
Abstract
The freshwater snail Oncomelania hupensis is the unique intermediate host of the blood fluke Schistosoma japonicum, which is the major cause of schistosomiasis. The snail inhabits two contrasting environments: the hilly and marshland regions. The hilly snails are smaller in size and have the typical smooth shell, whereas the marshland snails are larger and possess the ribbed shell. To reveal the differences in gene expression between the hilly and marshland snails, a total of six snails, three per environment, were individually examined by RNA sequencing technology. All paired-end reads were assembled into contigs from which 34,760 unigenes were predicted. Based on single nucleotide polymorphisms, principal component analysis and neighbor-joining clustering revealed two distinct clusters of hilly and marshland snails. Analysis of expression changes between environments showed that upregulated genes relating to immunity and development were enriched in hilly snails, while those associated with reproduction were over-represented in marshland snails. Eight differentially expressed genes between the two types of snails were validated by qRT-PCR. Our study identified candidate genes that could be targets for future functional studies, and provided a link between expression profiling and ecological adaptation of the snail that may have implications for schistosomiasis control.
Similar content being viewed by others

Introduction
The blood fluke Schistosoma japonicum (Platyhelminth: Trematoda) occurs in China and, to a lesser extent, in the Philippines and parts of Indonesia, and human infection by the blood fluke causes a major public health problem especially in lake and marshland regions1,2. In China, up to 11.6 million people from 12 provinces have been infected by this pathogen since 19491,2,3. Although over 40 species of mammals were identified to be definitive hosts of the blood fluke1,4, only one species of animal is its intermediate host: the freshwater snail Oncomelania hupensis (Gastropoda: Pomatiopsidae)2,3. Since mid-1950s, concerted control efforts, including molluscicide treatment, biological control and social intervention, have obtained remarkable achievements in decreasing the prevalence of infected people by the blood fluke in China5,6. By contrast, the construction of the Three Gorges Super Dam has dramatically changed the natural environmental condition in southern and central China, which would greatly impact the distribution of O. hupensis and the transmission of S. japonicum 7,8. Currently, humans infected by the blood fluke were concentrated along the Yangtze River’s middle and lower reaches and around great lakes in the central China9. Because of the intimate connection between the transmission of schistosomiasis and the geographical range of O. hupensis 10,11, the control of O. hupensis remains critical in prevention of schistosomiasis7,12.
As an amphibious animal, the schistosome-transmitting snail O. hupensis mainly inhabits two contrasting environments: the marshland region and the hilly region13,14. The hilly snails have the typical smooth shell and are located in hilly/mountainous regions along the middle and lower reaches of the Yangtze River, while the marshland snails are morphologically characterized by the ribbed shell and distribute in low-lying lake/marshland regions along the middle and lower reaches of the Yangtze River13,14. Snails in hilly region possess the smaller size with a height from 5.8 to 6.9 mm, whereas those in marshland regions have the larger size with a height about 7.5 mm and sometimes over 10.0 mm14. A total of four subspecies of O. hupensis were recognized in China, namely O. hupensis hupensis, O. hupensis robertsoni, O. hupensis tangi and O. hupensis guangxiensis 9,15. However, the snails occurring along the middle and lower reaches of the Yangtze River belong to the same subspecies O. hupensis hupensis 16.
Adults of the snail O. hupensis frequently occur in fertile soils with luxuriant growth of weeds14. Different ecological factors, such as the latitude, humidity, temperature, water level, soil and vegetation, would have critical impacts on the distribution of O. hupensis. In particular, water is one of the essential factors for development and reproduction of the snails, which are difficult to survive in dry conditions17,18. Indeed, the ratio of days in waterlogging to days in bottomland is a key factor to restrict the density and distribution of O. hupensis 16. Specifically, the emergence in bottomland for months from February to May permits O. hupensis to lay eggs, and the waterlogging after May is required for their egg-hatching and the development of young snails16. As another major ecological factor affecting the development of O. hupensis, vegetation not only keeps snails warm in winter and supplies shelter from the blazing sunlight in summer, but also provides plenty of nutrients during their life cycles14.
Owing to the apparent morphological differences and ecological variations, the snails inhabiting the marshland and hilly regions are expected to respond differentially by increasing or decreasing the expression levels of genes associated with morphological and ecological variations, as shown in other aquatic animals19,20,21. Thus, transcriptome responses to environmental changes in the snail would help understand how they adapt to local environments, which may have important implications for controls against S. japonicum infection.
In this study, we used RNA sequencing technology (RNA-seq) to characterize the transcriptome profiling of the snail O. hupensis from the two distinct habitats: the hilly region and the marshland region. Our study provided a link between gene expression profiling of the snail and its ecological adaptation, and identified candidate genes that could be targets for future studies.
Results
Molecular identification of specimens
Morphological identification of the snails is straightforward, because the hilly snails are smaller (5.8–6.9 mm in height) and have the typical smooth shell, while the marshland snails are larger (7.5–10 mm in height) and have the ribbed shell13,14. To validate our morphological diagnosis of specimens, we undertook the molecular identification using the 13 mitochondrial coding sequences. Specifically, we took the 13 mitochondrial genes from each of the four subspecies of O. hupensis in China and one subspecies in Philippines (O. hupensis quadrasi) downloaded from the GenBank database, and extracted the same genes in the six snails from our transcriptome assemblies. The 13 genes were concatenated and aligned, and the resulting alignment was used to conduct phylogenetic analysis by the maximum likelihood and Bayesian approaches. Both phylogenetic approaches revealed that all hilly snails or all marshland snails formed a monophyletic group, and that all snails examined in this study were clustered with the subspecies O. hupensis hupensis rather than other subspecies (Supplementary Fig. S1 ). Moreover, the phylogenetic relationships among the four subspecies of China recovered in this study were exactly same to that inferred from 16SrRNA sequences22. Thus, our molecular evidence unambiguously suggests that the six snails studied here belong to the same single subspecies O. hupensis hupensis.
