
Abstract
A synthetic method for diversely substituted tetrahydropyrrolo[1,2-a]quinolines was developed via CuCl-catalyzed cascade transformation of internal aminoalkynes with alkynes under microwave- irradiation.
Similar content being viewed by others
Introduction
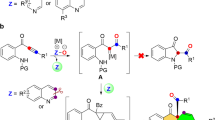
The substituted tetrahydropyrrolo[1,2-a]quinoline scaffold is found in a variety of biologically active compounds. For example, the treatment of cells with compound A (Fig. 1) results in reduced sensitivity to the toxicity of anthrax1. In addition, compound B exhibits potent malarial cysteine protease inhibitory activity2 and compound C was regarded as a selective Aurora B-INCENP interaction inhibitor3.

Selected biologically active molecules containing the tetrahydropyrrolo[1,2-a]quinoline scaffold.
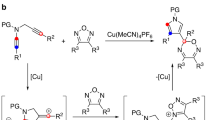
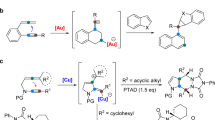
In the past few years, several synthetic methods have been explored on the construction of the tetrahydropyrrolo-[1,2-a]quinoline scaffold4,5,6. In particular, Liu et al. developed a gold catalyzed tandem reaction using terminal aminoalkynes and alkynes as starting material, which can establish the polyheterocycle scaffold simultaneously under mild reaction conditions (Fig. 2a)4. Consequently, Zhou et al. extended this reaction using less active terminal amidoalkynes and alkynes with similar conditions (Fig. 2b)5. In these reactions, only terminal aminoalkynes or amidoalkynes were used as substrates, and expensive gold catalysts were required with long reaction times, these factors restrict the exploration of larger panels of substituted tetrahydropyrrolo[1,2-a]quinoline substrates. Recently, Wasilewska et al. reported the rearrangement of N-(ortho-vinylphenyl) azabicyclo-[3.1.0]hexane derivatives to obtain the target compounds. However, the tedious procedure to prepare starting materials also restricts its widespread application (Fig. 2c)6.

Synthesis of tetrahydropyrrolo[1,2-a] quinolines.
The multiple biological activities of tetrahydropyrrolo-[1,2-a]quinoline attracted our interest and we try to search for a more convenient and efficient method. In 2010, Han et al. reported the Cu(I) catalyzed intramolecular amination of internal aminoalkynes to prepare N-heterocycles under microwave irradiation conditions7, where the copper catalyst was more tolerant towards basic amines than gold catalysts for Snogashira-type reactions. Inspired by this discovery, we envisioned that the tetrahydropyrrolo-[1,2-a] quinolines could be produced via a Cu(I) catalyzed tandem reaction using internal aminoalkynes and alkynes (Fig. 2d).
Results and Discussion
To validate the feasibility of the proposed process, aminoalkyne (1a) and phenylacetylene (2a) were chosen as the starting materials with different catalytic reaction conditions. Firstly, we tried the similar condition as Han’s method, in whose catalytic system CuBr can increase the reaction rate7. Thus, CuBr (10 mol %) was used as catalyst in dioxane under MW irradiation at 150 °C. To our delight, the desired product (3aa) can be produced with a relatively low yield of 16% (Fig. 3, Entry 1). Intriguingly, the replacement of dioxane with DMF and DMSO led to a remarkable increase in yield of desired product (74% and 55%, Entry 2 and 6), probably due to its excellent polarity and good stability under high temperature. Further screening of polar solvents (ethanol, methanol and water, Entry 3,4,7) revealed that protonic solvent ethanol, methanol and acetonitrilewere not suitable for this reaction. Additionally, the use of acetonitrile (Entry 3–5) also only produced trace of target compound (5%, Entry 7), maybe owing to the poor solubility of CuBr in thisese solvent. Subsequently, different copper catalysts were investigated, and the data showed that CuSCN, CuI, Cu[CH3CN]4PF4 and Cu[CH3CN]4BF4 provided moderate to good yields of 3aa (40–87%, Entry 8, 10–12), CuCl displayed the most potent catalytic efficiency with a yield of 90% (Entry 9). Further modification of the ratio of 1a to 2a revealed that 1:3 was optimal (Entry 9, 13–14). To confirm the use of MW irradiation is necessary or not in this reaction8,9,10, we also tried the reaction without microwave irradiation, only affording the desired product with 4% yield (Entry 15). Therefore, the optimal conditions for this copper catalyzed reaction between internal aminoalkynes and alkynes is CuCl (10 mol %), in DMF under microwave irradiation at 150 °C for 15 minutes.

