
Abstract
E-M183 (E-M81) is the most frequent paternal lineage in North Africa and thus it must be considered to explore past historical and demographical processes. Here, by using whole Y chromosome sequences from 32 North African individuals, we have identified five new branches within E-M183. The validation of these variants in more than 200 North African samples, from which we also have information of 13 Y-STRs, has revealed a strong resemblance among E-M183 Y-STR haplotypes that pointed to a rapid expansion of this haplogroup. Moreover, for the first time, by using both SNP and STR data, we have provided updated estimates of the times-to-the-most-recent-common-ancestor (TMRCA) for E-M183, which evidenced an extremely recent origin of this haplogroup (2,000–3,000 ya). Our results also showed a lack of population structure within the E-M183 branch, which could be explained by the recent and rapid expansion of this haplogroup. In spite of a reduction in STR heterozygosity towards the West, which would point to an origin in the Near East, ancient DNA evidence together with our TMRCA estimates point to a local origin of E-M183 in NW Africa.
Similar content being viewed by others
Introduction
The male-specific region of the Y chromosome (MSY) is one of the workhorses of human population genetics. The avoidance of recombination of this region implies that haplotypes are passed almost intact from generation to generation. As a result, variation is introduced into this region only by mutation1. Single nucleotide polymorphisms (SNPs) and short tandem repeats (STRs) are the most commonly used type of variation when working with the Y chromosome. Because of their low mutation rate (10−9 substitutions/site/year)2, SNPs (and some short indels) tend to be unique events in human evolution and can be easily combined into haplotypes, known as haplogroups. These haplogroups have been used to build consistent phylogenies that show a particular ethno-geographical distribution3,4,5.
Until recently, the main problem with SNPs was their ascertainment bias produced by the narrow range of populations (or even of individuals) assessed for variation. Now, the advent of next generation sequencing (NGS) of the MSY has solved the problem of ascertainment bias and has allowed the systematic discovery of thousands of new SNPs from worldwide populations. Several studies6,7,8,9,10,11,12,13 have provided refined Y chromosome phylogenies in which branch lengths are proportional to time, permitting the direct estimation of the time to most recent common ancestor (TMRCA) of nodes, and the coalescence of their branches can be used to trace effective population size back in time. Despite the improvements of NGS, further work is needed to better understand the internal diversity of specific haplogroups. On the other hand, STRs are also commonly used in population genetic studies; because of their higher mutation rate (10−3–10−4)14, Y-STRs exhibit a higher degree of variation and are thus an optimal tool for discriminating between closely related Y chromosomes. Although their mutation processes can be rather complex and can saturate much faster than SNPs, STRs can also provide good time estimates for relatively recent events1.
Here, by using whole Y chromosome sequences, we intend to shed some light on the historical and demographic processes that modelled the genetic landscape of North Africa. Previous studies suggested that the strategic location of North Africa, separated from Europe by the Mediterranean Sea, from the rest of the African continent by the Sahara Desert and limited to the East by the Arabian Peninsula, has shaped the genetic complexity of current North Africans15,16,17. Early modern humans arrived in North Africa 190–140 kya (thousand years ago)18, and several cultures settled in the area before the Holocene. In fact, a previous study by Henn et al.19 identified a gradient of likely autochthonous North African ancestry, probably derived from an ancient “back-to-Africa” gene flow prior to the Holocene (12 kya). In historic times, North Africa has been populated successively by different groups, including Phoenicians, Romans, Vandals and Byzantines. The most important human settlement in North Africa was conducted by the Arabs by the end of the 7th century. Recent studies have demonstrated the complexity of human migrations in the area, resulting from an amalgam of ancestral components in North African groups15,20.
Besides geography, cultural diversity must also be considered. Two branches of languages belonging to the Afro-Asiatic family define two major groups in North Africa: Arabs and Berbers. Arabic languages and culture, as well as the Islamic religion, were brought from the Near East during the Islamic expansion. The Berber people, characterized for speaking Berber languages, are considered the direct descendants of the ancestral pre-Arabic peoples of North Africa20. However, the Berber language and ethnicity should not be equated: many Berber speakers live in large cities, particularly in Morocco and Algeria, and some populations with traditional lifestyles, such as the Reguibates, speak Arabic dialects. In spite of their cultural differences, Y-chromosome SNPs and STRs21, and autosomal haplotype-based methods20 have demonstrated the absence of strong genetic differences between Berbers and Arabs.
Studies based on the Y chromosome have highlighted E-M78 and E-M81 as the most frequent paternal lineages in North Africa, although they showed different distribution patterns. Whereas the frequency of E-M78 declines towards Northwest Africa, E-M81 has been found at high frequencies (71%) in Northwestern Africa and its frequency decreases towards the East; it is found sporadically in S Europe and E Africa, and it is practically absent elsewhere. These evidences suggest that E-M81 must be considered to explore the historical and demographical processes that gave rise to current North African populations. However, little is known about the phylogeographic structure of this haplogroup and its origin and emergence are still very controversial. While some studies pointed to a Palaeolithic origin21, other authors claimed that E-M81 may have a Neolithic origin22. The most likely scenario, as suggested by Fadhlaoui-Zid et al.17, is that the origin of E-M81 is more recent than previously reported.