RNA-seq de novo assembly
The total number of singleton reads from all six individuals of the snail O. hupensis (three from hilly region and three from marshland region) (Fig. 1 ) was 258,878,390, with the number of singleton reads from each individual ranging from 38 to 51 million (M) (Table 1). After trimming, a total number of 3,735,164 reads with the length less than 25 base pair (bp) were discarded from all samples and the discarded reads for each sample was concentrated on a small percentage (1.3-1.56%). More than 98.4% of total reads from each sample were retained for de novo transcriptome assembly (Table 1). The de novo transcriptome assembly was generated by all retained reads from the six samples. The assembly contained a total of 564,625 contigs with an average length of 553 bp and an N50 value of 667 bp (Table 2 ). After removing the redundancies, we retained contigs with an FPKM (fragments per kilobase per million fragments mapped) value no less than two in at least two samples from either habitat. To avoid the mapping bias caused by incomplete fragements, we filtered the shorter fragmented contigs that were annotated by the same proteins, and retained the longest contigs. As such, we derived 34,760 unigenes (i.e. unique putative genes) from these contigs (Table 2 ). All unigenes have an N50 value of 1,243 bp with a mean length of 711 bp (Table 2). To visualize global transcriptomic patterns, we undertook hierarchical clustering of expression levels for all unigenes (Supplementary Fig. S2 ). It appears that all hilly samples formed a cluster while all marshland samples formed another (Supplementary Fig. S2 ). Raw transcriptome data of the six snails are available from the NCBI Sequence Read Archive under the accession number SRP103982.

Geographical locations of the sampling regions. (a) Location of Anhui province in China; The map was generated using ArcGIS version 10.2 (ESRI, Redlands, CA, USA); (b) Sampling sites in Wuhu City and Nanling County within Anhui province. The satellite images were taken from Google Earth http://earth.google.com/.
Functional annotation of the transcriptome
Of all unigenes, a total of 8,584 (24.69%) or 6,080 (17.49%) have a significant BLAST hit when annotated against NR (non-redundant) or Swiss-prot database. For GO (gene ontology) classification, NR annotated unigenes were converted to produce 3,262 GO terms via Blast2Go program23. Of the remaining 26,176 unigenes that are unannotated, 4,823 harbored a predicted open reading frame (ORF) with a length longer than 150 nucleotides. Through annotating these ORFs with the InterPro protein signature databases, we found 175 unigenes that were assigned to 82 GO terms. After removing the replicated terms generated by the Blast2Go program23 and InterProScan24, additional 16 GO terms were obtained. All these GO terms were categorized into 106 ancestral classes, and were depicted in detail (Fig. 2). Molecular functions were predominant at 37.13%, with the major categories encompassing “binding”, “catalytic activity”, “hydrolase activity” and “nucleotide binding”. Biological processes accounted for 35.17%, with “metabolism”, “protein metabolism”, “biosynthesis” and “nucleobase, nucleoside, nucleotide and nucleic acid metabolism” most presented. Cellular components possessed the lowest proportion at 27.71%, and mainly included “cell”, “intracellular”, “cytoplasm” and “nucleus” (Fig. 2).

The gene ontology categories in the transcriptome of Oncomelania hupensis. Each of three major categories was shown in bold, while all subcategories within each major category were shown in plain on the y-axis. The bar length represents the log10 transformation of unigene number, and the actual number of unigenes is shown on the x-axis. The percentage of unigene counts is presented on the right.
SNP calling and population clustering
A total of 326,685 single nucleotide polymorphisms (SNPs) were identified from the six samples. Two distinct population clusters, the marshland type and the hilly type, were recognized by the principal component analysis (PCA) (Fig. 3a) and the neighbor-joining (NJ) tree (Fig. 3b) based on all SNPs. In the PCA, the first principal component explained 29.52% while the second principal component explained 20.13% of the genetic differences (Fig. 3a). In the NJ tree, the two populations were completely separated (Fig. 3b).

Population clustering analysis. (a) Principal component analysis (PCA) of the snails. Black squares (H1-H3) refer to three individuals of hilly snails; Red triangles (M1–M3) indicate three marshland snails. (b) Neighbor-joining (NJ) tree based on all single nucleotide polymorphisms (SNPs). Black, hilly snails; Red, marshland snails.
For comparison, we generated a Multi-Dimension Scale (MDS) plot that shows the expression divergence of snails inferred from RNA-seq read counts. Consistent with the PCA plot inferred from SNPs (Fig. 3a), the MDS plot also revealed two distinct clusters of hilly and marshland snails (Supplementary Fig. S3 ). In contrast to the PCA analysis showing lower genetic divergence in hilly snails and higher divergence in marshland ones (Fig. 3a), the MDS analysis identified higher expression divergence within hilly snails and lower divergence within marshland ones (Supplementary Fig. S3 ). This disparity suggests that coding-sequence divergence and expression divergence are not coupled in the snails.
Differential gene expression between two habitat types of snails
A total of 3,456 unigenes were differentially expressed in the snails from the marshland region when compared with those from the hilly region, making up 9.94% of the total 34,760 unigenes. The numbers of upwardly expressed and downwardly expressed unigenes in the marshland and hilly snails were 1,064 and 2,392, respectively (Fig. 4a), and all differentially expressed genes were visualized by the hierarchical clustering of expression levels (Fig. 4b). Among all differentially expressed unigenes, 121 in hilly snails and 111 in marshland snails could be successfully annotated by the Swiss-prot database, respectively.

