
Abstract
Follicular development has a major impact on reproductive performance. Most previous researchers focused on molecular mechanisms of follicular development. The genetic architecture underlying the number of follicle, however, has yet not to be thoroughly defined in chicken. Here we report a genome-wide association study for the genetic architecture determining the numbers of follicles in a large F2 resource population. The results showed heritability were low to moderate (0.05–0.28) for number of pre-ovulatory follicles (POF), small yellow follicles (SYF) and atresia follicles (AF). The highly significant SNPs associated with SYF were mainly located on GGA17 and GGA28. Only four significant SNPs were identified for POF on GGA1. The variance partitioned across chromosomes and chromosome lengths had a linear relationship for SYF (R2 = 0.58). The enriched genes created by the closest correspondent significant SNPs were found to be involved in biological pathways related to cell proliferation, cell cycle and cell survival. Two promising candidate genes, AMH and RGS3, were suggested to be prognostic biomarkers for SYF. In conclusion, this study offers the first evidence of genetic variance and positional candidate genes which influence the number of SYF in chicken. These identified informative SNPs may facilitate selection for an improved reproductive performance of laying hens.
Similar content being viewed by others

Introduction
The reproductive performance of hens is an indicator of egg production, which occurs following a strict follicular hierarchy resulting from a process of orderly growth which begins with recruitment, followed by selection and, lastly, ovulation. During reproduction period, the majority of small yellow follicles (<8 mm in diameter) become atretic and are reabsorbed, with only 5% of follicles further developing into pre-ovulatory follicles (8–40 mm in diameter). Typically, a single SYF is selected daily to join the hierarchy POF for ovulation during peak laying periods1,2. Therefore, thoroughly understanding the process of follicular development and its mechanisms could aid in significantly improving egg production.
In chicken, most previous studies focused on the molecular mechanisms of follicular growth in vitro 3, which were associated with numerous candidate genes including FSHR, ESR, PRL and PRLR3,4. Follicle numbers were also a key determinant of laying performance and were affected by breed5, age6, and nutrition7. For instance, researchers found a big difference in follicle numbers between old laying hens (e.g. >1 year of age) and laying hens in peak stage8. Recently, a couple of genome-wide association studies (GWAS) conducted to study follicle numbers revealed that they are associated with menopausal age9 in humans, and ovarian function and fertility in cattle10. However, no genetic variants influencing follicular numbers have been reported in chicken thus far, especially using GWAS to investigate them directly.
In the present study, we conducted a GWAS to discover genomic regions explaining variations in follicle numbers of White Leghorn and a Chinese indigenous chicken, the Dongxiang Blue-shelled Chicken, using the 600 K Affymetrix Axiom Chicken Genotyping Array. The objective of this study was to investigate the genetic architecture underlying follicles and identify genetic markers of follicle numbers which would employ molecular marker-assisted selection on laying performance.
Results
Phenotypic measurements and genetic parameters
The overall means, standard errors, coefficients of variation, and heritability are illustrated in Table 1. The mean of the number of POF, SYF, and AF was 3.84, 16.36, and 4.91, respectively, with small standard errors (Table 1). The numbers of the three types of follicles had large coefficients of variation that ranged from 30.13% to 55.97% (Table 1). SYF had a moderate heritability (0.28), while POF and AF had low heritability of 0.13 and 0.05, respectively (Table 1). Both genetic and phenotypic correlation between each two of the three types of follicles were positive under bivariate GCTA analyses (Table S1).
Genome-wide association study (GWAS)
By analyzing a total of 1456 hens, we identified 55 genome-wide significant SNPs and 55 suggestive significant SNPs associated with SYF via univariate analyses in GEMMA (Table S2). Four significant SNPs on GGA1 passed the threshold of strong association (8.43 × 10−7) for POF (Table S3). However, no SNPs displayed significant genome-wide association with AF. The Manhattan plot (Fig. 1 left) shows the P values for all SNPs affecting each trait, across all chromosomes, while the quantile-quantile (QQ) plot (Fig. 1 right) displays significant deviations of observed P values from those expected. The deflation factor (λ) was estimated at 1.0402, 0.9884, and 1.0446 for POF, SYF, and AF, respectively, indicating a good consistency between the observed and expected P values (Fig. 1 right).