Screening of reaction conditions.
Next, the substrate scope of aminoalkynes 1 was investigated. As shown in Fig. 4, both products with electron-donating groups (3aa, 3ba) and electron-withdrawing groups (3ca, 3da) were obtained in excellent yield (84–91%), indicating the electronic effect was of little influence on this tandem reaction.

The scope of aminoalkynes 1. Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), catalyst (10 mol %), DMF (3.0 mL), at the corresponding temperature under argon. aIsolated yields are shown.
Reaction conditions: 1a (0.1 mmol), 2a (0.3 mmol), catalyst (10 mol %), solvent (3.0 mL), at the corresponding temperature under argon. a)Determined by 1H NMR spectroscopy using CH2Br2 as internal standard. b) 1a (0.1 mmol), 2a (0.2 mmol). c) 1a (0.1 mmol), 2a (0.4 mmol). d)The reaction was carried out without microwave irradiation.
Then, the effect of the substituent position in compound 1 on this tandem cyclization reaction was investigated. When 3-methyl-N-(pent-3-yn-1-yl)-aniline 1f was used as the substrate, cyclization was observed at the 6-position to produce 3fe in 70% yield. Although there were two possible positions where cyclization could occur with the alkyne moiety (2- and 6-position), none of the product with cyclization at the 2-position was detected. These results show that good regio-selectivity was achieved in this tandem cyclization, probably due to the steric hindrance effect. In addition, aminoalkynes with different R2 (ethyl, propyl and butyl) can also perform this reaction smoothly to give 3ga, 3ha and 3ia with yields ranging from 54% to 91%, suggesting that this method can be used for various aminoalkynes.
We have also investigated the substrate scope of the alkynes 2, including the presence of electron-donating groups (EDG) and electron-withdrawing groups (EWG) substituted phenylacetylenes (2a-2d), hexyne (2e), 3-phenyl-1-propyne (2 f), 4-phenyl-1-butyne (2g), N-methyl-N-(prop-2-yn-1-yl)aniline (2 h) and 4-(prop-2-yn-1-yloxy) anisole (2i). The data in Fig. 5 shows that all these alkynes can undergo this tandem cyclization reaction smoothly with moderate to good yields (41–90%).

The scope of alkyne 2. Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), catalyst (10 mol %), DMF (3.0 mL), at the corresponding temperature under argon. aIsolated yields are shown.
With the successful tandem cyclization of internal aminoalkynes with alkynes, we postulated the terminal aminoalkynes also could carry out this reaction with our method. The reaction between 4-methoxy-N-(pent-4-yn-1-yl)-aniline and phenylethylene under optimized condition also gave desired product 3aa in excellent yield (Fig. 6, 86%), which confirmed that both terminal aminoalkynes and internal aminoalkynes are suitable for this tandem reaction. Therefore, our method provides a more efficient and feasible choice for the construction of the tetrahydropyrrolo[1,2-a]quinoline scaffold than those previously reported4.

The reaction of terminal aminoalkyne 1 and alkyne 2a under optimized conditions.
Through examination of the reaction conditions, a possible mechanism of the copper-catalyzed tandem cyclization reaction was proposed in Fig. 7. Similar to that of the gold-catalyzed reaction4,5, the internal aminoalkyne 1 is first activated by the Cu catalyst to generate intermediate A, which is then converted to enamine intermediate B via intramolecular hydroamination. Then, alkyne 2 reacted with anamine B to provide propargylamine C, which was cyclized intramolecularly to give product 3 in the presence of copper catalyst. The mechanism is also supported by reported reference, in where the intermediate similar to compound C was synthesized and cyclized to target compound via copper catalysis11.