Analysis of differentially expressed unigenes. (a) Comparison between the hilly and marshland snails via RNA-seq measured in fragments per kilobase per million fragments mapped (FPKM). Each dot represents a unigene, and two green lines bracket unigenes having less than 4-fold differences in expression between environments; and the unigenes with ≥ 4-fold differences in expression were considered to be differentially expressed in this study. A total of 1,064 genes were upwardly expressed and 2,392 were downwardly expressed in hilly snails relative to marshland snails; (b) Heatmap of all 3,456 differentially expressed unigenes between the hilly and marshland snails.
For the upwardly expressed genes in the marshland snails, 65 GO terms were significantly enriched with a p-value less than 0.05 (Supplementary Table S1). Specifically, 42 GO terms such as “translation”, “peptide biosynthetic process”, “ribosome assembly” and “organonitrogen compound metabolic process” were over-represented in the “biological process” category; 11 GO terms including “structural constituent of ribosome”, “structural molecule activity”, “rRNA binding” and “translation elongation factor activity” were over-represented in the “molecular function” category (Supplementary Table S1). For the upwardly expressed genes in the hilly snails, a total of 70 GO terms were significantly enriched with a p-value less than 0.05 (Supplementary Table S2). In the “biological process” category, 36 GO terms including “immune system process”, “protein metabolic process”, and “development” were over-represented. The GO terms such as “structural constituent of ribosome”, “rRNA binding”, and “structural molecule activity” were over-represented under the “molecular function” category (Supplementary Table S2).
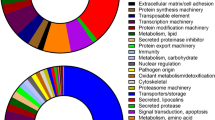
Functional enrichment analyses of GO terms for differentially expressed genes were compared between the two habitats (Fig. 5). The analysis of differentially expressed genes indicated that a large number of GO terms associated with metabolic processes and enzyme activities were enriched in snails from both habitats (Fig. 5). Furthermore, several GO terms related to larval development and immunity were significantly enriched in hilly snails (Fig. 5). By contrast, only four GO terms relating to reproduction were enriched in marshland snails (Fig. 5). The highly expressed unigenes in each habitat that could be annotated by the Swiss-prot database were listed (Table 3 ). Most genes that are highly expressed in each habitat were involved in metabolic activities (Table 3). Notably, nine genes associated with reproduction were significantly upregulated in marshland snails (Table 3). By contrast, in hilly snails, 18 and 14 upregulated genes were related to immunity and development, respectively (Table 3).

Gene ontology (GO) enrichment analysis of differentially expressed unigenes between the hilly and marshland snails. Gene number is provided adjacent to each GO term.
Validation of RNA-seq data by quantitative real-time PCR (qRT-PCR)
To verify the expression pattern shown by RNA-seq data, a total of eight differentially expressed genes between the two types of snails were selected for validation using qRT-PCR. Among these genes, four (LRP5, FAP, TEN-1, and P4HA2) were found to be highly expressed in hilly snails whereas the other four (AS3MT, ATHL1, BHMT, and PABP4) were identified to be upregulated in marshland ones by RNA-seq data (Table 3 ). Functionally, one gene (LRP5) is involved in immunity, three genes (FAP, LRP5, and TEN-1) are asscociated with development, and five genes (AS3MT, ATHL1, BHMT, LRP5, P4HA2, and PABP4) are related to metabolism (Table 3 ). Our qRT-PCR measurements for the eight genes revealed similar trends of expression changes estimated from the RNA-seq data (Fig. 6).

Expression profiles of eight differentially expressed genes. Expression levels in marshland snails were calculated as the control for each gene. Red bars represent the relative expression levels in hilly snails by RT-PCRs and grey bars indicate the relative expression levels in hilly snails by transcriptome sequencing. All relative expression values were shown with log2 (fold change) on the Y axis.
Discussion
This study characterized the de novo transcriptome assembly of the snail O. hupensis. GO term classification indicated that many unigenes are indispensable in metabolism and catalytic activities. SNP calling and population clustering suggested that genetic variation between the two habitat types within a relatively smaller geographical area was higher than that within population, which implied that habitat type might play a vital role in shaping the genetic differentiation of O. hupensis. Analysis of expression profiles showed that the majority of differentially expressed genes were related to metabolism. In addition, our study also revealed that upregulated genes involving in reproduction were enriched in marshland snails, whereas those genes relating to immunity and development were over-represented in hilly snails.
The RNA-seq generated high-quality reads for de novo transcriptome assembly of O. hupensis. For each contig cluster, the isoform with the highest expression level, measured by FPKM values, was selected as a unigene. We next filtered the shorter fragmented contigs that were annotated by the same proteins, and retained the longest contigs. The obtained contigs were selected as unigenes for subsequent analyses. Only 8,584 and 6,080 of the total 34,760 unigenes were annotated by the NR database and Swiss-prot database, accounting for 24.69% and 17.49%, respectively. The number of annotated unigenes was considerably limited, possibly because functions of many unigenes are usually specific to a certain lineage or a particular environment25. Furthermore, there were only five species in Gastropoda that had draft genome sequences. Specifically, two of the five species belong to the order Basommatophora, namely Lymnaea stagnalis 26 and Biomphalaria glabrata 27. The remaining three species Conus tribblei 28, Aplysia californica 29 and Lottia gigantea 30 belong to the orders Neogastropoda, Anaspidea, Patellogastropoda, respectively. No genome data for Pomatiopsidae had been published thus far, which may also limit the number of annotated unigenes. “Molecular function” made up the majority of GO categories that the unigenes in O. hupensis fell into, followed by “biological process” and “cellular component” (Fig. 2). While in another gastropod mollusc, the wandering snail Radix balthica, “biological process” represented the largest category; “cellular component” is the second largest category, and “molecular function” occupied the smallest proportion of unigenes31.
