
Abstract
Neoceratitis asiatica (Becker) (Diptera: Tephritidae) is one of the most important fruit pestsof wolfberry which is a traditional Chinese medicinal herb. We characterized the complete mitochondrial genome of N. asiatica and described its organization in this study. This mitogenome had a total length of 15,481 bp, consisting of 13 protein-coding genes, 2 rRNA genes, 22 tRNA genes and a non-coding region (A + T-rich control region). The overall base composition of N. asiatica in descending order was 40.6% A, 8.5% G, 38.4% T and 12.6% C. The phylogenetic relationships shows that Ceratitis capitata and N. asiatica may be sister taxa. This is the first report of the complete mitochondrial genome of a member of the Neoceratitis Genus and the complete mitochondrial genome sequence may provide useful information for phylogenetic analysis and studies between the genera Ceratitis and Neoceratitis.
Similar content being viewed by others


Introduction
The genus Neoceratitis Hendel is a predominantly afrotropical group with one species in Asia1, which partly distribute in Northwest China (Ningxia, Qinghai, Xinjiang and Inner Mongolia), Kazakhstan and Turkmenistan2. Neoceratitis asiatica (Becker) (Diptera:Tephritidae) is one of the most economically important fruit pests damaged the fruit of the Lycium turcomanicum Turcy (Solanaceae)2. The majority host plant, wolfberry, is a traditional Chinese medicinal herb and local cash crop3. The female adults only lay one egg in an unripe fruit, which exacerbates the destructive power of N. asiatica. The larvae feed on the wolfberry and develop with the ripening of wolfberry fruit. Once be damaged, the damaged maggot fruits cannot be used as a commodity, so maggot fruits rate can represent the loss rate. Wolfberries damage rate will reach 22–55% if not controlled by using pesticide4. In view of the seriousness of the damage to wolfberry, the research on N. asiatica (Becker) should be increasingly extensive and in-depth. However, the research on the genus Neoceratitis Hendel is very limited.
Mitochondrial genomes of insects have been very extensively studied. They have been applied particularly to studies regarding phylogeny and evolution5,6,7. To date there are fifty-seven complete mitogenomes of 23 Tephritidae species in GenBank (Supplementary Table S1).
Currently, studies on the mitochondrial genome of the genus Neoceratitis are mainly limited on the species N. cyanescens by fragments of four mitochondrial genes and one nuclear gene (COI, 16S, tRNA pro, ND6, period)8,9,10, while another important species N. asiatica (for this study) have not been published yet. Based on the research of N. cyanescens, we found that the genus Ceratitis has a close relationship to the genus Neoceratitis 8,9,10, but the phylogenetic status of the two genera cannot be explained very well.
In this study, we reported the first complete mitogenome of Neoceratitis species-N. asiatica and compared the mitogenome data with other tephritid species, aiming to providing more data to study the molecular phylogeny of Ceratitidinain particular.
Results
Mitochondrial genome sequencing and assembly
An Illumina library of N. asiatica was sequenced on a run of Hiseq 2500. After excluding the low quality value reads (lower than Q20), 466,428 read-pairs were generated finally. Through “map to reference” strategy to map all cleaned NGS reads to part of cox1 gene by Geneious R10.0., 58,875 reads were assembled to get the target sequence. After generating all assembled reads, a consensus sequence length 16,074 bp was generated. Then we manually examined for repeats at the beginning and end of the sequence to form a circle to gain the complete mitochondrial genome sequence of N. asiatica which was 15,481 bp.