In the present project, we analyse whole Y chromosome sequences from 32 North African individuals selected by carrying the derived allele at M183. M183 was first described by Karafet et al.5, and appears to be an extremely dominant subclade within E-M81, to the point that E-M81*(xM183) individuals are very rare. Since we found no samples derived for E-M81 and ancestral for E-M183, we selected our individuals on the basis of E-M183. The aim of the present study is to provide a phylogeographic refinement of this paternal lineage in order to shed light on the human population history of North Africa. By using whole Y chromosome sequences, we have been able to describe E-M183 subbranches that will be used to define whether this lineage presents any geographical substructure. Next, by using STRs we will interrogate the genetic diversity within E-M183 subclades in a larger dataset. Finally, both SNP data and STRs will be used to provide updated time constraints of the spread of E-M183.
Results
In the present study, we analysed whole Y chromosome sequences from 32 North African males who belong to the most frequent haplogroup in North Africa: E-M81 (E-M183). Using the samples genotyped for the present study we constructed an updated map showing the paternal lineage distribution in North African populations compared to neighbouring European and Near Eastern populations (Supplementary Fig. S1). And as previously reported by Fadhlaoui-Zid17, E-M81 is predominant in Northwestern Africa and almost absent elsewhere.
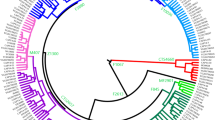
The phylogenetic relationships among the E-M183 carriers and other African and non-African lineages are shown in Fig. 1a (see also Supplementary Table S1). The tree was consistent with the previous independent haplogroup assignation and the known phylogeny of the Y chromosome5,7. Moreover, the analysis of whole Y chromosome data enabled the characterization of five new subclades within this Y chromosomal branch (Fig. 1b). Although four of these five SNPs had previously been described, to the best of our knowledge this is the first academic publication describing their phylogenetic relationships and population frequencies. The first variant, named SM001, clusters together all E-M183 samples but one, a sample from Lebanon. Another subclade is defined by Z5009, which groups two Moroccan samples, and one sample each from the Western Sahara, Libya, Palestine and France. CTS12227 is shared by three Berber samples (two Tunisian Berbers and one Zenata Berber), two Arab Algerians and a sample from Saudi Arabia. Finally, three Libyan samples, two Moroccan and one Tunisian are derived for PF6794, and all but one of these samples are also derived for PF6789.

Maximum parsimony tree. Phylogenetic relations among (a) all samples included in the project (in blue the E-M183 branch) and (b) individuals within E-M183 branch are shown, coloured by its North African origin. Branch lengths are proportional to the number of SNPs on the branch.
In order to study the geographical structure and phylogenetic robustness of E-M183, we genotyped the five subclades described using whole Y chromosome data in 250 North African samples (see methods). We also analysed the genetic diversity within and between North African populations by genotyping the set of Y-STRs contained in the AmpFlSTR®YFiler® kit.
Differences between and within populations
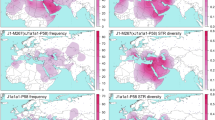
The distribution of each subhaplogroup within E-M183 can be observed in Table 1 and Fig. 2. Indeed, different populations present different subhaplogroup compositions. For example, whereas in Morocco almost all subhaplogorups are present, Western Sahara shows a very homogeneous pattern with only E-SM001 and E-Z5009 being represented. A similar picture to that of Western Sahara is shown by the Reguibates from Algeria, which contrast sharply with the Algerians from Oran, which showed a high diversity of haplogroups. It is also worth to notice that a slightly different pattern could be appreciated in coastal populations when compared with more inland territories (Western Sahara, Algerian Reguibates). And indeed, when we tested by AMOVA based on Y-STRs whether the coastal populations are different to more inland areas, we observe that 16.5% (P < 10−5) of the variance can be explained by differences among these two groups. Finally, surprisingly, Iberian samples showed the highest proportion of E-M183*, with a frequency over E-M183 chromosomes of 20%, whereas in North Africa the frequencies of M183* range from 0 to 7%. However, note that if these frequencies were given over all individuals (and not only over those carrying E-M183), then E-M183* would represent just 0.5% of all Iberian Y chromosomes, but it reaches 7.7% in Libyans.

Distribution of E-M183 subclades among North Africa, the Near East and the Iberian Peninsula. Pie chart sectors areas are proportional to haplogroup frequency and are coloured according to haplogroup in the schematic tree to the right. n: sample size. Map was generated using R software65.
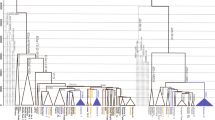
Median-joining networks based on Y-STR haplotypes data and coloured by population were used to investigate whether a particular geographical structure could be defined within this Y chromosomal lineage. As shown in Fig. 3a, apparently, no cluster by population can be distinguished. To better investigate the differences among populations we performed an AMOVA analysis based on Y-STRs. Our results showed that 8.1% (P < 10−5) of the overall genetic variation was explained by differences among populations. As a reference, F ST based on Y-STRs among a similar set of N. African populations, irrespectively of haplogroup, was 11.2%17, suggesting that paternal structure in North Africa does not seem to correlate with population ascription. Moreover, principal component analysis based on Y-STRs and coloured by population (Fig. 4a) showed that no geographical cluster can be distinguished within the PCA. However, when we plotted the mean values of each component in the PCA, we observed that all populations clustered together except those from West Asia, and apparently the second component was driving this split (Fig. 4a and Supplementary Fig. S2b). Differences in average PC2 value were marginally statistically significant (ANOVA, P = 0.042, Supplementary Table S2). Taken together, our results pointed that little or no population-based structure can be found within E-M183.