The transcriptome GO terms of O. hupensis were partially overlapped with those of another snail Radix balthica and several species of crustaceans31,32,33,34,35. Specifically, within the category “molecular function”, the subcategories “catalytic activity” and “binding” with the most mapped transcripts were also identified in R. balthica 28 and some crustaceans32,33,34,35. Under the category “biological process”, two subcategories “metabolism” and “cell organization and biogenesis” with a large amount of mapped transcripts were also observed in R. balthica 28. However, the subcategory “biological regulation” with a great many mapped transcripts in R. balthica 28, Oncopeltus fasciatus 30 and Benista tabaci 35 was not present in O. hupensis (Fig. 2). Of note, we cannot rule out that differences observed between O. hupensis and other species in transcriptome GO terms may have resulted from the small number of annotated unigenes derived from the transcriptome assembly.
The high-quality paired-end reads from the six snails were mapped to the reference transcriptome to identify SNPs. SNPs were then compared among samples and used to conduct the phylogenetic reconstruction and principal component analysis. According to the principal component analysis, the individuals from the same environment were clustered, implying that genetic difference between the two habitat types was relatively higher than that within one habitat type. Indeed, populations of O. hupensis from different areas had different degrees of genetic variation36. Likewise, O. hupensis from various habitats had an obviously different susceptibility against S. japonicum 37,38,39,40. Our study also indicated that the three samples from the marshland region harbored higher genetic difference than those from the hilly regions, which was consistent with earlier genetic evidence16,41. For the hilly snails, our principal component analysis showed that the hilly individuals formed a compact group (Fig. 3a ), which indicated that there was a considerable degree of sequence similarity among the hilly snails. However, the bootstrap values within the hilly snails were lower compared with those in the marshland snails, which were 100% at each node (Fig. 3b ). This observation suggests that in spite of the higher sequence similarity within the hilly snails, the number of phylogenetically informative sites in hilly snails may be smaller than that in marshland snails.
Because ecological factors, such as water level, humidity, temperature, soil and vegetation, were significantly different between the hilly and marshland environments42, regulation in gene expression can be the result of animals reacting to the environmental stresses43,44. Upwardly expressed genes in hilly snails are mainly involved in several physiological activities, including metabolism, immunity and development. Upwardly expressed genes in marshland snails are mostly concentrated on terms relating to metabolism and reproduction. A large amount of highly expressed genes in both types of the snails were enriched in GO terms relating to metabolism (Fig. 5 ). Four enriched GO terms, including “cellular amide metabolic process”, “organonitrogen compround metabolic process” and “peptide metabolic process”, are shared in both types of the snails (Fig. 5 ), which demonstrated that many genes were participated in macromolecule metabolic processes. Furthermore, several GO terms such as “carbohydrate metabolic process” and “polysaccharide metabolic process” were enriched in marshland snails, whereas GO terms such as “serine hydrolase activity” and “endocrine process” were over-represented in hilly snails (Fig. 5 ). The marshland snails harbor more highly expressed genes associating with metabolism than the hilly snails (Fig. 5 ), possibly because the larger snails in size from the marshland region tend to have a higher basal metabolic rate45.
A total of 18 genes related to immunity were highly expressed in hilly snails (Table 3). Previous studies revealed that snails from different habitats had a significant difference in the susceptibility against S. japonicum 39,40. For the subspecies O. hupensis hupensis, the marshland snails harbored a higher infection rate to the local S. japonicum than the hilly snails42. For CHIA gene, as a member of an atypical glycoside hydrolase family 18, were proved to possess a chitin-binding Peritrophin-A domain to bind and break down chitin, but also play a role in host defense against fungi, bacteria, and other pathogens in many species46,47,48. The tissue-specific expression of chitinase in triangle snail mussel Hyriopsis cumingii suggested that a chitinase-3 gene were highly expressed after shell damage49. In Crassostrea gigas, two chitinase-like proteins (CLPs) were transcriptionally stimulated in haemocytes when confronted with bacterial lipopolysaccharide challenge, also suggested that these two CLPs may play a role in immunity50.
Nine genes related to reproduction were found to be highly expressed in the marshland snails (Table 3 ). Since O. hupensis is dioecious and has an oviparous reproductive system, various environmental factors may play vital roles in mating and egg-laying42. For example, highly expressed gene ACR is a typical serine proteinase present in acrosome of mature spermatozoa and play a vital role in penetrating the zona pellucida of the ovum51. Highly expressed ACR may help the marshland snails maintain a relative higher fertilization rate than the hilly snails.
A total of 14 upregulated genes had been enriched in 13 GO terms associated with organismic development in hilly snails (Table 3 ). For example, LRP5, encoding the low-density lipoprotein receptor-related protein 5, plays an essential role in canonical Wnt pathway and skeletal homeostasis52. Several studies had indicated that LRP5 in mice and humans controls bone formation by inhibiting the expression of TPH1 (Tryptophan hydroxylase 1), a rate-limiting biosynthetic enzyme for serotonin in enterochromaffin cells of the duodenum53,54,55,56. Another highly expressed gene, NOTC3, was found to inhibit the osteoblast differentiation57,58, and also function in prenatal skeletal development and postnatal bone remodeling58. Considering the smaller size and thinner shell in hilly snails when compared with the rib-shelled marshland snails59, upregulated genes such as LRP5 and NOTC3 might play a role in the shell formation. This hypothesis awaits experimental validation in future.
Taken together, we characterized the differences in gene expression of the schistosome-transmitting snail O. hupensis inhabiting the hilly and marshland environments by RNA sequencing technology. The majority of differentially expressed genes between environments were involved in metabolism, and upregulated genes relating to development and immunity were enriched in hilly snails, while those associated with reproduction were over-represented in marshland snails. Our study identified candidate genes that could be targets for future functional studies, and provided a link between expression profiling and ecological adaptation of the snail that may have implications for schistosomiasis control.