Mitogenome features
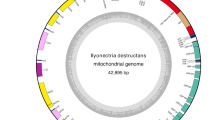
The complete mitogenome of N. asiatica was 15,481 bp in length. The gene content was typical of other ancestral insect mitochondrial genomes (Fig. 1 and Table 1): 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes and a non-coding region (A + T-rich control region). Nine PCGs (ND2, COI, COII, COIII, ATP6, ATP8, ND3, ND6 and CYTB), 14 tRNAs (tRNA Ile, tRNA Met, tRNA Trp, tRNA Leu(UUR), tRNA Lys, tRNA Asp, tRNA Gly, tRNA Ala, tRNA Arg, tRNA Asn, tRNA Ser(AGN), tRNA Glu, tRNA Thr and tRNA Ser(UCN)) and the control region were located on the major strand (J-strand). Four PCGs (ND5, ND4, ND4L and ND1), eight tRNAs (tRNA Gln, tRNA Cys, tRNA Tyr, tRNA Phe, tRNA His, tRNA Pro, tRNA Leu(CUN) and tRNA Val) and two rRNAs (lrRNA and srRNA) were located on the minor strand (N-strand).

Mitochondrial genome map of Neoceratitis asiatica.
Spacing sequences in 19 regions ranged from 2 to 54 bp, the longest located between tRNA Cys and tRNA Tyr. The overlapping sequences ranged from 1 to 32 bp in 10 regions, the longest was between tRNA Leu(CUN) and lrRNA.
Contrary to other insect mitogenomes11, the nucleotide composition of N. asiatica was negative AT skews in the control region, while the rest was all AT biased and positive AT skews and negative GC skews in the whole mitochondrial genome, PCGs, rRNAs, tRNAs and the control region (Table 2). The A + T content of the non-coding control region was 88.2%.
The commonest start codon was ATG (in 6 PCGs –COII, ATP6, COIII, ND4, ND4L, CYTB), followed by four for ATT (ND2, ATP8, ND5 and ND6), followed by two for ATA (ND1 and ND3) and one for TCG (COI). Ten PCGs (ND1, COI, COII, ATP8, ATP6, COIII, ND3, ND4, ND4L and ND6) had TAA stop codon, one PCG (ND3) had TAT, one PCG (CYTB) had TAG, while ND1 had incomplete stop codons T.
The size of 22 tRNAs ranged from 64 bp (tRNA Arg and tRNA Thr) to 72 bp (tRNA Pro). Most tRNAs could be folded into the cloverleaf structure except for tRNA Ser(AGN), which lacked the D-loop(Fig. 2). The number of base pairs in the DHU-stem ranged from 3 to 4 (Fig. 2). Most of the TΨ C-stems had 5 base pairs while 7 tRNAs (tRNA Ile, tRNA Lys, tRNA Arg, tRNA Ser(AGN), tRNA Thr, tRNA Cys, tR NA His) had 4 bp in the TΨ C-stems. The number of bases in the D-loop and TΨ C-loop was variable.

Putative secondary structures of tRNAs found in the mitochondrial genome of Neoceratitis asiatica.
The two genes encoding the small and the large ribosomal subunits were located between tRNA Leu(CUN) and tRNA Val, and between tRNA Val and the control region. The lrRNA was 1,359 bp long with an A + T content of 82.6%, and the srRNA was 790 bp long with an A + T content of 79.5%.
The control region (397 bp) was flanked by srRNA and tRNA Ile and was highly enriched in AT (88.2%).
Phylogenetic relationships
Six datasets were used to build phylogenetic trees: 1) PCG123: 13 protein-coding genes (all three codon positions included) with 11,048 nucleotides; 2) PCG123 + rRNA: 13 protein-coding genes and 2 rRNA genes with 12,834 nucleotides. 3) PCG123 + rRNA + tRNA: 13 protein-coding genes, 2 rRNA genes and 22 tRNA genes with 14,186 nucleotides. 4) PCG12: 13 protein-coding genes (first two codon positions included) with 7,342 nucleotides; 5) PCG12 + rRNA: 13 protein-coding genes and 2 rRNA genes with 9,117 nucleotides. 6) PCG12 + rRNA + tRNA: 13 protein-coding genes, 2 rRNA genes and 22 tRNA genes with 10,473 nucleotides.
