
Abstract
To further obtain insights into the Rhipicephalus microplus transcriptome, we used RNA-seq to carry out a study of expression in (i) embryos; (ii) ovaries from partially and fully engorged females; (iii) salivary glands from partially engorged females; (iv) fat body from partially and fully engorged females; and (v) digestive cells from partially, and (vi) fully engorged females. We obtained > 500 million Illumina reads which were assembled de novo, producing > 190,000 contigs, identifying 18,857 coding sequences (CDS). Reads from each library were mapped back into the assembled transcriptome giving a view of gene expression in different tissues. Transcriptomic expression and pathway analysis showed that several genes related in blood digestion and host-parasite interaction were overexpressed in digestive cells compared with other tissues. Furthermore, essential genes for the cell development and embryogenesis were overexpressed in ovaries. Taken altogether, these data offer novel insights into the physiology of production and role of saliva, blood digestion, energy metabolism, and development with submission of 10,932 novel tissue/cell specific CDS to the NCBI database for this important tick species.
Similar content being viewed by others
Introduction
The cattle tick Rhipicephalus microplus is a one-host tick, which preferentially feeds on bovines, and it is considered the most harmful cattle parasite1,2. The economic losses associated with R. microplus parasitism are due to direct effects of the tick itself, which causes skin injuries and long-standing blood loss, leading to anemia and reduction of both weight gain and milk production, or are produced indirectly via transmission of tick-borne pathogens such as Babesia spp. and Anaplasma marginale3. In spite of its huge impact on the economy, current tick control strategies still rely mostly on the use of chemical acaricides, even though selection of resistant tick populations to major used acaricides has been confirmed4,5. This is recognized as a worldwide drawback to successful tick control. Immunization of cattle against R. microplus and other ticks has been recognized as an alternative approach against chemical control strategy6. Thus, a deeper understanding of tick physiology is needed as a means to find molecular targets that can be useful in the development of novel tick control methods.
Transcriptomic analyses have contributed with valuable information regarding the physiology of several parasites, including ticks7,8. The genomes of Ixodes persulcatus, Ixodes scapularis, Haemaphysalis longicornis, Dermacentor silvarum, Hyalomma asiaticum, Rhipicephalus sanguineus, and Rhipicephalus microplus have been sequenced and now are helpful resources for studying tick physiology and biology9,10,11. The genome of the cattle tick R. microplus was estimated to be 7.1 Gbp in length and consists of approximately 70% repetitive DNA12. Recently, a cattle tick genome draft was reported13 using a hybrid sequencing and assembly approach to capture the repetitive fractions of the genome. According to this study, the R. microplus genome assembly is composed of 51.4% repetitive sequences, containing 38,827 putative R. microplus gene loci, of which 24,758 are protein coding genes13. In spite of the large number of loci and of the significant advance provided by this genome assembly, based on the proportions of highly conserved single copy genes found in all arthropods, the degree of completeness of this assemblage was estimated to be about 41%, still leaving a vast open field for gene discovery in this organism. Recently, a remarkable publication reported the genome assembly of six tick species, including that of R. microplus11. A BUSCO analysis of the predicted proteome of this tick indicated a 51.8% of complete and single BUSCOs, an improvement over the assembly of Barrero et al.13.
Currently, there are several studies describing the genome9,10,11,13, transcriptomes14,15,16,17, and proteomes of ticks18,19. Based on these datasets, many different experiments have been carried out that elucidated tick-host20 or tick-pathogen interactions18,19, and can support the development of new control strategies15. Taken together, these reports highlighted the complexity of tick physiology mechanisms comprising both known and novel proteins participating in multiple cellular pathways. Recently, one study in Ixodes ricinus transcriptome shows that the genes involved in cuticle formation, chitin metabolism, and blood digestion enzymes are more related to fed stages. The basic energy metabolism pathway genes are more actively expressed during unfed stages, egg development, and embryogenesis21. In another recently published study, an integrated R. microplus transcriptome and proteome analyses of larvae and salivary glands of nymphs, males and females feeding on susceptible and resistant bovine hosts showed that the expression of genes involved in the host-parasite interaction are associated with host immune activation profile22.
Here, aiming to obtain a further insight into R. microplus transcriptome, we used RNAseq to carry out a study of tissue differential gene expression: (i) embryos from 1, 3, 5, 7, 9, 11, 13 day-old eggs; (ii) ovaries from partially and fully engorged females; (iii) salivary glands from partially engorged females; (iv) fat body from partially and fully engorged females; and (v and vi) digestive cells from partially and fully engorged females. Reads from each library were mapped back into assembled transcriptome, giving us a view of gene expression in different tissues. Taken altogether, this study offers additional transcriptomic data for this important tick species. The data illustrate the dynamic gene expression changes during the parasitic phase and the embryo development of R. microplus and reveals sets of genes that have a highly tissue-specific expression profile, pointing to pathways that may perform essential roles in the physiology of these tissues. These findings could be useful for future studies of tick physiology, contributing to the development of new tick control methods, as well as new biotechnological and pharmacological applications.
Results and discussion
An overview of R. microplus transcriptome
In this study, aiming to obtain an insight into the differential gene expression profiles associated with major aspects of the physiology of Rhipicephalus microplus, we used RNA-seq to carry out a transcriptome study:(i) embryo from 1, 3, 5, 7, 9, 11, 13-day-old eggs (EMB); (ii) ovaries (OV) from partially and fully engorged females; (iii) salivary glands (SG) from partially engorged females; (iv) fat bodies (FB) from partially and fully engorged females; and (v) digestive cells from partially engorged females, and (vi) fully engorged females (DIG-P and DIG-F, respectively). We obtained > 500 million raw reads, and after removal of Illumina primers and trimming of low quality bases, we obtained > 190 million high quality reads (Table 1). High-quality clean reads were assembled de novo using Abyss and Trinity assemblers. The resulting assemblies were then assembled together using a pipeline of iterative and parallelized BLASTn and CAP3; where BLASTn with decreasing word sizes (from 300 to 60) fed the CAP3 assembler through 15 iterations, producing 179,859 contigs longer than 150 bp and with more than 5 reads (Table 2). CDS were extracted based on similarities to known proteins on public databases or by presence of a signal peptide sequence14. Using this strategy, a total of 20,900 CDS larger than 150 nucleotides (nt) were obtained (Table 2), and 18,857 of which were annotated in different functional categories (Table 3 and Table S2). While some contigs were truncated and/or fragmented, 10,932 sequences were over 50% full length (based on their similarities to available Acari proteins) and were submitted to the NCBI (GHWJ01000000). A BUSCO analysis23 of the transcriptome indicated 54% complete and single copy BUSCOs, a better result than the 41% found for the assembly of Barrero et al.13 and 51.8% found for the assembly of Jia et al.11.