Manhattan plot (left) displaying the association of all SNPs with preovulation follicle (POF; (A), small yellow follicle (SYF; (B), and atresia follicle (AF; (C) from GEMMA GWAS. The x-axis indicates the position of SNPs on each chromosome. The y-axis is the −log10 (observed P values) for genome-wide SNPs. The horizontal solid black and dash black line indicate the genome-wide significant (8.43 × 10−7) and suggestive significant (1.69 × 10−5) association thresholds, respectively. The quantile-quantile plots (right) shows the observed distribution of P values against the expected P values under the null hypothesis of no association.
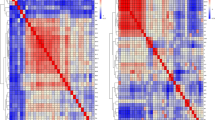
The genome-wide significant association SNPs for SYF were located in a 1.25 Mb region spanning from 1.25 Mb to 2.5 Mb on chromosome GGA28. The most significant SNPs associated with SYF on chromosome GGA28 spanned from 1.4 to 2.5 Mb and showed substantial linkage disequilibrium (LD; Fig. 2). A total of 64 small scale blocks were observed in this region. The identified SNPs with the strongest association with SYF were rs317889060 and rs316038837 (Table S2).

Linkage disequilibrium plots of significantly associated genetic and functional regions. D’ ≥ 0.8 indicates the strong LD block. The horizontal solid black and blue line indicate the genome-wide significant (8.43 × 10−7) and suggestive significant (1.69 × 10−5) association thresholds, respectively.
When analyzing the genomic selection, the Bayes B method in GenSel used to identify significant loci associated with SYF generated similar results. The significant variation threshold for the Bayes B method was selected as 0.5% for each window. The highest effect of SNPs on the SYF trait was listed in Table S4, for each significant window obtained via GenSel. A total of 42 SNPs were identified as significantly associated with the SYF trait. From these, twenty-eight significant SNPs were also detected in GEMMA (Table 2), indicating a high correlation (>50% common loci) between the two methods.
Annotation of significant SNPs and in silico pathway analysis
The four detected SNPs for POF was mapped to DCLK1, NBEA, and nearby SMAD9. The detailed annotations of the 28 SNPs significantly associated with the SYF trait, identified using both methods, were listed in Table 2. Six intergenic variant SNPs were associated with the SYF trait (Table 3). In addition, thirteen SNPs were located up- or down-stream of genes including TCF3, SCAMP4, OAZ1, DUS3L, ZAP70, ACSBG2, REXO1, NRTN, NMRK2, ABHD17A, ANP32B, and SNORD37 (Table 3). Two SNPs were mapped to non-coding transcript region and 1 SNP was synonymous variant. Also of note, six SNPs were encompassed in the introns of the DOT1L, MYO1F, ADAMTS10, SH3GL1, and RGS3 genes (Table 3).
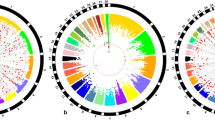
To explore the potential functional implications of the detected SNPs and identify the biological pathways and processes that these SNPs affect, the in silico pathway analysis and the Cytoscape method were used (Table 4). The significant SNPs were marked abundantly in genes participating in cell development, survival, differentiation, and apoptosis, which is in line with our expectations. The three identified clustered gene networks were involved in general molecular and cellular processes such as cell cycle, cell survival, cell development, and organismal injury and abnormalities (Fig. 3). The first network is composed of genes with transcriptional control (MBD3, TCF3, DOT1L) and the AMH, ZAP70, RGS3, and OAZ1 enzymes. The second informative network is formed of network interacting molecules, including many enzymes such as ACSBG2, ADAMTS10, ABHD17A, DAPK3, and REXO3. The third cluster contains genes involved in cellular development and cell cycle, such as SH3GL1, SCAMP4, ANP32B, DUS3L, and NMRK2 etc.

Network of interactions between GWAS positional candidate genes. The network shows interactions between the candidate genes enriched for significantly associated SNPs. (A) Tissue morphology, organismal injury and abnormalities. (B) Cell death and survival, organismal injury and abnormalities, cellular development. (C) Tissue morphology, cellular development, cell cycle. Green box indicates the detected candidate genes in current study, while blue box represents the genes identified from previous studies. The solid arrow line indicates direct relationship while the dash arrow line means indirect relationship.