Based on the datasets, the topology structures conducted from Bayesian and ML analyses were very similar (Fig. 3). From our results, the genera Ceratitis and Neoceratitis are sister groups in the trees with high posterior probabilities (1.0) and ML bootstraps (100).

Phylogenetic tree of Tephritidae family based on mitochondrial genomes.
Discussion
In this study, we are reporting the first complete mitochondrial genome of Neoceratitis species –N. asiatica (Becker) in Tephritidae. The mitochondrial genome of N. asiatica is a closed circular molecule of 15,481 bp, which is the shortest one among the other 22 tephritid mitogenomes available with the size ranging from 15,687 bp in B. tau to 16,253 bp in D. longicornis. The control region of N. asiatica mitogenome is 397 bp in length, which is also the shortest one in the other published tephritid mitogenomes with the size ranging from 801 bp in B. tau to 1,343 bp in D. longicornis (Supplementary Table S2).
The A + T contents of the whole mitogenome, PCGs, tRNAs, rRNAs and CR in N. asiatica are 79.0%, 77.8%, 77.9%, 81.5% and 88.2%, well in the range of amongst all reported tephritid mitogenomes, which range from 67.28% (B. minax) to 80.83% (P. utilis) in the whole mitogenome, from 64.30% (B. minax) to 78.90% (P. utilis) in PCGs, from 72.31% (B. minax) to 80.61% (P. utilis) in tRNAs, from 73.71% (B. minax) to 85.69% (P. utilis) in rRNAs and from 77.65% (B. minax) to 91.14% (C. capitata) in CR (Supplementary Table S2).
The AT skews and GC skews of N. asiatica in the whole mitogenome, PCGs, tRNAs, rRNAs and CR are0.028 (from 0.021 in C. capitata to 0.131 in B. minax) and −0.194 (from −0.175 in P. utilis to −0.316 in B. minax), 0.026 (from 0.019 in C. capitata to 0.148 in B. minax) and −0.180 (from −0.170 in P. utilis to −0.319 in B. minax), 0.024 (from 0.005 in P. utilis to 0.055 in B. minax) and −0.131 (from −0.074 in B. cucurbitae to −0.182 in B. minax), 0.048 (minimum) and −0.297 (from −0.263 in C. capitata to −0.356in B. minax), −0.045(minimum) and −0.576 (from −0.354 in D. longicornis to 0.04 in B. cucurbitae), respectively. The rRNAs and CR of N.asiatica shows the most marked AT skews compared with the other tephritid mitogenomes, which are significant parallels with the feature in C. capitata and C. fasciventris. The CR of N. asiatica, C. capitata and C. fasciventris all show negative AT skews, while that of the other tephritid mitogenomes show positive AT skews (Supplementary Table S2).
Seven PCGs in all Tephritidae species have the same start codons (ATG in ATP6, COII, CYTB, ND4 and ND4L, ATT in ND2, TCG in COI), and five PCGs (ATP6, ATP8, COIII, ND4L and ND6) have the same stop TAA codons (Table 3). In ND5, the TAT stop codon of N. asiatica is different from all the other Tephritidae species with TAA or T stop codon.
Phylogenetic relationship of Tephritid fruit flies based on molecular data has been reported by several researchers and there exist some arguments for a long period.