Median-joining network based on 13 Y-STRs: DYS19, DYS389I, DYS390, DYS391, DYS393, DYS438, DYS439, DYS437, DYS448, DYS456, DYS458, Y GATA H4, and DYS635. Each circle represents a haplotype with its size proportional to frequency and has been coloured according to (a) populations and (b) haplogroups (see key). The lines between them indicate the Y-STR mutational steps.

Principal component analysis based on Y-STRs. Coloured by (a) population and (b) by haplogroup (see key). Squares represent mean values of each component in the PCA coloured by (a) population and (b) haplogroup.
Age estimates
We have estimated the divergence of the E-M183 branch from its sister, E-M78, around 9,700 ya (Table 2) when using a fast mutation rate and ~12,700 ya when a slow mutation rate is considered (see methods). Both a frequentist (ρ) and a Bayesian method gave similar results. The TMRCA (Time to Most Recent Common Ancestor) of a certain haplogroup can provide some constraints on the time of its spread. Here, we used both Y chromosome SNP and STR data to obtain those estimates. Table 2 shows the TMRCA obtained for E-M183 and its sister clade (E-M78) by using SNP data from whole Y chromosome sequences, as well as the coalescence time for E-M183 and its subclades computed with Y-STR data. Regardless of using a Bayesian or a Rho-based approach, our findings when using SNP data suggest that E-M183 originated around 2,000 years ago (ya). It is worth to notice that when the tree is calibrated with a slow mutation rate23,24, the TMRCA of E-M183 given by BEAST reaches ~3,000 ya. However, age estimates computed using STR data strongly support that the coalescence time for this haplogroup is around 2,000 ya. We have also computed the coalescence times for each subclade by using Y-STRs (Table 2). The TMRCAs of E-SM001, E-CTS1227, and E-Z5009 are all ~2,000 ya and their confidence intervals broadly overlap with each other and with that of the whole of E-M183, pointing to a rapid radiation. On the contrary, E-PF6794 and its subclade E-PF6789 appear to be more recent, at ~1,500 ya. Interestingly, E-PF6789 is present in most of North Africa and the Near East (Table 1); if, as discussed below, E-M183 may have expanded from East to West, then ~1,500 ya sets an upper limit for this expansion.
Origin and dispersion
The star-like structure observed in the median-joining network of E-M183 (Fig. 3), could shed some light on the dispersion of E-M183. We found that Y-STRs are extremely homogeneous across E-M183 subhaplogroups, with the same haplotype shared by samples belonging to different subclades (Fig. 3b). This extreme homogeneity could be attributed to a recent and rapid radiation of this Y chromosomal branch25,26, which is also seen in the fact that most of its subclades seem to have appeared almost simultaneously. An AMOVA analysis showed that 8.6% (P < 10−5) of the genetic diversity is explained by differences among subclades, while the same type of analysis across haplogroups27 yields much higher values. Furthermore, in a principal component analysis based on Y-STRs and coloured by subclade (Fig. 4b) apparently no haplogroup cluster could be observed. However, the mean value for the first PC for E-PF6789 has a different sign than the rest, and the overall differences are statistically significant (ANOVA, P = 0.0049). In addition, despite the homogeneity of Y-STRs across E-M183 subhaplogroups mentioned above, we found a particular allele of DYS458 associated with PF6789. The frequency of allele DYS458*17 in PF6789-derived individuals is 71%, while it is much rare in other E-M183 chromosomes (31%) where allele DYS458*18 predominates (50%). When we turned to genetic diversity of each haplogroup (Supplementary Table S3), no significant differences are observed when comparing gene diversity and heterozygosity values between the different subhaplogroups, which again, could be due to a recent expansion of E-M183. However, PF6789 shows the lowest STR-based variance values. Next, Bayesian skyline analysis based on sequence data are in agreement with a recent expansion of this haplogroup. As shown in Fig. 5, regardless of using a) slow 24 or b) fast 2 mutation rates, we see a slight reduction and a rapid increase of population effective size around 2,000 ya. Finally, despite any geographical structure seems to be present within this North African Y chromosomal branch, other studies have proposed a correlation between the longitude and genetic diversity. To further investigate this correlation, we plotted the variance, the average gene diversity and the heterozygosity against the longitude of each population (Supplementary Fig. S3). Although a slight correlation has been observed between longitude and gene diversity (Spearman’s ρ = 0.77), as well as between longitude and STR allele size variance (ρ = 0.6), they are not statistically significant. When we turned to STR heterozygosity, the correlation (ρ = 0.89) becomes slightly significant (P = 0.03). However, this pattern appears to be driven by the fact that Western Sahara is the least diverse but most western population in our set.