The 18,857 CDS identified in the current study were searched against the predicted translated sequences from the R. microplus genome13 (24,758 CDS) using BLASTP. Over 9198 (48%), 10,308 (54%), and 13,692 (72%) of protein-coding sequences in the transcriptome had at least one significant BLASTP hit when using E-values < 1e–10, < 1e–5, or < 1, respectively (Table S1).
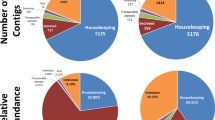
Reads from each library were mapped back into the assembled transcriptome and provided a view of gene expression in different tissues (Figs. 1, 2, and Table S2). Although reads from the synganglion were used for assembly, data from this tissue were not used for expression analysis in this study. Functional classification of the transcriptome by total number of CDS or according to normalized RPKM values for each class (Figs. 1, 2, and Tables S1–S8) highlighted the complexity of the resulting assembly and helped to visualize the significant metabolic alterations that occur in these tissues during feeding. Accordingly, the secreted class contained 17% of the total contigs and normalized reads per kilobase per million (RPKM). CDS without a significant match in all databases (referred to as unknown—UK—in Table S1) and transcripts with a significant match in at least one database, but that code a protein without a known function (referred to as unknown conserved—UC—in Table S1), were classified simply as unknown; this class contained 29.5% of the CDS and 35.1% of total normalized RKPM (Table 3).

Functional classification of extracted coding sequences (CDS) from the Rhipicephalus microplus transcriptome. In CDS, the proportion of the total number of different CDS encoding a polypeptide of the same predicted function are compared to the total number of CDS found in our transcriptome. In RPKM, the proportion of the total RPKM number of different sequence reads assigned to CDS encoding a polypeptide of the same predicted function are compared to the total RPKM number of mapped sequences in our transcriptome.

Differential expression of Rhipicephalus microplus transcriptome. Heatmap of normalized RPKM data from embryos from 1, 3, 5, 7, 9, 11, 13 day-old eggs (EMB), ovaries from partially and fully engorged adult females (OV), salivary glands from partially engorged adult females (SG), fat bodies from partially and fully engorged adult females (FB), digestive cells from partially engorged adult females (DIG.P), and digestive cells from fully engorged adult females (DIG.E). The color scale “Value” accounts for the Z-score deviation from the mean by standard deviation units.
To visualize the full breadth of differential expression between tissues and time points, a heatmap was constructed using normalized RPKM values for CDS with an average of total RPKM from all tissues ≥ 1 (considering the reads of all libraries), in order to avoid inclusion of poorly expressed contigs, totaling 16,240 CDS. The data presented a characteristic transcriptional pattern for each tissue, revealing a clear tissue-specific clustering of overexpressed genes in all samples (Fig. 2).
The cattle tick ovary and embryo transcriptome
Ovary, oogenesis and vitellogenesis
Embryo development of oviparous organisms relies on several different compounds, such as proteins, lipids, sugars and other molecules stored in yolk granules24. Oogenesis and vitellogenesis are essential processes for tick reproduction, including production of protein precursors and other molecules which are transported and accumulated inside the oocytes to ensure the embryo development25.
Data presented on a heatmap (Fig. 2) reveal the ovary as the tissue with a higher number of differentially expressed contigs (lower and upper right corner of the heatmap). To get a better insight on genes that are highly expressed in tick ovary, a total of 1091 transcripts, that are five times more expressed in ovary than in all combined tissues, were identified and categorized according to their putative function (Table S2). Transcripts coding for secreted proteins represent almost 17.4% of the total normalized RPKM in the ovary transcriptome. This functional class includes (i) proteins related to detoxification such as superoxide dismutase and arylsulfatase B, (ii) proteins with antimicrobial properties such as microplusin and ixoderin, (iii) proteases, (iv) proteins involved in signal transduction, and (v) proteins with unknown functions (Table S2).
Several transcripts coding for proteins with a well-known physiological role in oogenesis and vitellogenesis were identified in this study, including the predictable finding of several vitellogenin (Vg) transcripts (Rm-2788, Rm-23965, Rm-23966, Rm-23967, Rm-12079 and Rm-72548), which were highly expressed in fat body and in gut digestive cells26. This finding is consistent with the previous identification of the gut as an accessory site of Vg synthesis in ticks, in contrast to insects where Vg is mainly synthesized by the fat body. After being secreted to the hemolymph, the circulating Vg is uptaken by the growing oocytes, where it is named as vitellin (Vt) to highlight that some alterations are introduced in this storage lipoprotein after endocytosis. Vt is a major source of raw materials that support embryo growth. Proteases involved in Vt digestion in R. microplus have been described25,27,28,29,30,31,32,33. Interestingly, these enzyme transcripts are not abundant in ovaries, but are highly expressed in gut cells and fat body (Table S1, Tables S5–S8). This occurs because the ovary uptakes proteins synthesized and secreted in the hemolymph by other tissues. This phenomenon is suggested by data found here and is in accordance with previous studies25,31,32,34.
Transcripts coding for proteases involved in Vt digestion are overexpressed, such as the vitellogenin-degrading cysteine endopeptidase (VTDCE—Rm-15854), which was not significantly found in the ovary, and was overexpressed in DIG-F and FB (Table S1). The VTDCE synthesis by the gut and fat body, followed by protein export through hemolymph to the ovary was previously described25. Other examples are the tick heme-binding aspartic proteinase (THAP—Rm-14882) and Boophilus yolk-pro-cathepsin (BYC—Rm-849). From all the cathepsin-B coding transcripts with a role in oogenesis, only one (Rm-23945) is overexpressed in the ovary when compared with other tissues, suggesting a role in Vt degradation. A distinct pattern is observed for the Vg receptor (VgR—Rm-80856), since its transcript was expressed mostly in the ovary. The VgR is responsible for Vg uptake; the main reserve protein stored in oocytes for embryo development35.