The relationship between subgenus Zegodacus and other subgenus of Bactrocera is questionable. White suggested that subgenera Zeugodacus should split from Bactrocera to combine with Dacus genus to form a new genus—Zeugodacus from morphological evidence8. Latter, a lot of studies support the view from molecular level. Segura et al. reported the phylogenetic relationships among 23 tephritid species using the utilizing sequence of CYTB, tRNA Ser and ND1 genes. The result indicated Bactrocera cucurbitae is close to genus Dacus rather than other subgenus of Bactrocera 9. Krosch et al. rebuilt the phylogenetic tree of 125 species based on 16S rRNA, COI, COII and white eye genes to figure out the Tribe Dacini relationship and similarly the tree showed that Zeugodacus is the sister group to Dacus not Bactrocera. They suggested Zeugodacus should raise up to genus level10. Virgilio et al. also came to the result through the phylogenetic tree using two datasets. Dataset 1 was an alignment of 2,338 bp consisted of COI, 16S rRNA, tRNA pro, ND6 and period included 98 vouchers and dataset 2 was an alignment of 1,200 bp consisted of COI and 16 S rRNA included 159 vouchers11. In this study, we confirmed that subgenera Zeugodacus are closer to genus Dacus but distinct from other subgenera (Bactrocera, Daculus and Tetradacus) of Bactrocera genus from mitochondrial genome data level.
Han and Ro reconstructed the phylogeny of the family Tephritidae by mitochondrial 12 S, 16 S, and COII gene fragments using 79 tephritid species. Phylogenetic trees suggested that Dacini and Ceratitidini are sister group which both of them belong to Dacinae and have distance to Anastrepha which belong to Toxotrypanini12. While Krosch et al. found Anastrepha ludens which belongs to Trypetinae subfamily was closer to Dacini (Dacinae subfamily) than to C. capitata based on 16S rRNA, COI, COII and white eye genes10. Fernández et al. constructed the phylogenetic tree using the neighbour-joining method based on COII gene representing six genera (Ceratitis, Rhagoletis, Dacus, Bactrocera, Anastrepha and Toxotrypan) of the family. The result also showed that Anastrepha and Bactrocera cluster in one branch while Ceratitis formed another branch individually13. Nakahara and Murajiuse used a 1.3 kb portion of mitochondrial DNA containing the tRNA Leu and flanking COI and COII regions for phylogenetic analyses. The result also shows that Dacini seems more closely related to Anastrepha than to the Ceratitidini14. Our research also drew the same conclusion that Anastrepha fraterculus is closer to Dacini rather than to C. capitata using the published mitochondrial genome data (5 of 6 datasets posterior probabilities are 1.00 and ML bootstraps are 100 for Bayesian and ML analyses separately) which implicates that we should reconsider the phylogenetic relationships between Dacinae and Trypetinae according to the molecular evidence.
There is also an argument about the phylogenetic status of the genus Neoceratitis, most of which are sequenced by four mitochondrial and one nuclear gene fragment (COI, 16 S, tRNA Pro, ND6, period). Barr and McPheron investigated phylogenetic relationships within Ceratitidina and showed that Neoceratitis might be sister taxa to Ceratitis along with Carpophthoromyia and Capparimyia 15. Based on the gene fragments (COI, 16S, tRNA Pro, ND6, period), the study of Virgilio et al. strongly supported that the genera Ceratitis and Neoceratitis were sister taxa using Bayesian approach and maximum likelihood (ML) (Bayesian PP = 1.00, ML bootstrap support = 91)11. So far, various studies, all of which expounding with the sample Neoceratitis cyanescens, have shown the close relationship between the two genera, Ceratitis and Neoceratitis 9. Based on the previous studies mentioned above, the phylogenetic position between the genera Ceratitis and Neoceratitis was not well resolved. Thus we expected that the complete mitochondrial genome sequence of N. asiatica could make some contributions towards the phylogeny reconstruction of subtribe Ceratitidina.
In this study, the Bayesian and ML reconstructions place the two genera Ceratitis (C. capitata) and Neoceratitis (N. asiatica) together, which means they may be sister taxa. Limited to the data of complete mitochondrial genome in different Tephritidae species, exploring the relationship between the two genera Ceratitis and Neoceratitis still needs more researches.
Materials and Methods
Sample collection and DNA extraction
The N. asiatica samples were collected in Ningxia province, China and preserved in 100% ethanol. They were identified based on morphological characteristics. Genomic DNA was extracted from individual N. asiatica adult using the DNeasy DNA Extraction kit (QIAGEN).