Bayesian skyline plots. Two different mutation rates have been used: (a) a ‘slow’ rate24 of 6.17 × 10−10 substitutions/site/year and (b) a ‘fast’ rate2 of 10−9 substitutions/site/ year. Black lines indicate the median effective population size (Ne) and discontinuous grey lines the 95% higher posterior density intervals. Vertical grey line indicate the TMRCA estimated using each substitution rate.
Discussion
Several studies have explored the paternal structure of North Africa showing that E-M183 is the most frequent paternal lineage in North Africa17,22,28. However, these analyses focused on targeted SNPs of the Y chromosome, preventing the discovery of new variation within its sequence. Here, by using whole Y chromosome sequences, we have been able to increase the knowledge of internal new branches within E-M183, which has led to a refinement of the phylogeography of this lineage, and to shed light on the controversial dates for its origin.
Our results evidenced that Y-STR haplotypes within E-M183 individuals are strikingly similar to each other and thus, subhaplogroups within E-M183 cannot be distinguished from each other based on Y-STR differences. As proposed by Larmuseau et al.25, the scenario that better explains Y-STR haplotype similarity within a particular haplogroup is a recent and rapid radiation of subhaplogroups. Although the dating of this lineage has been controversial, with dates proposed ranging from Paleolithic to Neolithic and to more recent times17,22,28, our results suggested that the origin of E-M183 is much more recent than was previously thought. Whereas other studies have relied only on STR data to provide time estimates, here, for the first time, we have used Y chromosomal sequence data to calculate the TMRCA for E-M183. As a result, we have been able to update the TMRCA for this haplogroup by using both SNP and STR data, avoiding a possible bias introduced by inferring the TMRCA using only rapid mutation rates. In addition to the recent radiation suggested by the high haplotype resemblance, the pattern showed by E-M183 imply that subhaplogroups originated within a relatively short time period, in a burst similar to those happening in many Y-chromosome haplogroups23.
Regarding the geographical origin of E-M183, a previous study22 suggested that an expansion from the Near East could explain the observed east-west cline of genetic variation that extends into the Near East. Indeed, our results also showed a reduction in STR heterozygosity towards the West (Supplementary Fig. S3), which may be taken to support the hypothesis of an expansion from the Near East. In addition, previous studies based on genome-wide SNPs15,20 reported that a North African autochthonous component increase towards the West whereas the Near Eastern decreases towards the same direction, which again support an expansion from the Near East. However, our correlations should be taken carefully because our analysis includes only six locations on the longitudinal axis, none from the Near East. As a result, we do not have sufficient statistical power to confirm a Near Eastern origin. In addition, rather than showing a west-to-east cline of genetic diversity, the overall picture shown by this correlation analysis evidences just low genetic diversity in Western Sahara, which indeed could be also caused by the small sample size (n = 26) in this region. Alternatively, given the high frequency of E-M183 in the Maghreb, a local origin of E-M183 in NW Africa could be envisaged, which would fit the clear pattern of longitudinal isolation by distance reported in genome-wide studies15,20. Moreover, the presence of autochthonous North African E-M81 lineages in the indigenous population of the Canary Islands, strongly points to North Africa as the most probable origin of the Guanche ancestors29. This, together with the fact that the oldest indigenous inviduals have been dated 2210 ± 60 ya, supports a local origin of E-M183 in NW Africa. Within this scenario, it is also worth to mention that the paternal lineage of an early Neolithic Moroccan individual appeared to be distantly related to the typically North African E-M81 haplogroup30, suggesting again a NW African origin of E-M183. A local origin of E-M183 in NW Africa > 2200 ya is supported by our TMRCA estimates, which can be taken as 2,000–3,000, depending on the data, methods, and mutation rates used.
The TMRCA estimates of a certain haplogroup and its subbranches provide some constraints on the times of their origin and spread. Although our time estimates for E-M78 are slightly different depending on the mutation rate used, their confidence intervals overlap and the dates obtained are in agreement with those obtained by Trombetta et al.13 Regarding E-M183, as mentioned above, we cannot discard an expansion from the Near East and, if so, according to our time estimates, it could have been brought by the Islamic expansion on the 7th century, but definitely not with the Neolithic expansion, which appeared in NW Africa ~7400 BP and may have featured a strong Epipaleolithic persistence31. Moreover, such a recent appearance of E-M183 in NW Africa would fit with the patterns observed in the rest of the genome, where an extensive, male-biased Near Eastern admixture event is registered ~1300 ya, coincidental with the Arab expansion20. An alternative hypothesis would involve that E-M183 was originated somewhere in Northwest Africa and then spread through all the region. Our time estimates for the origin of this haplogroup overlap with the end of the third Punic War (146 BCE), when Carthage (in current Tunisia) was defeated and destroyed, which marked the beginning of Roman hegemony of the Mediterranean Sea. About 2,000 ya North Africa was one of the wealthiest Roman provinces and E-M183 may have experienced the resulting population growth.