Transcripts encoding for proteins involved in nuclear regulation and export from the nucleus such as cell division control protein, chromatin assembly and modification-related proteins, enzymes involved in DNA synthesis and repair, and histone-related proteins represent about 18% of total RPKM for the ovary. Similarly, proteins involved in signal transduction correspond to about 6% of total RPKM (Table S2), including enzymes such as acetylcholinesterase, juvenile hormone acid methyl transferase-like, kinases as serine/threonine-protein kinase and cyclin-dependent protein kinase, and amidases (Table S1). To date, juvenile hormones similar to those found in insects were not found in ticks29,36. However, a sequence coding to a transcript that is highly homologous to a juvenile hormone acid methyltransferase is overexpressed in the ovary (Rm-49336).
The analysis of expression of the detoxification genes (Fig. 3) in all organs revealed a specific profile for ROS metabolism in the tick ovary. Out of the 13 transcripts coding for cytosolic Cu, Zn superoxide dismutases (SODs), 12 showed a marked overexpression in the ovary. A similar profile was found for two (out of four) thioredoxin reductase transcripts, four thioredoxin peroxidases (out of five transcripts for this enzymatic activity), and one nucleoredoxin. Although highly expressed also in digestive cells, gamma-glutamyl cysteine synthetase and glutathione synthetase, together with thioredoxin and a phospholipid-dependent glutathione peroxidase, showed high expression in the ovary as well (Fig. 3). This suggests, that among all tick tissues analyzed in this study , the ovary presents an especially robust redox metabolism; the major effectors would be the Cu, Zn-SODs, thioredoxin, thioredoxin peroxidases, and a phospholipid hydroperoxide glutathione peroxidase, fueled by reductive potential provided by a glutathione/thioredoxin couple, which is two pools of energy bridged by thioredoxin reductase.

Expression analysis in the transcriptome of Rhipicephalus microplus. The graphs represents the antioxidant genes expression by each tissue. SG (salivary glands from partially engorged adult females) and FB (fat bodies from partially and fully engorged adult females). Transcript levels are expressed as RPKM.
The presence of a robust flow of antioxidant potential calls for an equally high source of reactive oxygen species. The fact that two transcripts coding for Duox type NADPH oxidases also have a predominant expression in ovaries make these proteins a candidate for this role. Interestingly, this enzyme has been ascribed a role as the source of hydrogen peroxide for chorion protein cross-linking in an insect37, a role that could well be assigned to these tick homologs.
Duox enzymes also have been ascribed a central role in the control of gut microbial symbionts in several insect species38,39. In ticks, the ovary is the residence of intracellular bacteria symbionts that are essential to tick development40,41,42. However, Guizzo et al.43 recently reported a quantitative analysis of the R. microplus microbial community revealing that, different from most animals, the tick gut harbors a very modest (if any) resident microbiota, in contrast to the ovary which has a large population of a Coxiella mutualist symbiont, that increase following the blood meal, paralleling the course of ovarian growth43. Interesting, Amblyomma sculptum and Amblyomma aureolatum have distinct transcriptional profiles of gut44 and microbiota composition45 that can be related to susceptibility to Rickettsia rickettsii.
Therefore, one possibility is that regulation of the expansion of the R. microplus microbial population in the ovary might also involve production of reactive oxygen species, making it essential for a panel of antioxidants to maintain intracellular redox homeostasis. This last hypothesis is in accordance with the identification of seventeen transcripts showing similarity to the antimicrobial peptide microplusin (Table S9). Levels of microplusin mRNA gradually increase along ovary development, reaching an impressive highest value three days after the adult females have dropped from the hots and start oviposition46. Moreover, microplusin is also present in hemolymph47. Importantly, it was recently reported that microplusin plays a role in protecting the tick Amblyomma aureolatum from infection with the tick-borne Rickettsia rickettsii48.
Research on insect reproduction has characterized many physiological and molecular properties of ovary and egg development49. By contrast, much less is known about these mechanisms in ticks19,50. In the ovary, this process is characterized by intensive cell division that determines oocyte and follicle cell development, and lately, the egg number. In this context, cell cycle regulation is well conserved in all organisms from yeast to humans51. Protein kinases participate in the regulation of cell division cycle controlling different pathways in this process. Cyclin-dependent kinase (CDK), aurora kinases, polo-like kinases, and checkpoint kinases (Bub1, BubR1, and Mps1) are the most important protein kinases related to cell division. Figure S1 shows the abundance of total CDK contigs by tissue. As expected, based on its function, the ovary has the highest abundance (55%) of CDK contigs. The CDKs 1, 5, and 10 are the most abundant sequences, while cyclin A-coding contigs represent the major cyclin observed in tick tissues, including ovaries (Figure S1). CDKs were previously characterized in R. microplus and I. persulcatus ticks52,53. Yet regarding control of cell fate and development, nucleoredoxin, a member of the thioredoxin family known to regulate the Wnt/beta-catenin signaling pathway in a redox-dependent way, also showed a high expression level in the ovary54.
Embryogenesis
Comparative analysis of embryo allowed the identification of 347 transcripts that are five times overexpressed in embryos than in other analyzed samples (Table S3). Interestingly, genes expressed in the embryo frequently were not five times more expressed than the sum of the other analyzed samples, possibly as a consequence that in embryo several different genes in diverse tissues and organs are being simultaneously expressed. Concerning the metabolism, 90 transcripts involved on metabolism of carbohydrates, lipids, and amino acids are transcribed in embryos. Among them, 25 transcripts are highly abundant, including enzymes of glucose metabolism; mainly glycolysis, gluconeogenesis, and pentose phosphate pathway. Interestingly, these genes were not highly expressed in ovaries, suggesting that these genes are transcribed during embryogenesis, and the mRNA or proteins are not maternally inherited.