Mitogenome sequencing and annotation
Genomic DNA library preparation and sequencing were carried out by Berry Genomics sequencing company (Beijing, China). Genomic DNA was fragmented with Bioruptor to an average insert size of 250 bp and sequenced on Illumina Hiseq 2500. Part of cox1 gene was sequenced as the “anchor” to reconstruct the mitochondrial genome of N. asiatica using a general insect primer pairLCO1490/HCO219816. We picked up the mitochondrial genome sequence with “map to reference” strategy and mapped all cleaned NGS reads to the “anchor” by Geneious R10.017. The parameters we set for assembly were: 1) minimum overlap identity 95%, 2) minimum overlap 50 bp, 3) maximum 5% gaps per read, and 4) maximum gap size 20 bp.
Thirteen protein-coding genes and two rRNA genes were identified by BLAST searches in NCBI (http://www.ncbi.nlm.nih.gov/) and then confirmed by alignment with homologous genes from other 22 Tephritid species available in GenBank. The tRNA genes were identified using the tRNAscan-SE18 and MITOS WebServer19. The circular map of N. asiatica complete mitochondrial genome was generated and annotated using Geneious. The start/stop codon usages were analysed by DNAMAN 8.0. The composition of skew was calculated manually based on the formula: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)20. The sequin file was edited and submitted to NCBI (NCBI GenBank accession number MF434829).
Phylogenetic analysis
A total of 25species of Diptera species were used in phylogenetic analysis, including 23Tephritidae and 2 outgroups species from Drosophilidae. Six datasets were used to build phylogenetic trees: 1) PCG123: 13 protein-coding genes (all three codon positions included); 2) PCG123 + rRNA: 13 protein-coding genes and 2 rRNA genes; 3) PCG123 + rRNA + tRNA: 13 protein-coding genes, 2 rRNA genes and 22 tRNA genes; 4) PCG12: 13 protein-coding genes (first two codon positions included) with; 5) PCG12 + rRNA: 13 protein-coding genes and 2 rRNA genes; 6) PCG12 + rRNA + tRNA: 13 protein-coding genes, 2 rRNA genes and 22 tRNA genes.
MrBayes v.3.2.521 and a PHYML22 online web server were used to analyze the six datasets under GTR + I + G model. The model was selected using Jmodeltest 2.1.723. In Bayesian analysis, two simultaneous runs of 1,000,000 generations were conducted for the matrix. Each one was sampled every 200 generations with a burn-in of 25%. Trees inferred prior to stationarity were discarded as burn-in, and the remaining were used to construct a 50% majority rule consensus tree. The ML analysis was conducted with 1,000 bootstraps. Phylogenetic trees were viewed and edited by FigTree v.1.4.324. Sequences were aligned using ClustalW with the default parameters implemented in MEGA 5.025. The ambiguous positions in the genes alignment were filtered with Gblocks v0.91b26. The aligned sequences of each gene were concatenated using SequenceMatrix v1.727.
References
Meyer, M. D. & Freidberg, A. Taxonomic revision of the fruit fly genus Neoceratitis Hendel (Diptera: Tephritidae). Zootaxa. 3223, 24–39 (2012).
Liang, G. Q. Fruit Flies (Diptera: Tephritidae: Dacinae: Trypetinae: Tephritinae). China Agriculture Press, Beijing, 10–11 (in Chinese) (2011).
Zhao, Z. H. et al. Risk assessment and control strategies of pests in Lyciumbarbarum fields under different managements. Chinese. Journal of Applied Ecology (in Chinese) 20(4), 843–850 (2009).
Wu, F. Z., Huang, R. X., Meng, Q. X. & Liang, Z. Q. Studies on the fruit fly Neoceratitis asiatica (Becker) (Diptera: Tephritidae). Journal of Plant Protection (in Chinese) (4) (1963).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Li, H. et al. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5, 8527 (2015).