Features of allelic contribution
The contribution to SYF manifestation, as well as the genotype-effect of the more prominent SNPs in Table 5, is that these SNPs have a strong impact (2.46–5.5%) on the phenotypic variation of trait. Notably, rs3160388837 is the most significant SNPs on GGA28, located up-stream of AMH, exerting a 5.5% contribution to the phenotypic variation of SYF. According to the GWAS data, the TT genotype is linked with a higher incidence of SYF, whereas CC is the risk genotype for low numbers of SYF (Table 5). Another particularly interesting SNP is rs312873273, located on chromosome GGA17, which explains 2.4% of variation in the SYF trait.
Genetic variation partition
The total genetic variance was partitioned onto individual chromosome, to dissect the genetic architecture of follicle development, by simultaneously fitting the genetic relationship matrix (GRM) of all the chromosomes. Only the SYF trait showed significant effects of individual chromosome on genetic variation (Fig. 4). Chromosome GGA28 and GGA17 explained the majority of the variation in SYF numbers, accounting for 5.7% and 3.9% of variance, respectively, which indicates different contributions of particular chromosomes to the SYF trait. Moreover, we observed a strong significant positive linear relationship (P = 1.8e-7, R2 = 0.58) between the estimated variance explained by each chromosome and the relative length of the chromosomes for SYF (Fig. 4).

Scatterplot showing the genome partitioning for small yellow follicle (SYF). Solid lines are shown for linear regressions, which were significant (R2 = 0.58; P = 1.8e-7). The characters in the circles are the chromosome numbers.
Discussion
The aims of this study were to identify the genetic architecture associated with the numbers of different types of follicles and explore the positional candidate genes for those traits using a novel F2 population and the 600 K SNP panel. To date, this is the first GWA study of follicle numbers in birds.
We used the commercial standard line White Leghorn and Chinese indigenous Dongxiang Blue-shelled Chicken to generate an F2 population, creating a unique opportunity to maximize the differences in traits, while increasing the power to identify SNPs for respective traits. Moreover, an extremely large population was used in this GWA study, as well as a higher density (600 K) SNP array, to increase the accuracy and reliability of the obtained data. Different types of follicles were selected for this GWA study, deepening our understanding of the genetic architecture underlying follicular development, from recruitment until ovulation.
Compared to a previous study of commercial hens11, the mean number of POF in the current study was slightly smaller, which may have been caused by the different breeds. In this study, identified heritability were low (0.13) for POF, which could be due to unavailability, using the clutch status of hens, once follicles in the hierarchy are selected for ovulation12. The extremely low heritability (0.05) of AF suggests that unknown internal and external factors may have affected this measurement. More precise methods and control mechanism to investigate POF and AF should be explored in a subsequent study. Heritability for SYF was moderate (0.28), which was similar to previously reported heritability of follicle numbers in cattle10. The heritability of SYF was higher than the ovary weight and POF weights in previous estimates13, but similar to the heritability of egg numbers14.
To accurately identify the potential loci affecting the number of chicken follicles, we used two different software systems –GEMMA and GenSel– to explore the observed associations for each trait. Very few SNPs associated with either POF or AF were identified using both software packages. However, a relatively high, consistent number of SNPs was identified genome-wide to significantly associate with SYF, using the two methods of analysis. Of the 55 (50.91%) most significant SNPs identified by GEMMA, 28 SNPs were also amongst the 42 (66.67%) most significant SNPs by GenSel. Both results showed evidence for association on chromosome GGA28 and GGA17 as multiple significant SNPs were located on these two chromosomes. Although most of the significant SNPs associated with SYF obtained in the current study were different from previous results of ovary weight and egg number13,14, the predicted positional candidate genes partially agree with previous reports13,14.
Two SNPs, rs16211213 and rs316413369, fall into non-coding regions of the genome, inferring that the function of these variations is the control of gene expression instead of variations in the gene product itself. The genes located near the significant SNPs were compiled as gene lists to perform an unbiased pathway analysis and build gene networks. All three resulting networks were tightly related to several of the genes (molecules) involved in cell development, cell cycle, cell death and survival. For example, MYO1F present in the first cluster network (Fig. 3A) has an ability to guide immune cell motility15; OAZ1 could regulate intracellular polyamine levels16; MBD3 is an essential requirement for pluripotency of embryonic stem cells17; DOT1L is over-expression in ovarian cancer, driving cell cycle progression18. Follicle growth results from a process of cell survival, differentiation, proliferation and apoptosis. Based on the present findings, it is possible to speculate that the genes within these networks play a key role in the genetic determination of SYF.