Among these 347 embryo-specific transcripts, those encoding transposons, lipocalins, glycine-rich proteins, and secreted proteins are the most abundant. These secreted proteins are homologous to genes previously described as defensins or genes involved in immune response. Some of these secreted molecules specifically expressed during embryogenesis could be trans-peptides involved in immune response during embryogenesis. Such a finding would be important for embryo survival, since chelicerates such as ticks do not display an extraembryonic enveloping protective layer such as the serosa, which is involved in embryonic immune response in insect egg. Conversely, protective layers have independently evolved in other chelicerates such as scorpions55,56,57. In insects, these protective cellular layers prevent the loss of water and secrete defensins against bacterial infections; these protective layers are absent in tick embryonic development. These secreted proteins may have a role in immune protection during the long embryogenesis of R. microplus, which takes about 21–22 days in a potentially harmful environment 58.
The cattle tick sialotranscriptome
As a blood-feeding parasite, R. microplus saliva is a complex mixture rich in bioactive compounds that modulate host hemostasis and immunological defenses to allow tick feeding activity59,60. In recent decades, cumulative information from transcriptomic and proteomic analyses of salivary glands and tick saliva of several tick species have provided insights into the immunological interactions at the tick–host interface. In addition to facilitating blood-feeding, the antihemostatic and immunomodulatory activities of tick saliva may also support survival and establishment of hemopathogens in the host61. R. microplus larvae attach to its host and then begin to feed and molt in nymphs before molting into immature adults, a process that takes around 12 days59. After mating, adult partially engorged females take larger blood meals to complete the maturation within 21–22 days and detach59,62. In the current study, SG were dissected from adult females feed on the host during 17 and 22 days. Therefore, data shown here combine part of the slow feeding phase and of the final rapid feeding phase. Consistent with reports that other tick species change salivary composition during feeding63,64,65,66,67, data in this study reveal that 320 transcripts are overexpressed in SG when compared to other tissues, including contigs encoding metalloproteases, proteinase inhibitors, and lipocalins (Table S1 and Table S4).
Tick metalloproteases have been suggested as participating in platelet disaggregation and blood coagulation by fibrinolytic and gelatinase activities, thereby facilitating blood feeding68. The observation that members of family M12 metalloproteases, similar to hemorrhagic proteases of snake venom, are overexpressed in the SG of R. microplus, suggest they have a role of this class of protein in tick feeding physiology. Snake venom M12 proteases were associated with hemorrhaging, edema, hypotension, hypovolemia, inflammation, and necrosis69. I. scapularis saliva has a specific metalloprotease similar to the hemorrhagic protease of snake venoms displaying gelatinase and fibrin(ogen)olytic activities68. In a related study, RNAi silencing of metalloproteases from I. ricinus impaired blood meal feeding and egg laying70. In R. microplus, vaccination with recombinant metalloprotease 4 (rBrRm‐MP4) significantly decreases the number of ticks that complete the life cycle on the host and egg hatching, providing an overall protection of 60% against tick infestation65.
Protease inhibitors, including Kunitz, TIL (trypsin inhibitor-like), and cystatin were overexpressed in SG (Table S1 and Table S4). Most Kunitz protease inhibitors target serine proteases, although some also block ion channels71. In ticks, some protease inhibitors have several Kunitz domains in tandem, and they are classified as monolaris, bilaris, trilaris, and so on, according the number of Kunitz domains. In R. microplus, 11 Kunitz protease inhibitors are overexpressed in the salivary glands, including nine monolaris and two bilaris (Table S1). Several members of Kunitz family have been functionally characterized as anti-blood clotting agents such as savignin72, the antithrombin boophilin73, as well as ixolaris, an anticoagulant that binds both factor VIIa and factor X74. Interestingly, in R. microplus transcriptome these contigs encoding Kunitz proteins are overexpressed mostly in salivary gland, as expected, but also showed high expression levels in digestive cells, which is in accordance with inhibition of blood clotting being supported not only by saliva but also being reinforced after blood ingestion by secretion by digestive cells. Cystatins are ubiquitous, reversible, and tight-binding inhibitors of cysteine proteases. Some tick salivary cystatins have immunosuppressive and anti-inflammatory activities75. A total of 27 cystatin coding sequences were identified in this study, and one contig is overexpressed in salivary glands. Proteins containing TIL (trypsin inhibitor-like) signature domains have been reported in blood-feeding mosquitoes and tick sialomes76. In R. microplus, TIL family members include peptides closely related to tick elastase inhibitors, which also have antimicrobial activity77, with a total of three contigs overexpressed in SG (Table S1 and Table S4).
Lipocalins are single modular proteins of around 200 amino acids that fold tightly in a β-barrel with potential for binding small hydrophobic molecules in a central pocket. Annotation of the most recently identified tick lipocalins is based on homology with annotated histamine-binding proteins from other tick species, displaying the characteristic PFAM tick histamine-binding domain (PF02098)78. Contigs matching the tick histamine-binding domain were identified being overexpressed in SG of R. microplus (Table S1 and Table S4). The high abundance of lipocalins was also confirmed in the proteome of R. microplus saliva60. Some of these R. microplus identified lipocalins have similarities with some lipocalins described in other ticks, which have antihemostatic and immunomodulatory activities79 such as the capacity to bind biogenic amines, which is compatible with tick saliva anti-hemostatic and immunomodulatory roles during blood feeding.
Digestive cells
A unique feature of tick physiology that differs from hematophagous insects is that blood digestion is entirely intracellular, occurring inside a differentiated cell lineage: the so-called digest cells (DIG)80. After erythrocyte lysis, blood proteins are taken up by endocytosis and digested in acidic vesicles by lysosomal hydrolases81. To address this singularity of the biology of ticks, we prepared cDNA libraries from DIG that were isolated from (1) partially engorged adult females (DIG-P) that were still engaged in the process blood feeding and were in the “big sip” stage, and (2) fully engorged females (DIG-F) that had already completed blood meal and have detached from the host. To our knowledge, this is the first transcriptomic analysis of this cell type, as other previous studies on gene expression in tick gut have used whole organ extracts16,82,83.