The genetic landscape of North Africa has been strongly influenced by geography. Past and recent historical migrations gave rise to a complex genetic landscape. A recent study on genome-wide data has shown a lack of correlation between geographical and genetic populations20. However, when we turn to the Y chromosome, it has been reported that the Y-chromosomal variation is strongly structured within the region22. Interestingly, this strong geographical structure becomes somehow diluted when we go deeply into a particular Y chromosomal branch, which could be attributed to the rapid radiation mentioned above. E-M183 subbranches have spread through all the area, and are now represented in all the populations sampled. Despite this lack of structure within E-M183, a different pattern could still be appreciated in coastal populations when compared with more inland territories (Western Sahara, Algerian Reguibates). This pattern has been observed with genome wide data20 and could be related to migrations along the coast. And as mentioned above, principal component analysis suggested that PF6789 could be driving this different component. Figure 2 shows that PF6789 is very frequent in Oran (Algeria), Tunisia and Libya, and is present in the Near East and the Iberian Peninsula. Finally, surprisingly the highest frequency of M183* is shown in the Iberian sample which, given the low frequency values of E-M183 in the area, could be attributed to genetic drift acting on a low-frequency variant.
The present study has provided a phylogeographic refinement of the North African lineage E-M183. Time estimates based on whole Y chromosome data have been contrasted to those obtained by Y-STRs in order to provide an updated TMRCA for E-M183 carriers. Despite the analysis of E-M183 and its subbranches has shed some light into the historical and demographic processes that could have given rise to the North African genetic landscape, more efforts should be done, probably by incorporating whole genome data, to better understand the North African genetic landscape. And indeed, our study strongly showcases the need of incorporating ancient DNA in the study of paternal North African structure, in order to understand the genetic background of the area before the coalescence time of E-M183.
Materials and Methods
Samples/ethics
We sequenced fifteen males from seven North African populations (Morocco, Libya, Algeria, Western Sahara, Tunisia, Berber Zenata and Berber Tunisia) by using whole-genome shotgun paired-end sequencing (Illumina HiSeq 2000) to a mean coverage of 30x. North African individuals were selected for belonging to the E-M183 haplogroup by using TaqMan (Life Technologies) probes. DNA donors were recruited with informed consent. All experimental protocols were approved by the Institutional Review Board of the Comitè Ètic d’Investigació Clínica-Institut Municipal d’Assistència Sanitària (CEIC-IMAS) in Barcelona (2013/5429/I) and were carried out in accordance with the approved guidelines. Moreover, 21 E-M183 Y chromosome sequences were supplied by YFull, which is a Y Chromosome sequence interpretation service (http://www.yfull.com/). We also included 32 additional Y chromosome sequences belonging to different haplogroups in order to build a reliable phylogeny. Some of these sequences were obtained from published studies32,33,34,35, other sequences were also supplied by YFull and some sequences are still unpublished (Lorente-Galdós et al., unpublished work). Overall, the initial Y chromosome dataset consists of 68 individuals (see Supplementary Table S1).
In order to validate the new variants within the E-M183 branch discovered by whole Y chromosome sequences and to study its genetic diversity at a population level, we used a larger dataset that includes 454 males from North African, Iberian, and Near Eastern populations (Supplementary Table S4). Donor samples were collected with the appropriate informed consent.
Data analysis, Variant Calling and Filtering
All sequence reads, both those obtained by whole-genome shotgun paired-end sequencing (Illumina HiSeq technology) and those supplied by other sources, were mapped to the human Y chromosome reference sequence (hg19/GRCh37) with BWA36. After that, we removed PCR duplicates, recalibrated base quality scores and performed an indel realignment applying the Genome Analysis Toolkit (GATK) v3.4–4637.
Variant calling was performed across the 68 initially selected samples simultaneously using the GATK UnifiedGenotyper tool37 leading to a raw call set of 107,672 SNPs. Then, we applied hard filtering parameters according to GATK best practices recommendations38,39 and ended up with 23,542 variants. After that, we considered only those sites supported by ≥5 reads, what we called the callable region, which reduced the dataset to 7,535 variant sites (see Supplementary Fig. S4).
Since the Y chromosome is especially rich in repeats, duplications and low-quality regions, we restricted our analysis to high quality regions defined by Wei et al.8, in order to avoid biases introduced by repeated sequences. After excluding ampliconic segments and heterochromatic, pseudoautosomal and X-transposed regions (8.97 Mb) from our dataset, we obtained 4.88 Mb of unique Y chromosomal sequence (<9% of the total Y chromosome length; see Supplementary Table S5 and Supplementary Fig. S4).
Finally, we used AWK in-house scripts to remove heterozygous calls, and VCFtools40 to discard those samples with ≥5% missing calls and/or a sequence read depth of <10X. At the end, our dataset contained 62 individuals and 4,269 variable sites.
Phylogenetic inference and dating
Haplogroups were assigned using AMY-tree v1.241. We considered an updated version of the Y chromosome haplogroup tree, as well as the Phylotree and ISOGG databases and the hg19 Y chromosome FASTA sequence (obtained from the UCSC genome browser, http://hgdownload.cse.ucsc.edu/goldenpath/hg19/chromosomes/). Results obtained are consistent with prior haplogroup assignation32,33,34,35 and, in the case of samples sequenced for this study, haplogroups are consistent with the experimental validation.
