG) does not create a CpG sequence (Figure S1). As shown in the LDLR CpG island map, exon 17 was not included in any CpG island (Figure S2). The literature reported 12 CpGs in the LDLR promoter in atherosclerosis patients36. However, it can be seen by inspection of the gene sequence at this point that the mutation is due to the duplication of a G in a homonucleotide tract of 5 Gs, which is frequently a mutational cause. At the present time it is not possible to distinguish between these two possibilities.
Four of the identified mutations in this study have been reported in the UCL-FH mutation database, and are predicted to be pathogenic. Two identified mutations are nonsense mutations: p.(Ser130*) (exon 4) is located in repeat three of the ligand binding domain of the receptor and p.(Trp533*) (exon 11) encodes the EGF-like domain. Both mutations create a premature stop codon resulting in peptide truncation and non-functional protein products. Also one missense mutation p.(Trp577Arg) was identified in the region of the gene that encodes the EGF-like domain. Variants in this domain block receptor dissociation in the endosome and this influences the machinery responsible for recycling receptors to the cell surface, which causes the disease phenotype. The exon 12 mutation p.(Trp577Arg) is located in the third Y-repeat (five repeats containing a YWTD motif) of the EGF-like domain of the LDL-R, where other missense mutations have been functionally characterized to be class 2 mutations (transport-defective alleles)1,37. Thus, we propose that the p.(Trp577Arg) mutation reported here would cause the same class of deficiency due to its location. The fourth mutation was identified in Exon 17 p.(Val806Glyfs*11) which encodes membrane-anchoring and cytoplasmic tail domains which are vital for locating the receptor correctly in the cell membrane, thus the truncated peptide at this position affects this machinery and leads to the disease phenotype.
Two novel mutations have been identified in LDLR that were not reported in the UCL-FH mutation database: a missense mutation in exon 10 p.(Leu479Gln) and a truncated peptide in exon 14 p.(Glu668*). Both mutations are encoded in the EGF-like domain of LDLR, which is known to be important for receptor dissociation in endocytosis and receptor recycling to the cell surface, and are thus pathogenic. Analysis of the proband family members carrying p.(Leu479Gln) showed that the mutation is heritable and causing FH.
Forty-three percent of subjects examined in this study were FH-mutation negative. It has been demonstrated that the elevated LDL-C levels in mutation negative subjects with a clinical diagnosis of FH are due to accumulation of LDL-C raising variants “polygenic FH”38. There are 12 common “polygenic” LDL-C raising variants described in the European population (MAF > 0.05%), however, the frequency of these variants in the Iranian population has not yet been studied, However, based on data from other countries, it is likely that the elevated levels of LDL-C seen in at least some of the patients who are FH- mutation negative have a polygenic and not a monogenic explanation, and this may be a possible explanation for the high cholesterol phenotype in the two non HoFH additional child probands FH9-P1 and FH10-P2. Studies have also showed mutations in ABCG5/8 may cause the phenotype of very elevated cholesterol levels39, and these genes could be examined in the mutation-negative patients in the future. The other possible explanation for the lack of a mutation in the three known FH genes could be that the hypercholesterolaemia is due to a mutation in an undiscovered gene(s). Recently, mutations in Signal-transducing adaptor protein 1 (STAP1) gene have been reported in Dutch families who had the FH phenotype but were mutation-negative for the three FH genes40, and this gene could also be examined in the future.
Two probands (7 and 12 years) suspected of having FH were included in this study, although their cholesterol levels did not exceed the FH cut-off, but they reported a strong family history of hyperlipidemia and MI. No mutations were found in any of the three tested FH genes in these patients. In both patients the presence of TX, a family history of CHD and hyperlipidaemia led to testing with a further lipoprotein screen: high levels of Lipoprotein(a) (Lp(a)) were found in both probands (Lp(a) > 100 mg/dL but with LDL-C levels within the normal range (~3.3 mmol/l) when undergoing lipid-lowering drug therapy). An elevated plasma concentration of Lp(a) has been reported as an independent risk factor for CVD41,42,43. The clinical phenotype of Lp(a) hyperlipidemia (Lp(a)-HLP) shows great variability, as does FH, however the diagnosis of Lp(a)-HLP can be determined by the plasma concentration of Lp(a)44.