Taken together, the analysis of these libraries showed 634 sequences that are overexpressed in DIGs (Table S6). From those, only three specific groups of functionally assigned transcripts accounted for more than 70% of the RPKMs from the most overexpressed contigs, namely secreted (61%), immunity-related (5%), and unknown conserved (6%). This marked departure from the class distribution profile obtained when all libraries are pooled together is probably a consequence of the unique physiology of this cell type exclusively found in ticks. Interestingly in DIG-P (Table S7), the most abundant contigs are classified as secreted (78.5%), unknown (12.1%), unknown conserved (3.2%), and immunity-related (2.4%). However, this profile changes to secreted (46.1%), oxidant/detoxification metabolism (14.3%), protease inhibitor (12.6%), and immunity-related (11.4%) in DIG-F (Table S8), possibly reflecting the alteration in metabolism and functions of the DIG, which must deal with a pro-oxidant menace represented by hemoglobin proteolysis and heme release. The most highly expressed transcript (Rm-24035) in this group is homologous to the second most highly expressed transcript in the transcriptome of the Dermacentor variabilis gut84 and accounts alone for close to 20% of the total digestive secretome RPKM of R. microplus (Table S1, Tables S6–S8).
Vertebrate blood is ~ 85% protein in dry weight, and therefore proteases are expected to be highly expressed in DIG. As a consequence of the intracellular mode of digestion of the blood meal, proteases found highly expressed in the transcriptome are acid lysosomal hydrolases such as legumains, cysteine, and aspartic proteinases, coherently with previous reports80,85,86. Notably, among all tissues analyzed here, the most highly expressed proteases (with signal peptide, suggesting that targets the protein to a secretory pathway) were found in the DIGs libraries. Noticeably, although several inhibitors of cysteine proteinase (cystatins) were identified, these were nearly ten times less expressed than inhibitors of serine proteases. These inhibitors can be related to the inhibition of mammalian proteases involved in blood coagulation and complement cascades87, acting as a regulator of blood digestion in gut88 and modulation of the tick immune proteolytic cascades, that have been ascribed relevant roles in the interaction with the microbiota and with pathogens89. However, it is also possible to speculate that part of these inhibitors might be negative regulators of hemoglobin degradation, as the proteolytic activity involves the release of large amounts of heme, a pro-oxidant molecule90. Keeping the pace of hemoglobin degradation with heme detoxification should be of paramount importance for this cell. Regarding this topic, it is relevant to observe that in DIGs a significant expression of three transcripts coding for an aspartic proteinase, whose activity was shown to be inhibited by heme (THAP, for tick heme-binding aspartic proteinase), was previously described as related to oogenesis28. In this study, THAP was found almost exclusively in the gut (97% of the RPKM); a result that makes it very likely that THAP may also play a relevant role in dietary hemoglobin degradation. However, we also identified the presence of two other contigs of probable paralogous enzymes (Rm-14881 and Rm-14882, Table S1) that are predominantly expressed in the fat body, which might code for the protein originally found in eggs and for this reason ascribed a role only in yolk degradation28,32.
The detoxification-related functional class is one of the most overexpressed class in DIGs (Table S1 and Tables S6–S8). For most hematophagous insects, it is assumed that the main dietary pro-oxidant component is the abundant heme and iron from the vertebrate blood, but transcriptomes of mosquitoes, triatomine bugs, and other tick species also showed high expression levels of cytochrome P-450 (CYP 450) and glutathione S-transferases (GSTs)64,91,92. These classical phase I and phase II detoxification enzymes belong to large gene families with multiple copies in the genome of insects and ticks, having essential roles in different pathways, especially in lipid metabolism. Taken all together, the 121 CYP 450s transcripts show a high RPKM value in all tissues analyzed here, including the DIG libraries. Several individual transcripts, however, showed a very tissue-specific expression pattern, including 29 CYP 450 transcripts that are at least fivefold more expressed in the DIG than in all other libraries taken together (Tables S6–S8). This finding highlights that each tissue needs a CYP 450 composition fitted to its own metabolic demands (Fig. 4A). In contrast, 80% of the total RPKM for all 59 GST transcripts are expressed in the DIG libraries, particularly in DIG from fully engorged females (Fig. 4B). This is largely due to only two GST transcripts that are members of the N delta epsilon subfamily, are very highly expressed in the digestive cells (Rm-18369 and Rm-2328), and show increased expression after the blood meal. Similarly, two GST transcripts were found in the gut and ovaries of the tick D. variabilis, and they were up-regulated in Rickettsia-infected and upregulated upon tick feeding93. Interestingly, a microarray analysis of the effect of Babesia bovis infection on whole gut gene expression83 identified one CYP 450 as the most highly upregulated transcript and a GST as one of the down-regulated genes.

Expression analysis in the transcriptome of Rhipicephalus microplus. The graphs represents the expression of detoxification-related genes in each tissue. SG (salivary glands from partially engorged adult females) and FB (fat bodies from partially and fully engorged adult females). Transcript levels are expressed as RPKM.
A putative secreted ferritin (Rm-68449) was highly transcribed in DIGs, similar to I. ricinus Fer 2, which knockdown leads to high tick lethality94, revealing an essential role in the absorption and transport of iron. It was previously shown that ticks do not synthesize heme95 and also do not have heme oxygenase, which cleaves the heme porphyrin ring and releases the iron atom in most organisms, rendering it available. As a consequence, ticks are not able to obtain iron from heme breakdown, relying solely on dietary iron to make their essential ferroproteins96, highlighting the central role played by this secreted ferritin in the tick life cycle.
Analysis of transcripts coding for classical antioxidant enzymes showed a common core of antioxidant potential sustained by glutathione synthesis (GCS, GS), thioredoxin, and two transcripts coding for thioredoxin reductases, a phospholipid-dependent glutathione peroxidase, and the mitochondrial Mn-SOD that are expressed in all tissues. DIG libraries, however, showed particularly high expression levels of specific isoforms for the cytosolic Cu, Zn SOD, and a thioredoxin peroxidase (2-cys family), plus a selenium glutathione peroxidase and catalase, highlighting a focus on hydrogen peroxide detoxification by this cell type. A result that is in line with previous experimental data reporting the severe effect of chemical inhibition of catalase on blood fed ticks97.