Materials and Methods
Sample preparation and RNA isolation
Wild uninfected adults of Oncomelania hupensis used in this study were collected in Anhui Province, southeast China in October 2015. Samples were obtained from the Yangtze River marshland region in Wuhu city (geographical coordinates: 31°20′N, 118°21′E) and the hilly region in Nanling county (30°48′N, 118°13′E), respectively (Fig. 1 ). For each habitat, three individuals of the snails were used as three biological replicates in transcriptome profiling. We examined microscopically in the laboratory to check infection in each individual of snails, and those without helminthic infection were used for RNA isolation. Following the manufacture’s protocol, total RNAs were isolated using Trizol (Invitrogen, CA, USA) and were subsequently quantified by a NanoDrop spectrophotometer (NanoDrop Technologies, DE, USA).
Library preparation and RNA sequencing
Six snails were individually used to construct RNA-seq libraries using the Illumina Truseq RNA Sample Preparation Kit (San Diego, CA, USA), and then quantified by Qubit (Life Technologies) accordingly to the manufacturer’s protocol. Details of library preparation for RNA-seq were described elsewhere60,61. Six paired-end libraries with an insert size of approximately 200 bp were sequenced to generate 125 bp paired-end reads on the Illumina HiSeq. 2500 sequencing platform.
Molecular identification of specimens
To clarify the phylogenetic relationships among the six snails and determine which subspecies these samples belong to, the de novo transcriptome assembly was conducted separately for each snail. All raw sequence reads were assessed by FastQC version 10.1 (www.bioinformatics.bbsrc.ac.uk/projects/fastqc) and were processed using Trimmomatic version 0.3262 to remove the residual adaptors and low quality sequences as well as ambiguous sequences. Trimming parameter settings were detailed as follows: we removed a total of 13 bp from the start of sequence reads, with a Phred quality score of leading and trailing bases < 4, and performed a sliding window approach once the average Phred quality score dropped below 15, and discarded those sequence reads < 25 bp. Six transcriptome assemblies were processed by the program Trinity version 2.2.063 with default parameters.
TBLASTN searches64 were applied to identify the 13 mitochondrial protein coding genes from each of the six transcriptome assemblies. The same genes of four subspecies in China and one subspecies in Philippines were downloaded from the GenBank database (http://www.ncbi.nlm.nih.gov/genbank) under accession numbers as follows: O. hupensis hupensis, JF284687; O. hupensis tangi, JF284695; O. hupensis guangxiensis, JF284696; O. hupensis robertsoni, JF284697 and O. hupensis phillipine, JF284698. The 13 genes were concatenated and aligned by PRANK version 10080265 and poorly aligned position and gaps were removed by GBLOCKS version 0.91b66. According to the Akaike information criterion (AIC) and Bayesian information criterion (BIC)67, we ran the program jModelTest version 2.1.1068 to select the best-fit model for concatenated mitochondrial sequences. PhyML version 3.769 were used to reconstruct for the ML tree under the recommended GTR model with 100 replicates, and MrBayes version 3.2.670 were used to reconstruct the Bayesian tree under the GTR + I + G model with 300 million generation.
De novo transcriptome assembly and unigene annotation
Paired-end reads from all six libraries were pooled and a de novo transcriptome assembly was conducted by Trinity version 2.2.0 program63 with default parameters, and generated a considerable amount of contigs. In order to reduce the effects of erroneous contigs, we separately mapped all filtered paired-end reads from each sample to these contigs by using software RSEM version 1.1.2171. We next defined a minimum expression filter of two fragments per kilobase per million fragments mapped (FPKM), and retained contigs with FPKM greater than two in at least two samples from either habitat. For contigs annotated as the same proteins, we removed the shorter fragmented transcripts and retained the longest contigs. The obtained contigs were chosen as a reference unigene set and were remapped by all filtered paired-ended reads from each sample to quantify gene expression level. The FPKM heatmap of all unigenes from six individuals was generated using the gplots package72.
The transcriptome-derived unigene set was annotated against NR (NCBI non-redundant) and Swiss-prot databases via BLASTX64, with an e-value threshold of 1e-5. GO (gene ontology) annotation was performed by the Blast2GO program23 with the NR annotation results in XML format as the input file. For those unannotated unigene, the perl script TransDecoder in the Trinity program package was conducted to extract the predicted open reading frame (ORF) at a minimum peptide length threshold of 50. With ORFs as inputs, the program InterProScan version 4.824 was applied to search the InterPro protein signature databases and predict the possible GO terms. GO terms generated by Blast2Go23 and InterProScan24 were grouped and categorized into three main biological terms, namely “cellular component”, “molecular function” and “biological process”, using a web-based program CateGOrizer with the GO-SLIM classification method73.
SNP calling and population structure analysis
Single nucleotide polymorphisms (SNPs) were identified from filtered paired-end sequence reads by mapping each sample to the final transcriptome-derived unigene set. The mapping was undertaken using the program Burrows-Wheeler Aligner (BWA) version 0.7.874 with the “mem” method and default parameters, after the alignments were converted to the SAM format. The program SAMtools version 1.3.175 with the “view” method was used to convert the SAM format to the BAM format for downstream analyses, with removal of the potential PCR duplicates using the “rmdup” option. All alignment files in BAM format were sorted with the “sort” option, and then grouped together to identify SNPs via the “mpileup” option in SAMtools, with parameters set as “-q 20 -Q 20 -C 50 -t DP SP -m 2 -F 0.002”. The likelihood of each possible genotype was calculated and stored in BCF format via SAMtools. All retained variant sites were filtered by scripts in the BCFtools package75, with parameters set as “-d 20 -D 140 -w 5”, in order to acquire variant sites in higher quality. Subsequently, SNPs were captured through the program VCFtools version 0.1.15 package75 with the “-remove-indels” option.