Other most promising candidate genes for SYF were AMH and RGS3. AMH, a glycoprotein, is a member of the TGF- β family, involved in tissue growth, cell differentiation and follicle development. Moreover, previous studies demonstrate that this gene is the essential in determining the number of primordial follicle19. Plasma AMH level is one of the criteria used to diagnose polycystic ovary syndrome (PCOS)20, which results in an anovulation, and subsequent infertility, in women of reproductive age. Besides, AMH is an excellent marker of ovarian reserve, as a decrease in AMH level is typically accompanied by ovarian senescence21,22. Also, AMH can inhibit the process of primordial follicle recruitment and follicle development22.
In this GWA study, we identified the significant SNP rs316038837 associated with the SYF trait, which is 14 kb up-stream AMH. In chicken, the expression level of AMH is strongly related to follicular growth, especially for small follicles23. Previous studies have demonstrated that AMH expression level in SYF, together with other genes such as BMP6, FSH, BMP15, played an essential role in follicle development23,24,25. Also, it was discovered that broiler ovaries had a higher AMH expression level than the standard commercial layer ovaries5, suggesting the inhibitory action of AMH in chicken follicle selection. The present GWAS results, along with the crucial role of AMH in chicken follicle development, indicate AMH might be an excellent candidate gene for SYF numbers and could have a similar role in mammalian ovarian folliculogenesis.
Another interesting positional candidate gene, identified in the present study, is RGS3 (rs312873273 associated with SYF) on chromosome GGA17. RGS3 encodes for a regulator of G-protein signaling26. Previous studies showed that RGS3 had negative regulatory functions on FHSR and LHR signaling in rats27. In human, this gene is involved in GnRH responsiveness in granular cells28. Moreover, many research studies on chicken showed the important roles of FSHR, FSH, and GnRH in follicular growth: the expression level of FSHR is tightly related to the number of pre-hierarchy follicles; FSH is critical to the recruitment of follicles; GnRH has an established role on the FSH synthesis1. The aforementioned information, leads to concluding that RGS3 might be an essential factor in follicular growth and have a potential inhibitory effect on follicle number.
Furthermore, the gene network analysis showed that the AMH and RGS3 genes can active NFκB (Fig. 3). NFκB is a transcription regulator, which can be induced by various stimuli, like AMH. The NFκB pathway may be involved in the basic control of ovary proliferation, apoptosis, and hormone release29. In swine, the NFκB signaling cascade is a mediator of FSH action which regulates AMH expression in ovarian cells30. In humans, AMH had an inverse relationship with breast tumor development and growth31,32,33,34. Based on the current results and previous studies, it is reasonable to speculate that AMH and RGS3 can regulate follicle development and growth via activation of the NFκB signaling pathway.
In summary, we have identified the first genetic variants influencing normal variation in the numbers of the different types of chicken follicles, especially SYF. The present GWAS results suggest that genetic variants of AMH (rs316038837) and RSG3 (rs312873273) genes could be used as prognostic biomarkers for SYF development. However, it will be important to further evaluate the role of these regions in follicular development. Since follicular development is strongly related to egg production, it will be interesting to examine whether variations in these genomic regions relates to reproductive performance. Also, these identified informative SNPs may facilitate selection for an improved productive performance of chicken.
Methods
Ethics statement
All animal care, slaughtered and experimental procedures were approved by the Institutional Animal Care and Institutional Ethic Committee of Jiangsu Institute of Poultry Science. All methods were performed in accordance with guidelines of the Jiangsu Institute of Poultry Science.
Resource population
In brief, six males from each of White Leghorn (WL) and a Chinese indigenous chicken, the Dongxiang Blue-shelled Chicken (DX), were mated to 133 and 80 females from DX and WL, respectively, to generate the F1 generation. Unrelated F1 chickens were selected to breed the F2 generation: 25 males × 406 females from WL/DX parental and 24 males × 233 females from DX/WL parental. A total of 1893 F2 hens were generated. Detailed information about the resource population can be found in previously published studies35,36.
Phenotypic trait
Hens were euthanized with 60–70% carbon dioxide gas at 72 weeks of age. A total of 1456 hens were used to record the different follicle numbers, including number of pre-ovulatory hierarchical follicles (POF) (>8 mm in diameter), number of small yellow follicles (SYF) (5–8 mm), and number of atresia follicles (AF). If the initial phenotypic data were not normally distributed, they were transformed by using box-cox method in the JMP software to obtain a normal distribution for all subsequent analyses. Means, standard errors, fixed effects, and covariates were calculated using the JMP statistical software37. Heritability was estimated by ASReml38.