Limitation
There were some limitations in this study. One was the small sample size, which means we cannot extrapolate with any certainty to the overall prevalence of the detected mutations in FH patients in Iran. Also, in many countries where this has been examined, 5–10% of FH patients carry a gross insertion or deletion of the LDLR gene and in order to obtain a full molecular characterization of the mutation spectrum in the Iranian population screening for this type of defect should have been carried out. However at the current time an appropriate method to do this is not up and running in our laboratory, and unfortunately we do not have sufficient DNA to carry this out. The NGS method should have allowed us to look for any such gross deletions and insertions but again the quality of the DNA was inadequate to allow us to perform this with accuracy. Also it was not possible to perform segregation analysis for all patients with novel variants due to communication difficulties with their relatives living in different parts of the country. Another limitation was that polygenic FH was not examined in the mutation negative group due to lack of reports about polymorphism frequency in the Iranian population, and the gene encoding Apo(a) was not screened because of grant and time limitations.
Conclusion
Although more than 1,700 variants have been reported in LDLR 45, there are still novel variants being found, proving the heterogeneity of FH. In this work we showed how we selected 16 FH patients as probands out of 57 based on DLCN clinical criteria, and then 14 probands were diagnosed as HOFH based on cholesterol levels. To confirm the clinical diagnoses genetic tests were performed, which indeed confirm the clinical diagnosis in 57% of the subjects. Among the seven mutations identified in this study, two variants were novel (previously unreported). Also we show that for a novel variant it is important to run family genetic analysis to confirm co-segregation of the variant and high cholesterol levels, and thus the pathogenicity of the mutation. We think our work gives a guideline for physicians for how to integrate clinical observations and genetic tests for patient benefit. It is recommended that FH-mutation negative subjects with a clinical diagnosis of FH are examined for 12 common “polygenic” LDL-C raising variants. Also, a genetic screening for the Apo (a) gene in patients with Lp(a)-HLP is recommended. Further and larger studies are needed to more fully elucidate the frequency of FH causing mutations in Iran and to identify common and unique mutations in subjects of Persian ancestry.
Methods
Subjects
Fifty-seven samples were collected from 16 child probands and family members from different paediatric and endocrinology clinics in five different cities (Ahvaz, Bandar Abbas, Esfarayen, Mashhad and Torbat Heydariye), which represents three regions of Iran. Clinical diagnosis was made according to the Dutch Lipid Clinic Network Criteria (DLCNC) as previously described12. The probands were diagnosed with clinical definite FH (DFH) if they had a total cholesterol >6.7 mmol/l or LDL-C > 4.0 mmol/l if <16 years and, TX or TX in a first or second degree relative. They were diagnosed with clinical probable FH (PFH) if they had a high cholesterol as DFH and a family history of myocardial infarction (MI) in a first degree relative <60 years or a family history of total cholesterol l >7.5 mmol/l in a first or second degree relative. Also, any child who was suspected of having FH but who did not meet the criteria still underwent analysis for LDLR, APOB and PCSK9 mutations because if the child was from a family with PFH, there is a high probability that he/she has FH. For this reason, samples were collected from the proband and any first degree relatives.
Blood sampling and DNA isolation
This study was performed on 16 children and their family members from five different cities of Iran. All families were diagnosed for FH according to the DLCNC criteria. Information on consanguinity was obtained by questionnaire. The study procedures were approved by the Ethics committee (approval code #IR.MUMS.fm.REC1394142) of the Mashhad University of Medical Sciences, Mashhad, Iran. All patients’ parents and/or their guardians gave their written informed consent to participate in this study.
Blood samples (3 mL) were obtained from all patients and their available parents were collected in EDTA tubes. Genomic DNA was extracted from peripheral blood samples using the standard salting-out method46 with the High Pure PCR Template Preparation Kit (version 20).
Next generation sequencing (NGS)
A targeted-NGS method was used. Primers to amplify coding regions (±25 base pairs (bp)) of the three autosomal dominant FH genes (LDLR, APOB, PCSK9), and the autosomal recessive FH gene (LDLRAP1) for targeted sequencing were designed using the Illumina Design Studio47. Amplicon length was set at 250 bp. The library preparation was performed using the TruSeq Custom Amplicon (TSCA) v1.5 kit (Illumina, San Diego, CA) and sequencing (in both directions) was done using the Illumina MiSeq platform. All experiments were performed in accordance with relevant guidelines and regulations.