Proposed mechanism for copper-catalyzed tandem cyclization.
In order to support the proposed mechanism, we performed LC-HRMS analysis of the reaction mixture of 1b and 2a in DMF after stirring for 10 min at 150 °C under microwave irradiation12,13,14. Three peaks were detected with m/z = 347.2493, 276.1748, 276.1737 at retention times of 6.47, 8.47 and 8.87 min, respectively. The first is the [2 M + H]+ of the starting material 1b, the last peak is confirmed as 3ba by comparison with the obtained product, and the middle peak can be attributed to the intermediate C (Fig. 8). In addition, we performed reaction of 1b and 2a in DMF after stirring for 2 h at 150 °C and isolated the intermediate C. (The structure of C was confirmed by 1H NMR spectra, see SI).

LC-HRMS analysis of the reaction mixture of 1b and 2a to identify key intermediate C.
Moreover, these synthesized tetrahydropyrrolo[1,2-a]quinolines were submitted to biologically evalution in in various phenotypic screening. To be of interest, several of them demonstrated cytotoxic activity against human pancreatic cancer cell CFPAC1 and CAPAN2 in vitro (see the Supporting Information, Table S1). For example, compound 3ha showed an inhibition ratio of 68.2% at 100 µM against pancreatic cancer cell capan-2 proliferation in vitro. Further cell cycle analysis using different concentrations of 3ha showed that the percentage of capan-2 cells in G0/G1 phase (44.63%) treated with compound 3ha (80 µM) was significantly higher than that of control (28.39%) (Fig. 9), indicating the growth of capan-2 cells can be arrested by compound 3ha at G0/G1 phase in various concentrations. (see the Supporting Information, Figure S1).