Another class of enzymes that appeared here highly expressed but are usually not particularly highly expressed in the gut of insect vectors, are sulfotransferases. They were included here in the general class of detoxification, because adding sulfate from a high-energy donor is a detoxification mechanism in some cells. On the other hand, one of the products of the reaction catalyzed by this enzyme, 3′-phosphoadenosine-5′-phosphosulfate (PAP) is a sulfate donor to the biosynthesis of sulfated polysaccharides, major components of extracellular matrix of the tick gut98. This hypothesis is consistent with some sulfotransferases (such as Rm-25779, Rm-18149, Rm-64114, and Rm-139775) decreased expression in digestive cells after blood meal, making it likely that those enzymes are acting as players in extracellular matrix production that might support the growth of cells (Fig. 4C,D). Sulfotransferases are also involved in the inactivation of octopamine99, which functions as a neuromuscular transmitter in insects100.
A major feature of tick gut DIGs is the presence of an organelle dedicated to heme detoxification, the hemosome. A heme transporting pathway directs heme derived from hemoglobin degradation in the acidic digestive vacuoles to the cytosol and then to the hemosome. Several ABC transporters have high expression levels in DIGs and therefore are likely candidates to function as heme transporters, including Rm-73791 that is almost exclusively expressed in DIGs. This is in accordance with previous biochemical data101, where ABC-like transporters were implicated on heme transport in digestive cells of R. microplus. In addition, although this transporter has considerable expression levels in gut, it is even more intensively expressed in the ovary, which raises the interesting possibility that these transporters may be involved in heme transport to the growing oocyte, where vitellin is a major heme-storage protein.
Among typical immune effectors, only defensins seem to be highly expressed in the DIG. However, transcripts receiving an immune label were mainly related to bacterial killing by phagocytosis (ixoderins, plus a set of proteins related to lysosomal function). This is consistent with the digestive function of these cells being exerted by means of digestion of blood proteins in a lysosomal acid compartment and can be related to very low bacterial levels observed in the gut, including during blood feeding43,102.
The cattle tick energy metabolism
Considering the relatively low amount of glucose ingested by the cattle tick, since the concentration in blood is around 50 mg/dL103, carbohydrate metabolism in this organism deserves some discussion concerning the correlation with this very rich protein diet. Two hexokinase coding transcripts were found in the six libraries analyzed. The isoform represented by Rm-54671, is almost 30 times more expressed than Rm-19014, but it is from six to seventy times less expressed in digestive cells than in any other tissue (Table S1 and Table S6). Interestingly, besides the role played in metabolism, hexokinases are also enrolled in the inhibition of the intrinsic pathway apoptosis, preventing the release of cytochrome c from the mitochondria104. Therefore, it is possible that hexokinases are also involved in controlling the apoptosis in R. microplus organs.
The essential enzymes needed to the synthesis and degradation of glycogen are present in this dataset. The sequences of phosphoglucomutase (Rm-32898), UDPG-pyrophosphorylase (Rm-37883), UTP-glucose-1-phosphate uridyltransferase (Rm-26034), glycogen synthase (Rm-18733), and glycogen phosphorylase (Rm-26011) are observed in all tissues. The enzymes involved in glycogen accumulation are poorly expressed in digestive cells of fully engorged females. The transcript coding for hexokinase, the first enzyme of the glycolytic pathway, has a very low expression in digestive cells of both partially and fully engorged females; we could not find a transcript coding for phosphofructokinase-1. In contrast, the glucose 6-phosphate-phosphatase transcript was highly expressed, especially in digestive cells of partially engorged females. The high abundance of this transcript in digestive cells led us to speculate that most of the glucose in these cells might not come from the blood meal but from gluconeogenesis, and later being exported to hemolymph and other tissues. The following six enzymes catalyze reactions that are reversible in cellular conditions; the direction of these reactions thus being governed by the law of mass action, and therefore participating both in glycolysis and gluconeogenesis: fructose-bisphosphate aldolase (Rm-1568), triosephosphate isomerase (Rm-4663), glyceraldehyde 3-phosphate dehydrogenase (Rm-10107), 3-phosphoglycerate kinase (Rm-26739), phosphoglycerate mutase (Rm-62495), and enolase (Rm-19039) were found. Their expression levels, although varying among organs, are higher than those of transcripts coding for enzymes that are participating only in glycolysis but not in gluconeogenesis (Table S1).
All subunits of pyruvate dehydrogenase were found: Rm-169115 is E1 alpha subunit; Rm-8313 is E1 beta subunit; Rm-56844 is dihydrolipoamide dehydrogenase; and Rm-12428 is dihydrolipoamide acetyltransferase. The tricarboxylic acid cycle is a central metabolic pathway. All transcripts coding enzymes and their subunits were identified, and the expression of these enzymes shows differences among different tissues; the TCA cycle appearing to be more active on the salivary glands, ovaries, and digestive cells of partially engorged females. Carnitine O-palmitoyltransferases I and II ORF, involved in fatty acids oxidation metabolism (Rm-12481 and Rm-18519, respectively), were expressed in all organs tested. Nevertheless, their expression is higher in salivary glands. Moreover, ORFs of all enzymes necessary for β-oxidation were found, showing that conversion of fatty acids into ketone bodies occurs in R. microplus (Table S1).
Enzymes coding for the pentose phosphate pathway are all present in all tissues but are particularly highly expressed in the ovary. This could account for the high demand for NADPH reducing power by the glutathione/thioredoxin dependent antioxidant mechanisms described above, as well as by the very pronounced concentration of the lipid biosynthesis in the ovary (fatty acid synthase transcripts Rm-43199, Rm-25088, and Rm-25089, and two for acetyl CoA carboxylase Rm-44844 and Rm-44843).