Population clustering analysis was conducted using two approaches. The first approach was performed to analyze the population structure based on neighbor-joining (NJ) method. The program TreeBeST version 1.9.2 (sourceforge.net/projects/treesoft/files/treebest) was used to constructed the NJ phylogenetic tree with 1,000 bootstraps, based on concatenated SNPs from all samples. The other approach was based on nonparametric principal component analysis (PCA)76. SNPs in the binary PED format were generated via VCFtools with the “-remove-indels-plink” option, and were next used for further analysis via the program PLINK version 1.07 (pngu.mgh.harvard.edu/purcell/plink/)77. PCA was conducted using the program Genome-wide Complex Trait Analysis (GCTA) version 1.25.278, with the “-pca 2” option.
In comparison with the population clustering inferred from genetic divergence, we also generated a Multi-Dimension Scale (MDS) plot that shows the expression divergence of snails inferred from RNA-seq read counts. Using the MDS function in edgeR79, a multi-dimensional scaling plot of six snails was generated based on contig counts, with the cutoff of one count per million (CPM) at 4.
Differential gene expression analysis
The filtered paired-end reads from the six samples were separately aligned back to the final transcriptome-derived unigene set with RSEM version 1.1.2171. Gene expression was quantified as the total number of fragment counts that uniquely mapped to the unigene set. By using the matrix of fragment counts from each sample, we conducted differential expression analysis via the edgeR program79, which is a Bioconductor software package80 performed in R environment. The edgeR program could calculate whether there are significant differences in gene expression between marshland and hilly snails, and differentially expressed transcripts resulting from pairwise comparisons were extracted and clustered via TMM (trimmed means of M values across samples) normalized FPKM values by a suite of scripts in Trinity package. Differentially expressed genes (DEGs) between the two environments were identified at a significance level of 0.05, with a false discovery rate (FDR) corrected method81. Meanwhile, the fold change between the two environments was no less than 4, namely an absolute minimum value of log2-transformed fold change (logFC) equaled to 2.
Along with the selection of differentially expressed genes, an FPKM heatmap was generated via the gplots program72 performed in R environment. Differentially expressed genes were annotated against the NR/Swiss-prot database using BLASTX. Functional enrichment tests of these unigenes were analyzed using the DAVID bioinformatics resources82. Benjamini-Hochberg multiple testing correction81 was used to adjust the significance of functionnal enrichment.
Validation by quantitative real-time PCR (qRT-PCR)
A total of eight differentially expressed genes between the two types of snails were selected for validation using qRT-PCR. Among these genes, four (LRP5, FAP, TEN-1, and P4HA2) were found to be highly expressed in hilly snails whereas the other four (AS3MT, ATHL1, BHMT, and PABP4) were identified to be upregulated in marshland ones by RNA-seq data (Table 3 ). All selected genes have significant BLAST hits against the homologs in NR and Swiss-prot database and have multiple exons for designing primers crossing exon-intron junctions, aiming to eliminate the impact of DNA interference. RNA samples from each individual were the same as those prepared for cDNA library construction. Equal amounts of mRNA were used to generate the first-strand cDNAs by the reverse transcriptase polymerase chain reactions (M-MLV Reverse Transcriptase; Thermo Fisher Scientific, Wilmington, DE). The SYBR Green real-time PCR reaction was executed using the Bio-rad CFX96TM system (Bio-Rad, USA), with the protocol as follows: 95 °C for 5 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The gene β-actin in O. hupensis was also parallelly amplified as the internal control for normalization, and primers of β-actin were taken from Zhang et al. 83. Real-time PCR reactions for each gene from six snails were run in triplicate and each reaction was repeated three times, then the standard curves were achieved. The expression levels of each gene from either habitat snails were normalized to β-actin. The fold-changes were calculated through comparing the hilly snails to the marshland snails using the 2−ΔΔCt method84.
Data availability
Raw RNA-seq data have been deposited to the NCBI Sequence Read Archive under the accession number SRP103982.
References
Li, Y. S. et al. Epidemiology of Schistosoma japonicum in China: morbidity and strategies for control in the Dongting Lake region. Int J Parasitol 30, 273–281 (2000).
Tang, F. Y. et al. Spatio-temporal trends and risk factors for Shigella from 2001 to 2011 in Jiangsu Province, People’s Republic of China. Plos One 9, e83487 (2014).
Zhou, Y. B. et al. Spatial-temporal variations of Schistosoma japonicum distribution after an integrated national control strategy: a cohort in a marshland area of China. Bmc Public Health 13, 297 (2013).
He, Y. X., Salafsky, B. & Ramaswamy, K. Host-parasite relationships of Schistosoma japonicum in mammalian hosts. Trends Parasitol 17, 320–324 (2001).
Ross, A. G. P. et al. Schistosomiasis in the People’s Republic of China: prospects and challenges for the 21st century. Clin Microbiol Rev 14, 270–295 (2001).
Yuan, H. C., Jiang, Q. W., Zhao, G. M. & He, N. Achievements of schistosomiasis control in China. Mem I Oswaldo Cruz 97, 187–189 (2002).
Yi, Y. A., Xu, X. J., Dong, H. F., Jiang, M. S. & Zhu, H. G. Transmission control of schistosomiasis japonica: implementation and evaluation of different snail control interventions. Acta Trop 96, 191–197 (2005).
Seto, E. Y. W. et al. Impact of changing water levels and weather on Oncomelania hupensis hupensis populations, the snail host of Schistosoma japonicum, downstream of the Three Gorges Dam. Ecohealth 5, 149–158 (2008).
Zhou, Y. B., Yang, M. X., Zhao, G. M., Wei, J. G. & Jiang, Q. W. Oncomelania hupensis (Gastropoda: Rissooidea), intermediate host of Schistosoma japonicum in China: Genetics and molecular phylogeny based on amplified fragment length polymorphisms. Malacologia 49, 367–382 (2007).
Wang, L. D., Utzinger, J. & Zhou, X. N. Schistosomiasis control: experiences and lessons from China. Lancet 372, 1793–1795 (2008).