Variant detection and analysis
The raw data acquired were aligned to human reference genome (Hg19). The criteria for the standard variant calling pipeline were: coverage ≥30×, a minimum of five reads for an altered allele, a Phred quality ≥20, and a strand bias filter. A sensitive pipeline was used to ensure that variants were not missed (a coverage ≥15×, a minimum of two reads for an altered allele, a Phred quality ≥ zero, and no strand bias filter). Variant annotation was carried out with Variant Effect Predictor (VEP) tool from Ensembl48 and based on the transcripts LDLR: ENTS00000558518, APOB: ENTS00000233242, PCSK9: ENTS00000302118, and LDLRAP1: ENTS00000374338. Reported variants were filtered based on their frequency and functional affects. Variants with minor allele frequency >1% according to the 1000 Genomes genotype data49 were considered non-pathogenic and excluded from further analysis. The remaining variants were flagged as rare (frequency = 0.005) or novel (frequency = 0). Also, variants were flagged as functional when they were most likely to affect a protein’s function, that is: non-synonymous, stop gain, stop loss, frame-shift deletions and insertions, and splice site variants. In-silico mutation prediction tools were also used to predict the possible impact of an amino acid substitutions on the structure and function of a human protein, including Sorting Intolerant Form Tolerant (SIFT)50, Polyphen-2 V251, and MutationTaster52. Also, the variants were described as causing FH if they were reported on the UCL-FH mutation database (http://www.ucl.ac.uk/ldlr/Current/)45,53, which is supported by evidence in the literature. While variants were described as possible FH-causing if they were unreported in the database but they were predicted to be pathogenic in-silico and highly conserved (the UCSC genome browser used to analyze the conservation level of the variant position). The variants that passed filtering pipelines were directly Sanger sequenced for validation, as described below.
Sanger sequencing
Variants were confirmed by Sanger sequencing. Oligonucleotide primers for PCR were designed to cover, where appropriate, intron–exon junctions and up to 60 bp of the introns54. Primers were designed to amplify the promoter and LDLR exons; a part of exon 26 of APOB that includes the p.(R3527Q) mutation and p.(D374Y) (exon 7) of the PCSK9 gene were included for screening (Table S1). PCR was carried out in the Rotor-Gene6000 (Qiagen Ltd, Crawley, West Sussex, UK). The PCR reaction mix comprised 1.5 ng of DNA, 0.05 µl from each primer, and 5 µl AccuMelt HRM SuperMix (2x) (Quanta Biosciences) to a total volume 10 µl. The PCR conditions were as follows; started at 95 °C for 5 minutes, followed by 40–45 cycles of denaturing at 95 °C for 5 seconds, annealing at 60 °C for 10 seconds, and extending at 70 °C for 20 seconds. The amplified fragment was sequenced using Sanger sequencing. The DNA sequence was assessed manually.
References
Hobbs, H. H., Brown, M. S. & Goldstein, J. L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Human Mutation 1, 445–466 (1992).
Benn, M., Watts, G. F., Tybjaerg-Hansen, A. & Nordestgaard, B. G. Familial Hypercholesterolemia in the Danish General Population: Prevalence, Coronary Artery Disease, and Cholesterol-Lowering Medication. The Journal of Clinical Endocrinology & Metabolism 97, 3956–3964 (2012).
Leitersdorf, E., V der Westhuyzen, D., Coetzee, G. & Hobbs, H. Two common low density lipoprotein receptor gene mutations cause familial hypercholesterolemia in Afrikaners. Journal of Clinical Investigation 84, 954 (1989).
Moorjani, S. et al. Homozygous familial hypercholesterolemia among French Canadians in Québec province. Arteriosclerosis, Thrombosis, and Vascular Biology 9, 211–216 (1989).
Sjouke, B. et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype–phenotype relationship, and clinical outcome. European heart journal 36, 560 (2015).
Walter, K. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Wald, D. S. et al. Child–Parent Familial Hypercholesterolemia Screening in Primary Care. New England Journal of Medicine 375, 1628–1637 (2016).
Zuliani, G. et al. Characterization of a New Form of Inherited Hypercholesterolemia: Familial Recessive Hypercholesterolemia. Arteriosclerosis, Thrombosis, and Vascular Biology 19, 802–809 (1999).
Humphries, S. et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. Journal of medical genetics 43, 943–949 (2006).
Soutar, A. K. & Naoumova, R. P. Mechanisms of Disease: genetic causes of familial hypercholesterolemia. Nature Clinical Practice Cardiovascular Medicine 4, 214–225 (2007).
Marks, D., Thorogood, M., Neil, H. A. W. & Humphries, S. E. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 168, 1–14 (2003).