DNA isolation, genotyping, quality control, and imputation
Blood was collected from the wing vein and genomic DNA was isolated using the standard phenol/chloroform method. Genotyping was performed on 1456 F2 hens, using the 600 K Affymetrix Axiom Chicken Genotyping Array (Affymetrix, Inc., USA). Quality control of genotyping data was initially conducted using the Affymetrix Power Tools v1.16.0 (APT) (http://affymetrix.com/) software with dish quality control >0.82, sample call rate >97%, and SNP call rate ≥95% for the downstream analyses. Further quality control and genotyping were implemented by the PLINK v1.90 software39 with minor allele frequency (MAF) ≥5% and Hardy-Weinberg equilibrium (HWE) test P ≥ 1 × e10−6. Before the GWA analysis, an imputation was performed using the BEAGLE v4.0 procedure40 to avoid some sporadic missing genotypes. Besides, principal component analysis and independent inspection number was implemented in the PLINK package to determine the thresholds for genome-wide significant and suggestive associations. Finally, a total of 59,286 independent tests were obtained. The cutoff threshold of genome-wide significant and suggestive P-values were 8.43 × 10−7 (0.05/59,286) and 1.69 × 10−5 (1.00/59,286), respectively.
Statistical analyses
The SNPs significantly associated with the respective phenotypic traits were identified using both genome-wide efficient mixed-model association (GEMMA)41 and the Bayesian methods in Gensel software42. The GEMMA model can be written as
where y represents a vector of phenotypic values for n individuals; W is a matrix of covariates (fixed effects with a column vector of 1 and top five PCs), α is a vector of the corresponding coefficients including the intercept; x is a vector of the genotypes of the SNP marker, β is the effect size of the marker; μ is a vector of random individual effects; ε is a vector of random error. Bayesian methods can be used to genomic prediction of breeding values and GWAS43,44,45.
The Bayes B model was described as
where y is a vector of phenotypes; x is an incidence matrix to account for fixed effects on phenotypes; b is a vector of fixed effects; z j is a vector of genotypes covariates at SNP j (AA = −10, AB = 0, and BB = + 10); α j is the allele substitution effect of SNP j; δ j is a parameter that indicates whether SNP j was included in the Markov chain Monte Carlo (MCMC) chain; ε is the error associated with the analysis.
Manhattan plots and quantile-quantile (QQ) plots of each trait were obtained by the “gap” package46,47 in R project. The GenABEL package was used to calculate the genomic inflation factor λ. Linkage disequilibrium (LD) analysis was determined by Haploview v4.248 and assessed by D′ ≥0.8 and r2 ≥1/3 indicating strong LD status. The contributions to phenotypic variance (CPV) of the significant SNPs were calculated in GCTA based on the genetic relationship matrix (GRM) built by PLINK.
Candidate genes
Variant Effect Predictor (VEP) and BioMart tools were performed to identify the candidate genes that located the significant SNPs49,50. A mutual information network was generated based on current results using minet package for R50 with the Aracne algorithm. The network graphs were exported from R for visualization in the MCODE package within Cytoscape 3.2.152,53. Using this visualization tool, we explored networks where one or more SNP was connected via genes.
References
Hocking, P. M. Biology of Breeding Poultry (ed. Dunn, I. C.) 75 (CAB International, 2009).
Gilbert, A. B., Perry, M. M., Waddington, D. & Hardie, M. A. Role of atresia in establishing the follicular hierarchy in the ovary of the domestic hen (Gallus domesticus). J Reprod Fertil 69, 221–227 (1983).
Hu, S., Duggavathi, R. & Zadworny, D. Regulatory mechanisms underlying the expression of prolactin receptor in chicken granulosa cells. PLoS One 12, e0170409 (2017).
Hernandez, A. & Bahr, J. Role of FSH and epidermal growth factor (EGF) in the initiation of steroidogenesis in granulosa cells associated with follicular selection in chicken ovaries. Reproduction 125, 683–691 (2003).
Johnson, P. A., Kent, T. R., Urick, M. E., Trevino, L. S. & Giles, J. R. Expression of anti-mullerian hormone in hens selected for different ovulation rates. Reproduction 137, 857–863 (2009).