Dai, J. R. et al. Resistance to niclosamide in Oncomelania hupensis, the intermediate host of Schistosoma japonicum: should we be worried? Parasitology 142, 332–340 (2015).
Yang, G. J. et al. Optimizing molluscicide treatment strategies in different control stages of schistosomiasis in the People’s Republic of China. Parasite Vector 5, 260 (2012).
Jiang, Q. W. et al. Morbidity control of schistosomiasis in China. Acta Tropica 82, 115–125 (2002).
Zhou, X. N. Science on Oncomelania snail. (Science Press, 2005).
Zhou, Y. B., Zhao, G. M. & Jiang, Q. W. Genetic variability of Schistosoma japonicum (Katsorada, 1904) intermediate hosts Oncomelania hupensis (Gredler, 1881) (Gastropoda: Rissooidea). Ann Zool 58, 881–889 (2008).
Wilke, T. et al. Oncomelania hupensis (Gastropoda: Rissooidea) in eastern China: molecular phylogeny, population structure, and ecology. Acta Trop 77, 215–227 (2000).
Wang, R. B. & Zheng, J. Three Gorges Dam project and the transmission of schistosomiasis in China. Chin J Schisto Control 15, 71–74 (2003).
Wu, C. G., Zhou, X. N. & Xiao, B. Z. The relationship between changes of ecological environment after build of Three Gorges Dam and transmission of schistosomiasis. Foreigh Med Sci Parasit Dis 32, 224–228 (2005).
Latta, L. C., Weider, L. J., Colbourne, J. K. & Pfrender, M. E. The evolution of salinity tolerance in Daphnia: a functional genomics approach. Ecol Lett 15, 794–802 (2012).
Fay, S. A., Swiney, K., Foy, R. & Stillman, J. H. De novo assembly of the Paralithodes camtschaticus (red king crab) transcriptome to inform its response to ocean acidification. Integr Comp Biol 53, 281–281 (2013).
Chu, N. D., Miller, L. P., Kaluziak, S. T., Trussell, G. C. & Vollmer, S. V. Thermal stress and predation risk trigger distinct transcriptomic responses in the intertidal snail Nucella lapillus. Mol Ecol 23, 6104–6113 (2014).
Li, S. Z. et al. Landscape genetics: the correlation of spatial and genetic distances of Oncomelania hupensis, the intermediate host snail of Schistosoma japonicum in mainland China. Geospatial Health 3, 221–231 (2009).
Gotz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36, 3420–3435 (2008).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res 33, 116–120 (2005).
Asselman, J. et al. Conserved transcriptional responses to cyanobacterial stressors are mediated by alternate regulation of paralogous genes in Daphnia. Mol Ecol 24, 1844–1855 (2015).
Davison, A. et al. Formin is associated with left-right asymmetry in the pond snail and the frog. Curr Biol 26, 654–660 (2016).
Adema, C. M. et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun 8, 15451 (2017).
Barghi, N., Concepcion, G. P., Olivera, B. M. & Lluisma, A. O. Structural features of conopeptide genes inferred from partial sequences of the Conus tribblei genome. Mol Genet Genomics 291, 411–422 (2016).
Moroz, L. L. et al. Sequencing the Aplysia genome: a model for single cell, real-time and comparative genomics. Submission of intent to complete sequencing project from the Aplysia Genome project. http://www.genome.gov/Pages/Research/Sequencing/SeqProposals/AplysiaSeq.pdf (2004).
Simakov, O. et al. Insights into bilaterian evolution from three spiralian genomes. Nature 493, 526–531 (2013).
Feldmeyer, B., Wheat, C. W., Krezdorn, N., Rotter, B. & Pfenninger, M. Short read Illumina data for the de novo assembly of a non-model snail species transcriptome (Radix balthica, Basommatophora, Pulmonata), and a comparison of assembler performance. Bmc Genomics 12, 317 (2011).
Ewen-Campen, B. et al. The maternal and early embryonic transcriptome of the milkweed bug Oncopeltus fasciatus. Bmc Genomics 12, 61 (2011).
Li, C. Z. et al. Analysis of Litopenaeus vannamei transcriptome using the next-generation DNA sequencing technique. Plos One 7, e47442 (2012).
Lenz, P. H. et al. De novo assembly of a transcriptome for Calanus finmarchicus (Crustacea, Copepoda) - the dominant zooplankter of the North Atlantic Ocean. Plos One 9, e88589 (2014).
Wang, X. W. et al. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. Bmc Genomics 11, 400 (2010).
Zhou, Y. B., Zhao, G. M., Wei, J. G. & Jiang, Q. W. Analysis of genetic diversity of AFLP marker among populations of Oncomelania hupensis. Chin J Parasitol Parasit Dis 24, 7–30 (2006).
Yuan, H. C., Upatham, E. S., Kruatrachue, M. & Khunborivan, V. Susceptibility of snail vectors to Oriental anthropophilic Schistosoma. Southeast Asian J Trop Med Public Health 15, 86–94 (1984).
Hong, Q. B. et al. Susceptibility of Oncomelania hupensis from different districts and different environmental type to Schistosoma japonicum in different districts. Chin J Schisto. Control 7, 83–86 (1995).
Liu, Z. C. et al. Susceptibility of Oncomelania snails collected from three regions of Hunan Province to Schistosoma japonicum isolated from Yueyang. Chin J Schisto. Control 21, 31–34 (2009).
Gong, Z. Q. et al. Survival of Oncomelania snails from lake regions and mountain regions and their susceptibility to Schistosoma Japonicum under simulated field conditions. Acta Academiae Medicinae Jiangxi 12, 9–12 (2014).