A FASTA formatted alignment with the high quality variable sites of all samples was built and used to produce a maximum parsimony tree using MEGA542 (Fig. 1).
We calculated the TMRCA and its standard deviation for E-M81, as well as the divergence time from and its sister clade (E-M78), with two independent methods: i) a Bayesian estimation of the node ages performed with BEAST v1.8.243. Markov chain Monte Carlo (MCMC) samples were based on 15,000,000 generations and the first 1,500,000 generations (10%) were discarded as burn-in. For the analysis, we combined the outputs of five independent runs using LogCombiner. In all runs, we used GTR as a substitution model under a strict clock. As a coalescent tree prior, we used an expansion model using the priors specified by Trombetta et al.13. Finally, in order to reduce the BEAST computational time, we included only variant sites and multiplied the substitution rate by the number of callable sites (4.88 Mb) and divided it by the number of variable sites (4,269 sites). And ii) the rho statistic44, as implemented in the Network 5.0 software (www.fluxus-engineering.com). This statistic is linearly related to time and mutation rate (ρ = μT), assuming rate constancy across the tree branches.
Given the uncertainty regarding the mutation rate, both for BEAST and rho estimates we used two different substation rates: i) a ‘fast’ rate2 of 10−9 substitutions/site/ year; and ii) a ‘slow’ rate24 of 6.17 × 10−10 substitutions/site/year.
For the analysis of the effective population size, Bayesian skyline plots were generated using BEAST v1.8.243. Markov chain Monte Carlo (MCMC) samples were based on 15,000,000 generations, and the first 1,500,000 generations (10%) were discarded as burn-in. Again, five different runs were combined using Logcombiner and, in all of them, we used GTR as a substitution model under a strict clock and a linear piecewise linear skyline model with 10 groups. We used the same fast and slow rates as for the dating methods described above.
Haplogroup frequencies and validation of new variants
An updated map of E-M81 haplogroup frequency distribution (Supplementary Fig. S1) was constructed using the Surfer Golden software v 10.0.500 (Golden Software, Golden, CO, USA), in which we included haplogroup frequencies obtained in this study, as well as frequencies obtained in other studies45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60. AWK in-house scripts have been used to define all variants within E-M183 (see Supplementary Fig. S5 and Supplementary Table S6). In particular, five of those variants are defining new subclades (SM001, PF6794, PF6789, CTS12227, Z5009) within the North African phylogenetic branch E-M183 (Fig. 1b, Supplementary Fig. S6). To study those subbranches at a population level, we validated them in a set of 454 North African, European, and Near Eastern individuals (see Methods; Supplementary Table S7). First, we selected samples with the derived allele at M183 by using single TaqMan (Life Technologies) probes. Overall, 250 individuals were derived for M183 and, subsequently, selected to validate the five new variants by Taqman (Life Technologies) assays by using the manufacturer’s protocols (Supplementary Table S8).
Y-STR analysis
The individuals that were derived for E-M183 were subsequently typed for the 17 Y-STRs (DYS19, DYS385a/b, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS438, DYS439, DYS437, DYS448, DYS456, DYS458, Y GATA H4, and DYS635) contained in the AmpFlSTR®YFiler® PCR Amplification kit (Life Technologies, Carlsbad, CA, USA)61 (Supplementary Table S9). However, the final dataset consists on 12 Y-STRs and 185 individuals to eliminate missing values. Diversity, average number of pairwise differences and heterozygosity were calculated using Arlequin 3.562. In order to explore whether E-M183 (and its subbranches) showed any signal of population structure, a hierarchical analysis of molecular variance (AMOVA) based on Fst was performed using Arlequin 3.562. We group the populations using two criteria: i) according to their geographical origin; and ii) according to their E-M183 subbranches. Median-joining networks were drawn with Network 5.0.0.1 (www.fluxus-engineering.com). Finally, the TMRCA has been estimated with the ρ approach, implemented within the program Network 5.0.0.163, and using a mutation rate of one mutation per haplotype per 858 years, obtained from the compilation in the YHRD database (www.yhrd.org, accessed on Dec. 14th, 2016) for this set of 13 Y STRs and using a generation time of 30 years64.
Data availability statement
The datasets generated and analysed during the current study are available in https://figshare.com/articles/North_African_Ychromosome_dataset_vcf/5537941.
References
Jobling, M. A. & Tyler-Smith, C. The human Y chromosome: an evolutionary marker comes of age. Nat. Rev. Genet. 4, 598–612 (2003).
Helgason, A. et al. The Y-chromosome point mutation rate in humans. Nat. Genet. 47, 453–457 (2015).
Hammer, M. F. A Recent Common Ancestry for Human-Y-Chromosomes. Nature 378, 376–378 (1995).
Underhill, P. A. et al. Y chromosome sequence variation and the history of human populations. Nat. Genet. 26, 358–361 (2000).
Karafet, T. M. et al. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 18, 830–838 (2008).
Francalacci, P. et al. Low-Pass DNA Sequencing of 1200 Sardinians Reconstructs European Y-Chromosome Phylogeny. Science 341, 565–569 (2013).
Poznik, G. D. et al. Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 341, 562–5 (2013).