Lillpers, K. & Wilhelmson, M. Age-dependent changes in oviposition pattern and egg production traits in the domestic hen. Poult Sci 72, 2005–2011 (1993).
McDerment, N. A., Wilson, P. W., Waddington, D., Dunn, I. C. & Hocking, P. M. Identification of novel candidate genes for follicle selection in the broiler breeder ovary. BMC Genomics 13, 494 (2012).
Ciccone, N. A., Sharp, P. J., Wilson, P. W. & Dunn, I. C. Expression of neuroendocrine genes within the hypothalamic-pituitary-ovarian axis of end of lay broiler breeder hens. In Meeting of the UK Branch of the World-Poultry-Science-Association. S11–S12 (2003).
Schuh-Huerta, S. M. et al. Genetic markers of ovarian follicle number and menopause in women of multiple ethnicities. Hum Genet 131, 1709–1724 (2012).
Walsh, S. W. et al. Heritability and impact of environmental effects during pregnancy on antral follicle count in cattle. J Dairy Sci 97, 4503–4511 (2014).
Lillpers, K. & Wilhelmson, M. Genetic and phenotypic parameters for oviposition pattern traits in three selection lines of laying hens. Br Poult Sci 34, 297–308 (1993).
Wolc, A., Bednarczyk, M., Lisowski, M. & Szwaczkowski, T. Genetic relationships among time of egg formation, clutch traits and traditional selection traits in laying hens. J Anim Feed Sci 463, 44 (2010).
Sun, C. et al. Promising loci and genes for yolk and ovary weight in chickens revealed by a genome-wide association study. PLoS One 10, e0137145 (2015).
Yuan, J. et al. Identification of promising mutants associated with egg production traits revealed by genome-wide association study. PLoS One 10, e0140615 (2015).
Kim, S. V. et al. Modulation of cell adhesion and motility in the immune system by Myo1f. Science 314, 136–139 (2006).
Xiao, S. et al. Genome-scale RNA interference screen identifies antizyme 1 (OAZ1) as a target for improvement of recombinant protein production in mammalian cells. Biotechnol Bioeng 113, 2403–2415 (2016).
Kaji, K. et al. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol 8, 285–292 (2006).
Zhang, X. et al. Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer. J Hematol Oncol 10, 29 (2017).
Hansen, K. R., Hodnett, G. M., Knowlton, N. & Craig, L. B. Correlation of ovarian reserve tests with histologically determined primordial follicle number. Fertil Steril 95, 170–175 (2011).
Dewailly, D. et al. Diagnosis of polycystic ovary syndrome (PCOS): revisiting the threshold values of follicle count on ultrasound and of the serum AMH level for the definition of polycystic ovaries. Hum Reprod 26, 3123–3129 (2011).
Xu, H. et al. Retrospective cohort study: AMH is the best ovarian reserve markers in predicting ovarian response but has unfavorable value in predicting clinical pregnancy in GnRH antagonist protocol. Arch Gynecol Obstet 295, 763–770 (2017).
Alvaro Mercadal, B. et al. AMH mutations with reduced in vitro bioactivity are related to premature ovarian insufficiency. Hum Reprod 30, 1196–1202 (2015).
Johnson, P. Follicle selection in the avian ovary. Reprod Domest Anim 47, 283–287 (2012).
Ocon-Grove, O. M., Poole, D. H. & Johnson, A. L. Bone morphogenetic protein 6 promotes FSH receptor and anti-Mullerian hormone mRNA expression in granulosa cells from hen prehierarchal follicles. Reproduction 143, 825–833 (2012).
Stephens, C. S. & Johnson, P. A. Bone morphogenetic protein 15 may promote follicle selection in the hen. Gen Comp Endocrinol 235, 170–176 (2016).
Koch, W. J., Hawes, B. E., Allen, L. F. & Lefkowitz, R. J. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc Natl Acad Sci USA 91, 12706–12710 (1994).
Castro-Fernandez, C., Maya-Nunez, G. & Mendez, J. P. Regulation of follicle-stimulating and luteinizing hormone receptor signaling by. Endocrine 25, 49–54 (2004).
Hamel, M. et al. Genomic assessment of follicular marker genes as pregnancy predictors for human IVF. Mol Hum Reprod 16, 87–96 (2010).