Shi, C. H., Qiu, C. P., Xia, M. Y., Feng, Z. & Davis, G. M. Preliminary study on cytochrome C oxidase 1 gene of Oncomelania hupensis from Miaohe area in Hubei Province. Chin J Parasitol Parasit Dis 19, 41–44 (2001).
Wu, J. Y. et al. Three Gorges Dam: impact of water level changes on the density of schistosome-transmitting snail Oncomelania hupensis in Dongting Lake area, China. Plos Neglect Trop D 9, e0003882 (2015).
Schoville, S. D., Barreto, F. S., Moy, G. W., Wolff, A. & Burton, R. S. Investigating the molecular basis of local adaptation to thermal stress: population differences in gene expression across the transcriptome of the copepod Tigriopus californicus. Bmc Evol Biol 12, 170 (2012).
De Wit, P. & Palumbi, S. R. Transcriptome-wide polymorphisms of red abalone (Haliotis rufescens) reveal patterns of gene flow and local adaptation. Mol Ecol 22, 2884–2897 (2013).
Marsden, I. D., Shumway, S. E. & Padilla, D. K. Does size matter? The effects of body size and declining oxygen tension on oxygen uptake in gastropods. J Mar Biol Assoc U.K. 92, 1603–1617 (2012).
Suzuki, M. et al. Cellular expression of gut chitinase mRNA in the gastrointestinal tract of mice and chickens. J Histochem Cytochem 50, 1081–1089 (2002).
Donnelly, L. E. & Barnes, P. J. Acidic mammalian chitinase - a potential target for asthma therapy. Trends Pharmacol Sci 25, 509–511 (2004).
Wiesner, D. L. et al. Chitin recognition via chitotriosidase promotes pathologic type-2 helper T cell responses to cryptococcal infection. Plos Pathog 11, e1004701 (2015).
Wang, G. L., Xu, B., Bai, Z. Y. & Li, J. L. Two chitin metabolic enzyme genes from Hyriopsis cumingii: cloning, characterization, and potential functions. Genet Mol Res 11, 4539–4551 (2012).
Badariotti, F., Lelong, C., Dubos, M. P. & Favrel, P. Characterization of chitinase-like proteins (Cg-Clp1 and Cg-Clp2) involved in immune defence of the mollusc Crassostrea gigas. Febs J 274, 3646–3654 (2007).
Tranter, R., Read, J. A., Jones, R. & Brady, R. L. Effector sites in the three-dimensional structure of mammalian sperm beta-acrosin. Structure 8, 1179–1188 (2000).
Williams, B. O. & Insogna, K. L. Where Wnts went: the exploding field of Lrp5 and Lrp6 signaling in bone. J Bone Miner Res 24, 171–178 (2009).
Yadav, V. K. et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell 135, 825–837 (2008).
Frost, M. et al. Patients with high-bone-mass phenotype owing to Lrp5-T253I mutation have low plasma levels of serotonin. J Bone Miner Res 25, 673–675 (2010).
Modder, U. I. et al. Relation of serum serotonin levels to bone density and structural parameters in women. J Bone Miner Res 25, 415–422 (2010).
Rosen, C. J. Breaking into bone biology serotonin’s secrets. Nat Med 15, 145–146 (2009).
Long, F. X. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Bio 13, 27–38 (2012).
Engin, F. & Lee, B. NOTCHing the bone: insights into multi-functionality. Bone 46, 274–280 (2010).
Zhao, Q. P., Jiang, M. S., Littlewood, D. T. J. & Nie, P. Distinct genetic diversity of Oncomelania hupensis, intermediate host of Schistosoma japonicum in mainland China as revealed by ITS sequences. Plos Neglect Trop D 4, e611 (2010).
Lin, G. H. et al. Transcriptome sequencing and phylogenomic resolution within Spalacidae (Rodentia). Bmc Genomics 15, 32 (2014).
Wang, K., Hong, W., Jiao, H. & Zhao, H. Transcriptome sequencing and phylogenetic analysis of four species of luminescent beetles. Sci Rep 7, 1814 (2017).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990).
Loytynoja, A. & Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320, 1632–1635 (2008).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17, 540–552 (2000).
Posada, D. & Buckley, T. R. Model selection and model averaging in phylogenetics: advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53, 793–808 (2004).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. j ModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9, 772–772 (2012).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321 (2010).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323 (2011).
Warnes, G. R. et al. gplots: various R programming tools for plotting data (2016).
Hu, Z. L., Bao, J. & Reecy, J. M. CateGOrizer: a web-based program to batch analyze gene ontology classification categories. Online Journal of Bioinformatics 9, 108–112 (2008).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Menozzi, P., Piazza, A. & Cavallisforza, L. Synthetic maps of human gene-frequencies in Europeans. Science 201, 786–792 (1978).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575 (2007).
Yang, J. A., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, 80 (2004).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57, 289–300 (1995).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57 (2009).
Zhang, S. H. et al. Three goose-type lysozymes in the gastropod Oncomelania hupensis: cDNA sequences and lytic activity of recombinant proteins. Dev Comp Immunol 36, 241–246 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25, 402–408 (2001).
Acknowledgements
The authors thank Kai Wang and Jin-Wei Wu for help in data analysis and are grateful to Hai-Ning Du and Wen-Jie Shu for technical assistance in the lab. This work was supported by the key scientific research fund from Wannan Medical College (WK2015Z02).
Author information
Authors and Affiliations
Contributions
J.S.Z. and A.Y.W. collected the samples and participated in data analysis; H.B.Z. designed the study and revised the manuscript; Y.H.C. analyzed the data and wrote the manuscript. All authors gave final approval for publication.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, JS., Wang, AY., Zhao, HB. et al. Transcriptome sequencing and differential gene expression analysis of the schistosome-transmitting snail Oncomelania hupensis inhabiting hilly and marshland regions. Sci Rep 7, 15809 (2017). https://doi.org/10.1038/s41598-017-16084-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-16084-z
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