Wei, W., Ayub, Q. & Chen, Y. A calibrated human Y-chromosomal phylogeny based on resequencing Accepted Email alerting service A calibrated human Y-chromosomal phylogeny based on resequencing. 388–395, https://doi.org/10.1101/gr.143198.112 (2013).
Scozzari, R. et al. An unbiased resource of novel SNP markers provides a new chronology for the human Y chromosome and reveals a deep phylogenetic structure in Africa. Genome Res. 24, 535–44 (2014).
Lippold, S. et al. Human paternal and maternal demographic histories: insights from high-resolution Y chromosome and mtDNA sequences. Investig. Genet. 5, 13 (2014).
Hallast, P. et al. The Y-Chromosome Tree Bursts into Leaf: 13,000 High-Confidence SNPs Covering the Majority of Known Clades. Mol. Biol. Evol. 32, 661–673 (2015).
Batini, C. et al. Large-scale recent expansion of European patrilineages shown by population resequencing. Nat. Commun. Commun 7152, https://doi.org/10.1038/ncomms8152 (2015).
Trombetta, B. et al. Phylogeographic refinement and large scale genotyping of human Y chromosome haplogroup E provide new insights into the dispersal of early pastoralists in the African continent. Genome Biol Evol 7, 1940–1950 (2015).
Gusmão, L. et al. Mutation rates at Y chromosome specific microsatellites. Hum. Mutat. 26, 520–8 (2005).
Henn, B. M. et al. Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet. 8, e1002397 (2012).
Botigué, L. R. et al. Gene flow from North Africa contributes to differential human genetic diversity in southern Europe. Proc. Natl. Acad. Sci. USA 110, 11791–6 (2013).
Fadhlaoui-Zid, K. et al. Genome-wide and paternal diversity reveal a recent origin of human populations in North Africa. PLoS One 8, e80293 (2013).
Smith, T. M. et al. Earliest evidence of modern human life history in North African early Homo sapiens. Proc. Natl. Acad. Sci. 104, 6128–6133 (2007).
Henn, B. M., Cavalli-Sforza, L. L. & Feldman, M. W. The great human expansion. Proc. Natl. Acad. Sci. USA 109, 17758–64 (2012).
Arauna, L. R. et al. Recent historical migrations have shaped the gene pool of Arabs and Berbers in North Africa. Mol. Biol. Evol. 34, 318–329 (2017).
Bosch, E. et al. High-resolution analysis of human Y-chromosome variation shows a sharp discontinuity and limited gene flow between northwestern Africa and the Iberian Peninsula. Am. J. Hum. Genet. 68, 1019–29 (2001).
Arredi, B. et al. A predominantly neolithic origin for Y-chromosomal DNA variation in North Africa. Am J Hum Genet 75, 338–45 (2004).
Poznik, G. D. et al. Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences. Nat. Genet. 12, 809–809 (2016).
Fu, Q. et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 514, 445–9 (2014).
Larmuseau, M. H. D. et al. Recent radiation within Y-chromosomal haplogroup R-M269 resulted in high Y-STR haplotype resemblance. Ann. Hum. Genet. 78, 92–103 (2014).
Solé-Morata, N., Bertranpetit, J., Comas, D. & Calafell, F. Recent Radiation of R-M269 and High Y-STR Haplotype Resemblance Confirmed. Ann. Hum. Genet. 78, 253–254 (2014).
Bosch, E. et al. Variation in short tandem repeats is deeply structured by genetic background on the human Y chromosome. Am. J. Hum. Genet. 65, 1623–1638 (1999).
Bosch, E. et al. Population History of North Africa: Evidence from Classical Genetic Markers. Hum. Biol. 3, 295 (1997).
Fregel, R. et al. Demographic history of Canary Islands male gene-pool: replacement of native lineages by European. 14, 1–14 (2009).
Fregel, R., Méndez, F. L., Bokbot, Y., Martín-socas, D. & María, D. Neolithization of North Africa involved the migration of people from both the Levant and Europe. 1–14 (2017).
Mulazzani, S. et al. The emergence of the Neolithic in NorthAfrica: A new model for the Eastern Maghreb. Quat. Int. 410, 123–143 (2016).
Altshuler, D. L. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–9 (2014).
Reich, D. et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 (2010).
Schuster, S. C. et al. Complete Khoisan and Bantu genomes from southern Africa. Nature 463, 943–7 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–8 (2011).
Van der Auwera, G. A. et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics. https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Van Geystelen, A., Decorte, R. & Larmuseau, M. H. D. AMY-tree: an algorithm to use whole genome SNP calling for Y chromosomal phylogenetic applications. BMC Genomics 14, 101 (2013).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Jobling, M., Hollox, E., Hurles, M., Kivisild, T. & Tyler-Smith, C. Human Evolutionary Genetics. Human Evolutionary Genetics, 2nd edition, https://doi.org/10.1017/CBO9781107415324.004 (Garland Science, 2014).
Cinnioǧlu, C. et al. Excavating Y-chromosome haplotype strata in Anatolia. Hum. Genet. 114, 127–148 (2004).
Cruciani, F. et al. Phylogeographic Analysis of Haplogroup E3b (E-M215) Y Chromosomes Reveals Multiple Migratory Events Within and Out Of Africa. The American Journal of Human Genetics 74 (2004).