Pavlová, S., Klucska, K., Vašíček, D., Kotwica, J. & Sirotkin, A. V. Transcription factor NF-κB (p50/p50, p65/p65) controls porcine ovarian cells functions. Anim Reprod Sci 128, 73–84 (2011).
Pavlova, S. et al. The involvement of SIRT1 and transcription factor NF-kappaB (p50/p65) in regulation of porcine ovarian cell function. Anim Reprod Sci 140, 180–188 (2013).
Gupta, V. et al. Mullerian inhibiting substance suppresses tumor growth in the C3(1) T antigen transgenic mouse mammary carcinoma model. Proc Natl Acad Sci USA 102, 3219–3224 (2005).
Hoshiya, Y. et al. Mullerian Inhibiting Substance induces NFkB signaling in breast and prostate cancer cells. Mol Cell Endocrinol 211, 43–49 (2003).
Segev, D. L. et al. Mullerian inhibiting substance inhibits breast cancer cell growth through an NFkappa B-mediated pathway. J Biol Chem 275, 28371–28379 (2000).
Segev, D. L. et al. Mullerian inhibiting substance regulates NFkappaB signaling and growth of mammary epithelial cells in vivo. J Biol Chem 276, 26799–26806 (2001).
Yi, G. et al. Genome-wide association study dissects genetic architecture underlying longitudinal egg weights in chickens. BMC Genomics 16, 746 (2015).
Shen, M. et al. Genome-wide association studies for comb traits in chickens. PLoS One 11, e0159081 (2016).
JMP®, Version 11. 2.1. SAS Institute Inc., Cary, NC, 1989-2007. 16.
Gilmour, A. R., Gogel, B., Cullis, B. & Thompson, R. ASReml user guide release 3.0. Hemel Hempstead: VSN International Ltd. (2009).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81, 559–575 (2007).
Browning, B. L. & Browning, S. R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. The American Journal of Human Genetics 84, 210–223 (2009).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44, 821–824 (2012).
Fernando, R. & Garrick, D. J. GenSel: user manual for a portfolio of genomic selection related analyses. Ames: Iowa State University: Animal Breeding and Genetics (2008).
Meuwissen, T. H. E., Hayes, B. J. & Goddard, M. E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829 (2001).
Schneider, J. F. et al. Genome-wide association study of swine farrowing traits. Part II: Bayesian analysis of marker data. J Anim Sci 90, 3360–3367 (2012).
Peters, S. O. et al. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J Anim Sci 90, 3398–3409 (2012).
Zhao, J. H. gap: Genetic Analysis Package. R package version 11–12. http://cran.r-project.org/web/packages/gap/index.html (2014).
Turner, S. D. qqman: an R package for visualizing GWAS results using Q-Q and manhattan plots. R package version 11–12. http://cran.r-project.org/web/packages/qqman/index.html (2014).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
McLaren, W. et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070 (2010).
Kinsella, R. J. et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford). 2011, 56–65 (2011).
Meyer, P. E., Lafitte, F. & Bontempi, G. minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinformatics 9, 461 (2008).
Shannon, P. et al. Cytoscape: a software environ- ment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2 (2003).
Acknowledgements
This study was supported in part by the China Agriculture Research Systems (CARS-41-K02), Integration Innovation Project of Layer Research Systems in Jiangsu Province (SXGC[2017]252) and Fund for Independent Innovation of Agricultural Sciences in Jiangsu Province (CX[16]1008). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would like to acknowledge the staff of China Agricultural University for involvement in this program.
Author information
Authors and Affiliations
Contributions
K.W. and N.Y. were responsible for the project design. L.Q., M.M., T.D., J.L., Y.H. and Y.L. collected the sample data. J.G., X.W., M.S. and H.S. coordinated data analysis. The manuscript was written through contributions of all authors. And all authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, M., Sun, H., Qu, L. et al. Genetic Architecture and Candidate Genes Identified for Follicle Number in Chicken. Sci Rep 7, 16412 (2017). https://doi.org/10.1038/s41598-017-16557-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16557-1
This article is cited by
-
Genome-wide selection signatures detection in Shanghai Holstein cattle population identified genes related to adaption, health and reproduction traits
BMC Genomics (2021)
-
Dynamic expression and functional analysis of circRNA in granulosa cells during follicular development in chicken
BMC Genomics (2019)
-
Genome-wide association studies for 30 haematological and blood clinical-biochemical traits in Large White pigs reveal genomic regions affecting intermediate phenotypes
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