Liyanage, K. E., Burnett, J. R., Hooper, A. J. & van Bockxmeer, F. M. Familial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distribution. Critical Reviews in Clinical Laboratory Sciences 48, 1–18 (2011).
Robinson, J. G. Management of Familial Hypercholesterolemia: A Review of the Recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Manag Care Pharm 19, 139–49 (2013).
Wiegman, A. et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. European Heart Journal 36, 2425–2437 (2015).
Al-Ashwal, A. et al. Identification and treatment of patients with homozygous familial hypercholesterolaemia: information and recommendations from a Middle East advisory panel. Current vascular pharmacology 13, 759–770 (2015).
Farrokhi, E. et al. Molecular Characterization of Iranian Patients with Possible Familial Hypercholesterolemia. Indian Journal of Clinical Biochemistry 26, 244–248 (2011).
Farrokhi, E., Shirani, M., Modarresi, M., Roghani, F. & Hashemzadeh, M. The study of mutations of the 9 exons of LDLR gene in patients with familial hypercholesterolemia in Cheharmahal Bakhtiari province. Arak Medical University Journal 13, 30–37 (2011).
Fard-Esfahani, P., Zeinali, C., Rouhi Dehboneh, S., Taghikhani, M. & Khatami, S. A Novel Mutation in exon 4 of the low density lipoprotein (LDL) receptor gene in an Iranian Familial Hypercholesterolemia patient. Iranian Biomedical Journal 9, 139–142 (2005).
Fard-Esfahani, P., Mohammadi-Torbati, P., Khatami, S., Zeinali, S. & Allahyari, M. T. M. Familial Defective Apoliporrotein B 100: Frequency of R3500q Mutation of Apoliporotein B Gene in Iranian Hypercholesterolemic Patients. Acta Medica Iranica 43, 193–196 (2005).
Ekrami, M. et al. Genetic Analysis of Iranian Patients With Familial Hypercholesterolemia. Iran Biomed Journal (2017).
Saadat, M., Ansari-Lari, M. & Farhud, D. Short report consanguineous marriage in Iran. Annals of human biology 31, 263–269 (2004).
Cuchel, M. et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. European heart journal 35, 2146–2157 (2014).
Nemati, M. H. Coronary revascularization in a child with homozygous familial hypercholesterolemia. Interactive cardiovascular and thoracic surgery 10, 131–132 (2010).
Nemati, M. H., Astaneh, B. & Joubeh, A. Triple coronary artery bypass graft in a 10-year-old child with familial hypercholesterolemia. General thoracic and cardiovascular surgery 57, 94–97 (2009).
Sözen, M. M. et al. The molecular basis of familial hypercholesterolaemia in Turkish patients. Atherosclerosis 180, 63–71 (2005).
Schmidt, H. H. J. et al. Liver transplantation in a subject with familial hypercholesterolemia carrying the homozygous p.W577R LDL-receptor gene mutation. Clinical Transplantation 22, 180–184 (2008).
Ramachandran, S. et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proceedings of the National Academy of Sciences of the United States of America 102, 15942–15947 (2005).
Nobe, Y. et al. Familial Hypercholesterolemia in Utah Kindred with Novel 2412-6 Ins G Mutations in Exon 17 of the LDL Receptor Gene. Japanese Heart Journal 40, 435–441 (1999).
Ekström, A., Wallmark, F. & Nilsson, E. Mutations in the low-density lipoprotein receptor gene in Swedish familial hypercholesterolaemia patients: clinical expression and treatment response. European Journal of Clinical Investigation 28, 740–747 (1998).
Kuhrová, V. et al. Spectrum of low density lipoprotein receptor mutations in Czech hypercholesterolemic patients. Human Mutation 19, 80–80 (2002).
Fouchier, S. W., Defesche, J. C., Umans-Eckenhausen, M. A. & Kastelein, J. J. The molecular basis of familial hypercholesterolemia in The Netherlands. Human Genetics 109, 602–615 (2001).
Yu, W. et al. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis 165, 335–342 (2002).
Myant, N. B., Forbes, S. A., Day, I. N. M. & Gallagher, J. Estimation of the Age of the Ancestral Arginine3500 → Glutamine Mutation in Human ApoB-100. Genomics 45, 78–87 (1997).
Ollila, J., Lappalainen, I. & Vihinen, M. Sequence specificity in CpG mutation hotspots. FEBS Letters 396, 119–122 (1996).