The cattle tick immune system
The transcriptome of R. microplus revealed 210 CDS classified in the immune system category (Table S1). Among them, important components of immune signaling pathways, antimicrobial peptides, and recognition proteins were identified. Additional CDS of proteins that belong to other functional categories, such as nuclear, protease and protease inhibitors, detoxification, and protein export and modification, but that still play a role in immune responses, were also identified (Table S9). Importantly, 43 of the 210 sequences in the immune system category were overexpressed (fold-change ≥ 5) in one specific organ of the tick (Table S9). Specifically, 24 were overexpressed in DIG, including one defensin (but not lysozymes nor microplusins, that were expressed in other tissues) and showing an immune profile dominated by ixoderins (fibrinogen-related proteins with lectin activity, involved in microorganisms phagocytosis, 9 transcripts)105,106, ML [(MD-2 (myeloid differentiation factor-2)-related Lipid-recognition)] domain containing proteins (involved in lipid trafficking and lysosomal function, 5 transcripts), and GILT (gamma-interferon inducible lysosomal thiol reductases, and 4 transcripts)107,108. GILT are the only molecules capable of keeping essential SH residues of cysteine proteinases reduced at acid pH, therefore preserving their activity inside the endosomal pathway109. As mentioned above, the R. microplus gut is inhabited by a very minute microbial population, probably reflecting the operation of a powerful microbe killing control, such as the production of hemocidins110,111. Data presented here suggest that the endosomal/lysosomal pathway also play a central role in the gut immunity of ticks (Table S9), which differs from the mode of operation of the insect gut. Indeed, it was previously reported that the immune responses of the tick gut is involved in delineating susceptibility to the zoonotic pathogen R. rickettsii44. While rickettsial infection downregulates immune-related genes in the gut of A. aureolatum, immune-related genes are upregulated in Amblyomma sculptum, which is much less susceptible to infection with R. rickettsii than A. aureolatum48. Interestingly, most of the immune-related transcripts show a very different pattern of expression between libraries from fully engorged and partially engorged ticks, suggesting the existence of different immune challenges between these two phases in the tick life cycle. A more detailed account of the immune-related transcripts found in this work follows.
Immune signaling pathways
Different from the vastly available information on the immune signaling pathways in insects, very little is known about this subject in ticks. Sequences encoding components of immune signaling pathways were identified in the genome of Ixodes scapularis9,112,113. A study on R. microplus immune signaling pathway components revealed that Toll and JAK/STAT pathways of ticks are closely related to those of insects and crustaceans114. Conversely, certain components of IMD pathway of apomorphic orders of insects (Diptera and Coleoptera) are missing in ticks, as well as in plesiomorphic orders of insects (Hemiptera) and crustaceans. However, it was observed that IMD pathway is functional in I. scapularis115 and in R. microplus116 , although little is known about its activation and effectors. Reports on functional characterization of components of tick immune signaling pathways are also scarce. It has been suggested that the JAK/STAT pathway plays a role in the control of infection by both Anaplasma phagocytophilum117,118 and Borrelia burgdorferi119 in I. scapularis. Moreover, it was reported that the host IFNγ upregulates a tick GTPase through the JAK/STAT pathway, limiting B. burgdorferi proliferation120. The IMD pathway was also enrolled in protection of ticks against infection. The knockdown of the transcription factor Relish increased the susceptibility of R. microplus to Anaplasma marginale116 and of I. scapularis to A. phagocytophilum115. Interestingly, it was shown that the activation of IMD pathway does not occur through recognition of bacterial peptidoglycans by PGRPs (peptidoglycan-recognition proteins) as in insects, but through recognition of bacterial lipids115.
In the current study, we identified five CDS of Toll-like receptors and two CDS of the cytokine spaetzle, which are components of Toll pathway (Table S1). Other components of this same pathway, such as the adaptor MyD88 and cactin, the interactor protein of the NF-kB inhibitor cactus (IkB), were also identified (Table S1). Regarding components of IMD pathway, we identified the NF-κB relish (Rel/NF-κB) and caudal, its inhibitory protein (Table S1). In addition, transcripts of the inhibitor proteins PIAS (protein inhibitors of activated STAT) and SOCS (suppressor of cytokine signaling) of JAK/STAT pathway were also detected. In relation to the JNK pathway, we identified a CDS of the transcription factor Jun (Table S1). Notably, all components of immune pathway components identified in the present study have been previously identified114. This result reinforces the constitutive expression of signaling pathway components in R. microplus, as previously suggested114.
Antimicrobial peptides
AMPs are important effectors of immune signaling pathways and represent pivotal factors of invertebrate immune system, acting directly against a wide range of microorganisms, such as protozoa, bacteria, and fungi121. Among the AMPs described in ticks, defensins are a prominent group. These small peptides are synthesized as preprodefensins and are present in several species of both soft and hard ticks122. In R. microplus, one defensin was found in hemocytes47. Importantly, the knockdown of varisin, a defensin from the tick D. variabilis, reduced in 50% the activity of hemolymph against the Gram-positive bacterium Micrococcus luteus123. On the other hand, silencing of this same gene intriguingly reduced the infection of the tick-borne pathogen A. marginale124. Also, defensins were reported to be differentially expressed upon infection with Rickettsia montanensis125. In addition to hemolymph, expression of defensins was also reported to occur in other tick organs, for example the gut and salivary glands. Nineteen defensin CDS were here identified in the R. microplus transcriptome. Among them, one CDS exhibited high transcript levels in digestive cells and another in salivary glands (Table S1). Defensin transcripts, besides transcripts of other AMPs, were also previously identified in the salivary glands of other tick species48,126.
The presence of AMPs as well as other immune compounds in the tick gut could be relevant because the slow intracellular digestion of nutrients, as well as neutral pH, may favor the proliferation of microorganisms. In addition to defensins, lysozymes were reported to be present in the gut of ticks, especially in the soft tick Ornithodorus moubata127,128. We identified five CDS of lysozymes in the R. microplus transcriptome (Table S1) without overexpression in a specific tick organ, and none of them showing expressions levels comparable to defensins or microplusins (see below). Interestingly, in addition to the endogenous AMPs, peptides derived from host hemoglobin (hemocidins) were also detected in the tick gut111. Production of hemocidins occur by action of aspartic (cathepsin D-type) and cysteine (cathepsin L-type) peptidases. Notably, transcripts of both cathepsins D- and L-types were detected in the current study (Table S1).
Histidine-rich peptides were identified in hemolymph47, ovaries and eggs46 of R. microplus (microplusin) and in synganglion of Amblyomma hebraeum129. It was shown that microplusin has the property of chelating copper, inhibiting the respiration of both bacteria and fungi130,131. Twenty-one microplusin CDS were detected in the present study (Table S1). Three of them presented high transcript levels in the fat body and two in the ovary (Table S5 and Table S2). Infections can affect tick fertility, since resources can be diverted from reproductive process to the synthesis of immune factors and tissue repair. Indeed, it was previously shown that R. rickettsii-infection diminish the reproduction rate in ticks132. As mentioned above, it was recently reported that the knockdown of microplusin increases the susceptibility of A. aureolatum to infection with R. rickettsii48. However, microplusin showed no effect on either the bacterial transmission to the host or the tick fitness.