Flores, C. et al. Reduced genetic structure of the Iberian peninsula revealed by Y-chromosome analysis: implications for population demography. Eur. J. Hum. Genet. 12, 855–863 (2004).
Alonso, S. et al. The place of the Basques in the European Y-chromosome diversity landscape. Eur. J. Hum. Genet. 13, 1293–1302 (2005).
Beleza, S. et al. Micro-phylogeographic and demographic history of Portuguese male lineages. Ann. Hum. Genet. 70, 181–194 (2006).
Battaglia, V. et al. Y-chromosomal evidence of the cultural diffusion of agriculture in Southeast Europe. Eur. J. Hum. Genet. 17, 820–30 (2009).
Ramos-Luis, E. et al. Phylogeography of French male lineages. Forensic Sci. Int. Genet. Suppl. Ser. 2, 439–441 (2009).
Brisighelli, F. et al. Uniparental Markers of Contemporary Italian Population Reveals Details on Its Pre-Roman Heritage. PLoS One 7 (2012).
Martínez-Cruz, B. et al. Evidence of pre-roman tribal genetic structure in basques from uniparentally inherited markers. Mol. Biol. Evol. 29, 2211–2222 (2012).
Boattini, A. et al. Uniparental Markers in Italy Reveal a Sex-Biased Genetic Structure and Different Historical Strata. PLoS One 8 (2013).
Bučková, J., Černý, V. & Novelletto, A. Multiple and differentiated contributions to the male gene pool of pastoral and farmer populations of the African Sahel. Am. J. Phys. Anthropol. 151, 10–21 (2013).
Karachanak, S. et al. Y-chromosome diversity in modern Bulgarians: new clues about their ancestry. PLoS One 8, e56779 (2013).
Rębała, K. et al. Contemporary paternal genetic landscape of Polish and German populations: from early medieval Slavic expansion to post-World War II resettlements. Eur. J. Hum. Genet. 21, 415–22 (2013).
Larmuseau, M. H. D. et al. Increasing phylogenetic resolution still informative for Y chromosomal studies on West-European populations. Forensic Sci. Int. Genet. 9, 179–185 (2014).
Santos, C. et al. Mitochondrial DNA and Y-chromosome structure at the mediterranean and Atlantic façades of the Iberian Peninsula. Am. J. Hum. Biol. 26, 130–141 (2014).
Solé-Morata, N., Bertranpetit, J., Comas, D. & Calafell, F. Y-chromosome diversity in Catalan surname samples: insights into surname origin and frequency. Eur. J. Hum. Genet. 23, 1549–1557 (2015).
Mulero, J. J. et al. Development and validation of the AmpFlSTR Yfiler PCR amplification kit: a male specific, single amplification 17 Y-STR multiplex system. J. Forensic Sci. 51, 64–75 (2006).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Bandelt, H. J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Fenner, J. N. Cross-cultural estimation of the human generation interval for use in genetics-based population divergence studies. Am. J. Phys. Anthropol. 128, 415–23 (2005).
R core Team, 1. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (2013).
Acknowledgements
We want to thank the hundreds of volunteers who made this work possible. We would also like to thank Lara R Arauna for helpful discussion and comments, Iñigo Olalde for assistance with the raw sequence data processing and Roger Anglada for technical support. Funding was provided by the Agencia Estatal de Investigación and Fondo Europeo de Desarollo Regional (FEDER) (grant CGL2016-75389-P), and by Agència de Gestió d’Ajuts Universitaris i de la Recerca (Generalitat de Catalunya) grant 2014 SGR 866. NS is supported by a Formació de personal Investigador (FI) fellowship from Generalitat de Catalunya (FI_B00685).
Author information
Authors and Affiliations
Contributions
N.S.M., D.C. and F.C. designed the study. V.U., A.B., K.F., P.Z., F.C. and D.C. collected the data. N.S.M., A.B. and C.G.F. performed the experiments. N.S.M., C.G.F., F.C. and D.C. analysed the data. N.S.M., C.G.F., V.U., A.B., K.F., P.Z., F.C. and D.C. contributed to the interpretation of the result. N.S.M., F.C. and D.C. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Solé-Morata, N., García-Fernández, C., Urasin, V. et al. Whole Y-chromosome sequences reveal an extremely recent origin of the most common North African paternal lineage E-M183 (M81). Sci Rep 7, 15941 (2017). https://doi.org/10.1038/s41598-017-16271-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16271-y
This article is cited by
-
Archaeological Research in the Canary Islands: Island Archaeology off Africa’s Atlantic Coast
Journal of Archaeological Research (2024)
-
Challenges in the recovery of the genetic data from human remains found on the Western Balkan migration route
International Journal of Legal Medicine (2023)
-
The genomic history of the indigenous people of the Canary Islands
Nature Communications (2023)
-
Population genetic study of 17 Y-STR Loci of the Sorani Kurds in the Province of Sulaymaniyah, Iraq
BMC Genomics (2022)
-
Insights into the Middle Eastern paternal genetic pool in Tunisia: high prevalence of T-M70 haplogroup in an Arab population
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