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics 13, 484–492 (2012).
Zhi, Y., Huang, Y., Li, Z., Zhang, R. & Wang, S. Hypermethylation in promoter area of LDLR gene in atherosclerosis patients. Journal of molecular cell biology 40, 419–427 (2007).
Hattori, H. et al. Eight novel mutations and functional impairments of the LDL receptor in familial hypercholesterolemia in the north of Japan. Journal of Human Genetics 47, 80–87 (2002).
Talmud, P. J. et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case–control study. Lancet 381 (2013).
Rios, J., Stein, E., Shendure, J., Hobbs, H. H. & Cohen, J. C. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Human Molecular Genetics 19, 4313–4318 (2010).
Fouchier, S. W. et al. Mutations in stap1 are associated with autosomal dominant hypercholesterolemia. Circulation research 115, 552–555 (2014).
Lamon-Fava, S., Diffenderfer, M. R. & Marcovina, S. M. Lipoprotein(a) metabolism. Current Opinion in Lipidology 25, 189–193 (2014).
Durrington, P. N., Schofield, J. D., Siahmansur, T. & Soran, H. Lipoprotein (a): gene genie. Current Opinion in Lipidology 25, 289–296 (2014).
Bos, S., Yayha, R. & van Lennep, J. E. R. Latest developments in the treatment of lipoprotein (a). Current Opinion in Lipidology 25, 452–460 (2014).
Orsó, E., Ahrens, N., ć, D. & Schmitz, G. Familial hypercholesterolemia and lipoprotein(a) hyperlipidemia as independent and combined cardiovascular risk factors. Atherosclerosis Supplements 10, 74–78 (2009).
Leigh, S. et al. The UCL low-density lipoprotein receptor gene variant database: pathogenicity update. Journal of Medical Genetics (2016).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research 16, 1215 (1988).
Futema, M., Plagnol, V., Whittall, R. A., Neil, H. A. W. & Humphries, S. E. Use of targeted exome sequencing as a diagnostic tool for Familial Hypercholesterolaemia. Journal of medical genetics 49, 644–649 (2012).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biology 17, 122 (2016).
Abecasis, G. R. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protocols 4, 1073–1081 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nature methods 7, 248–249 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods 11, 361–362 (2014).
Usifo, E. et al. Low-Density Lipoprotein Receptor Gene Familial Hypercholesterolemia Variant Database: Update and Pathological Assessment. Annals of Human Genetics 76, 387–401 (2012).
Whittall, R. A. et al. Development of a high-resolution melting method for mutation detection in familial hypercholesterolaemia patients. Annals of Clinical Biochemistry 47, 44–55 (2010).
Damgaard, D. et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis 180, 155–160 (2005).
Hartog, C., Fryer, A. & Upadhyaya, M. Mutation analysis of iduronate-2-sulphatase gene in 24 patients with Hunter syndrome: Characterisation of 6 novel mutations. Human Mutation 14, 87–87 (1999).
Nissen, H. et al. Mutation screening of the LDLR gene and ApoB gene in patients with a phenotype of familial hypercholesterolemia and normal values in a functional LDL receptor/apolipoprotein B assay. Clinical Genetics 54, 79–82 (1998).
Acknowledgements
RHF is funded by King Abdullah Medical City (KAMC) in Makkah, and the Ministry of Health, Saudi Arabia (KAMC6). SEH is a British Heart Foundation (BHF) Professor is funded by a BHF grant (BHF PG08/008) and by the NIHR UCLH BRC. MF is supported by (NIHR) University College London Hospitals Biomedical Research Centre (BRC68). RV is supported by the Research Council at the Mashhad University of Medical Sciences, Mashhad, Iran (grant code: 931094).
Author information
Authors and Affiliations
Contributions
R.H.F. wrote the manuscript. R.H.F., M.F. and S.E.H. interpreted the data. S.E.H. and A.S. jointly supervised the work. The other co-authors (R.V., M.R.A., S.H., M.A., H.Z., and M.M.) recruited and performed clinical diagnosis of patients, and all authors commented on the draft of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fairoozy, R.H., Futema, M., Vakili, R. et al. The Genetic Spectrum of Familial Hypercholesterolemia (FH) in the Iranian Population. Sci Rep 7, 17087 (2017). https://doi.org/10.1038/s41598-017-17181-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17181-9
This article is cited by
-
Molecular modeling of LDLR aids interpretation of genomic variants
Journal of Molecular Medicine (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