Cell cycle analysis. Cell cycle distribution upon treatment of 3ha varies between capan-2 cell lines. The cell cycle distribution of capan-2 cells was determined by flow cytometry.
Conclusions
In summary, using a cheap catalyst CuCl and easily available starting materials, we developed a convenient and efficient method for the synthesis of diversely substituted tetrahydropyrrolo[1,2-a]quinolines with excellent regio- and chemoselectivity. Further studies, including asymmetric variation of this tandem reaction and extensive biological evaluations on tetrahydropyrrolo[1,2-a]quinolines are currently underway in our laboratory.
Experimental Section
General procedure for the synthesis of diversely substituted tetrahydropyrrolo[1,2-a]quinolines under microwave irradiation
To a 5 mL Biotage Microwave vial equipped with a magnetic stir bar, CuCl (0.0100 mmol), aminoalkyne 1 (0.100 mmol), alkyne 2 (0.300 mmol) and DMF (3.00 mL) were added. The resulting mixture in sealed vial was stirred at 150 °C under microwave irradiation for 15 min, and water (10.0 mL) was added to the vial to quench the reaction. The mixture was extracted with AcOEt (3 × 10.0 mL) and the combined organic layers was washed with small amounts of water (5 × 5.00 mL) and dried with Na2SO4. The solvent was evaporated in vacuo and the residue was purified by silica gel column chromatography using n-hexane/EA as eluent to give the desired products.
Cell Viability assay
Cells were counted in logarithmic phase and 5,000 cells were placed in 96-well plates. After treatment with compounds (0.4, 20 and 100 μM), cells were incubated for an additional 2 h with CCK-8 reagent (100 μL/mL medium) and the absorbance was read at 450 nm using a microplate reader (Sunnyvale, CA, USA). Cell proliferation inhibition rates were calculated according to the following formula: the proliferation inhibition ratio (%) = 1 − [(A1 − A3)/(A2 − A3)] × 100, where, A1 is the OD value of drug experimental group, A2 is the OD value of blank control group, A3 is the OD value of the RPMI1640 medium without cells. Assays were performed on three independent experiments.
Apoptosis assay by flow cytometry
Exponentially growing cells were seeded in 6-well plates (5 × 104/well) and cultured overnight in a 5% CO2 atmosphere at 37 °C. After treatment with 3ha/3ah/DMSO for 24 h, cells were harvested and washed with PBS. Then cells were stained with Annexin V-FITC Apoptosis Kit according to the manufacturer’s instructions and analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, US). Assays were performed on three independent experiments.
References
Slater, L. H. et al. Identification of Novel Host-Targeted Compounds That Protect From Anthrax Lethal Toxin-Induced Cell Death. ACS Chemical Biology 8, 812–822 (2013).
Shah, F. et al. Identification of Novel Malarial Cysteine Protease Inhibitors Using Structure-Based Virtual Screening of a Focused Cysteine Protease Inhibitor Library. Journal of Chemical Information and Modeling 51, 852–864 (2011).
Unsal, E., Degirmenci, B., Harmanda, B., Erman, B. & Ozlu, N. A small molecule identified through an in silico screen inhibits Aurora B–INCENP interaction. Chemical Biology & Drug Design 88, 783–794 (2016).
Liu, X.-Y. & Che, C.-M. A Highly Efficient and Selective AuI-Catalyzed Tandem Synthesis of Diversely Substituted Pyrrolo[1,2-a]quinolines in Aqueous Media. Angewandte Chemie International Edition 47, 3805–3810 (2008).
Zhou, Y. et al. Gold-Catalyzed One-Pot Cascade Construction of Highly Functionalized Pyrrolo[1,2-a]quinolin-1(2H)-ones. The Journal of Organic Chemistry 74, 7344–7348 (2009).
Wasilewska, A. et al. Synthesis of Polycyclic Aminocyclobutane Systems by the Rearrangement of N-(ortho-Vinylphenyl) 2-Azabicyclo[3.1.0]hexane Derivatives. Chemistry – A European Journal 19, 11759–11767 (2013).
Han, J., Xu, B. & Hammond, G. B. Highly Efficient Cu(I)-Catalyzed Synthesis of N-Heterocycles through a Cyclization-Triggered Addition of Alkynes. Journal of the American Chemical Society 132, 916–917 (2010).
Larhed, M. & Hallberg, A. Microwave-assisted high-speed chemistry: a new technique in drug discovery. Drug Discovery Today 6, 406–416 (2001).
Lew, A., Krutzik, P. O., Hart, M. E. & Chamberlin, A. R. Increasing rates of reaction: microwave-assisted organic synthesis for combinatorial chemistry. Journal of Combinatorial Chemistry 4, 95–105 (2002).
Lidström, P., Tierney, J., Wathey, B. & Westman, J. Microwave assisted organic synthesis—a review. Tetrahedron 57, 9225–9283 (2001).
Hamann, L. G. et al. Synthesis and Biological Activity of a Novel Series of Nonsteroidal, Peripherally Selective Androgen Receptor Antagonists Derived from 1,2-Dihydropyridono[5,6-g]quinolines. Journal of Medicinal Chemistry 41, 623–639 (1998).
Guo, H., Qian, R., Liao, Y., Ma, S. & Guo, Y. ESI-MS Studies on the Mechanism of Pd(0)-Catalyzed Three-Component Tandem Double Addition-Cyclization Reaction. J. Am. Chem. Soc. 127, 13060–13064 (2005).
Meyer, S., Koch, R. & Metzger, J. O. Investigation of Reactive Intermediates of Chemical Reactions in Solution by Electrospray Ionization Mass Spectrometry: Radical Cation Chain Reactions. Angew. Chem. Int. Ed. 42, 4700–4703 (2003).
Sabino, A. A., Machado, A. H. L., Correia, C. R. D. & Eberlin, M. N. Probing the Mechanism of the Heck Reaction with Arene Diazonium Salts by Electrospray Mass and Tandem Mass Spectrometry. Angew. Chem. Int. Ed. 43, 2514–2518 (2004).
Acknowledgements
We thank Mrs. Jianyang Pan (Pharmaceutical Informatics Institute, Zhejiang University) for performing the NMR for structure elucidation.
Author information
Authors and Affiliations
Contributions
C.M., R.S., X.D., Y.H. designed the work. M.C., J.Z., Y.Y., M.Z., C.S. carried out the experiments, analyzed the data. C.M., R.S., X.D., Y.H. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, CL., Zhao, JH., Yang, Y. et al. A Copper-Catalyzed Tandem Cyclization Reaction of Aminoalkynes with Alkynes for the Construction of Tetrahydropyrrolo[1,2-a]quinolines Scaffold. Sci Rep 7, 16640 (2017). https://doi.org/10.1038/s41598-017-16887-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16887-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