Song, F. et al. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian Site-Heterogeneous Mixture Models. Genome Biol. Evol. 8(5), 1411–1426, https://doi.org/10.1093/gbe/evw086 (2016).
White, I. M. Taxonomy of the Dacina (Diptera: Tephritidae) of Africa and the Middle East. Afr. Entomol. Memoir. 2, 1–156 (2006).
Segura, M. D., Callejas, C., Fernández, M. P. & Ochando, M. D. New contributions towards the understanding of the phylogenetic relationships among economically important fruit flies (Diptera: Tephritidae). B. Entomol. Res. 96, 279–288 (2006).
Krosch, M. N. et al. A molecular phylogeny for the Tribe Dacini (Diptera: Tephritidae): Systematic and biogeographic implications. Mol. Phylogenet. Evol. 64, 513–523 (2012).
Virgilio, M., Jordaens, K., Verwimp, C., White, I. M. & De Meyer, M. Higher phylogeny of frugivorous flies (Diptera, Tephritidae,Dacini): Localised partition conflicts and a novel generic classification. Mol. Phylogenet. Evol. 85, 171–179 (2015).
Han, H. Y. & Ro, K. E. Molecular phylogeny of the family Tephritidae (Insecta: Diptera): New insight from combined analysis of the mitochondrial 12S, 16S, and COII genes. Mol. Cell. 27, 55–66 (2009).
Fernández, P. et al. A phylogenetic study of the family Tephritidae (Insecta: Diptera) using a mitochondrial DNA sequence. Proceedings of the 6th International Symposium on fruit flies of economic importance, Stellenbosch, South Africa, 6–10 May 2002. Isteg Scientific Publications. 439–443 (2004).
Nakahara, S. & Muraji, M. Phylogenetic analyses of Bactrocera fruit flies (Diptera: Tephritidae) based on nucleotide sequences of the mitochondrial COI and COII genes. Research Bulletin of Plant Protection Japan. 44, 1–12 (2008).
Barr, N. B. & McPheron, B. A. Molecular phylogenetics of the genus Ceratitis (Diptera: Tephritidae). Mol. Phylogenet. Evol. 38(1), 216–230 (2006).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology. 3, 294–299 (1994).
Kearse, M. et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12), 1647–1649 (2012).
Lowe, T. M. & Eddy, S. R. Trnascan-se: a program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Research. 25(5), 955–64 (1997).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2), 313–319 (2013).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Ronquist, F. & Huelsenbeck, J. P. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19(12), 1572 (2003).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Bio. 59(3), 307–21 (2010).
Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Rambaut, A. FigTree (version 1.4.3). Available online at: http://tree.bio.ed.ac.uk/software/figtree/ (2016).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17(4), 540 (2000).
Vaidya, G., Lohman, D. J. & Meier, R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 27, 171–180 (2011).
Acknowledgements
The work is supported by National Key R&D Program of China (No. 2016YFF0203202) and Science and technology innovation pilot Fund Project of Ningxia Academy of Agriculture and Forestry Sciences (NKYZ-16–0502).
Author information
Authors and Affiliations
Contributions
Y.S., Y.Z., S.-Q.F. and Z.-H.L. designed the study. J.H. and R.Z. collected samples.Y.S., Y.Z., Z.-Z.B. and L.-J.L. performed the molecular work. Y.S., Y.Z., S.-Q.F. and Z.-H.Z. analyzed data. Y.S. and Y.Z. wrote the manuscript with other authors. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, Y., Zhang, Y., Feng, S. et al. The mitochondrial genome of the wolfberry fruit fly, Neoceratitis asiatica (Becker) (Diptera: Tephritidae) and the phylogeny of Neoceratitis Hendel genus. Sci Rep 7, 16612 (2017). https://doi.org/10.1038/s41598-017-16929-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16929-7
This article is cited by
-
The first complete mitochondrial genome sequences for Ulidiidae and phylogenetic analysis of Diptera
Molecular Biology Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