In conclusion, this study described the transcriptome assembly of various tissues of R. microplus and expanded the number of genes annotated for this economically important parasite. Functional annotation and classification revealed that many genes related to blood digestion and host-parasite interaction are activated in gut cells compared with other tissues. Furthermore, essential genes for cell development and embryogenesis were overexpressed in ovaries. A number of candidate genes for important biochemical pathways were also identified. The availability of this transcriptome opens new perspectives in the study of biochemical role of gene products. Finally, these novel data provide useful information on the tick physiology and serve as a valuable platform to support the study and development of new control methods.
Material and methods
Ethics statement
All animals used in these experiments were housed at Faculdade de Veterinária, Universidade Federal do Rio Grande do Sul (UFRGS). This study was conducted considering ethic and methodological aspects in agreement with the International and National Directives and Norms by the Animal Experimentation Ethics Committee of the Universidade Federal do Rio Grande do Sul (UFRGS). The protocol was approved by the Comissão de Ética no Uso de Animais (CEUA) – UFRGS (project 14403).
Ticks
Rhipicephalus microplus ticks (Porto Alegre strain) free of Babesia sp. and Anaplasma sp. were maintained on Hereford bovines acquired from a naturally tick-free area (Santa Vitoria do Palmar, RS, Brazil). The bovines were housed in individual thick-proof pens on slatted floors and infested with 20,000 10-day-old R. microplus larvae per animal59. Partially engorged female (PEF) ticks were manually removed from cattle, while fully engorged female (FEF) ticks were obtained after spontaneous detachment from the host. After collection, ticks were washed with 70% ethanol and had the dorsal surface dissected with a scalpel blade.
Isolation of digestive cells
Digestive cells were isolated as previously described81. Essentially, ticks were dissected in Petri dish containing sterile PBS, guts were isolated, opened gently with tweezers, and cells were detached from the gut wall manually with tweezers. Subsequently, cells were carefully collected using a 1-mL pipette tip, washed three times in sterile PBS and placed in a 12-well culture plate. In order to reduce the amount of debris and gut soluble contents, cells were washed with a gentle flow of culture medium (L-15 Leibowitz’s medium supplemented with 150 mM NaCl) produced by a 1-mL pipette tip. During the isolation of the digestive cells, the gut was dissected and kept in an ice-cold medium. The procedure can be observed by the movie file included as supplemental material (Supplementary video 1).
RNA extraction, cDNA library construction and sequencing
Tissues (as described below) were separated with fine-tipped forceps and washed in ice-cold phosphate buffered saline pH 7.2 (PBS). For egg collection, FEF ticks were collected and incubated in Petry dishes at 28 °C and 85% relative humidity until oviposition. For embryos, eggs were kept during twelve days in disposable tubes under the same conditions as described above. Total RNA was extracted from the following tissues: (i) embryos (1, 3, 5, 7, 9, 11, 13 day-old eggs); (ii) ovaries from partially and fully engorged females; (iii) salivary glands from partially engorged females; (iv) fat bodies from partially and fully engorged females; (v and vi) digestive cells from partially and fully engorged females, using TRIZOL reagent according to the manufacturers’ instructions (Thermo Fisher Scientific, Waltham, MA, USA). A total amount of 10 µg RNA was used as input material for the library preparation. Sequencing libraries were generated using the IlluminaTruSeqTM RNA Sample Preparation Kit (Illumina, San Diego, USA) following the manufacturer’s recommendations.
Transcriptome assembly and bioinformatics
Bioinformatic analyses were performed as described previously14. Illumina adaptor sequences and low-quality bases were removed from the raw reads. The quality-filtered sequence reads of each sample were pooled to generate a single transcriptome assembly of the R. microplus. Reads were assembled with Abyss 1.3.3 software (https://www.bcgsc.ca/resources/software/abyss)133 with various k values (from 25 to 95 at five unit intervals). Because the Abyss software tends to miss highly expressed contigs, we have also run the Trinity 2.0 assembler (https://trinityrnaseq.github.io/)134 on the raw data. The resulting assemblies were joined by an iterative BLAST and CAP3 assembler14.
Coding sequences (CDS) were extracted using an automated pipeline14, based on similarities to known proteins, or by obtaining coding sequences from the larger open reading frame (ORF) of the contigs containing a signal peptide identified by version 3.0 of the SignalP software (https://www.cbs.dtu.dk/services/SignalP-3.0/)135. A non-redundant set of the coding sequences and their protein sequences were mapped into a hyperlinked excel spreadsheet. Signal peptide, transmembrane domains, furin cleavage sites, and glycosylation sites were determined with software from the Center for Biological Sequence Analysis (https://www.cbs.dtu.dk/services/). The automated annotation of the proteins was based on matches to various databases, including Gene Ontology (https://geneontology.org/)136, Pfam (https://pfam.xfam.org/)78, Swissprot (https://www.ebi.ac.uk/uniprot/), KOG (https://mycocosm.jgi.doe.gov/help/kogbrowser.jsf)137, SMART (https://smart.embl-heidelberg.de/)138, Refseq-invertebrates, and sequences containing Acari [organism] protein sequences obtained from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). The manual annotation was performed as detailed in previous article14.
To estimate the transcripts abundance, reads were mapped back into the CDS using BLASTn with a word size of 25 (-W 25 switch). The matches were used if they had less than two gaps and if their scores were equal to the best score. The resulting BLAST file was used to compile the number of reads each CDS received from each library, and to count the number of hits at each base of the CDS, allowing for the determination of the average CDS coverage, percent linear coverage, as well as maximum and minimum coverage. Mapping of the reads and RPKM values were included in an Excel spreadsheet.
Finally, tick genomes11,13 were used as reference to assess the assembly quality in terms of gene completeness using BUSCO v. 4.1.3 (https://busco.ezlab.org/)23.
Data availability
Raw sequence reads were deposited in the NCBI Sequence Read Archive (Biosample SAMN02463642 and Bioproject PRJNA232001). Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GHWJ00000000. The version described in this paper is the first version, GHWJ01000000.
References
Grisi, L. et al. Reavaliação do potencial impacto econômico de parasitos de bovinos no Brasil. Rev. Bras. Parasitol. Vet. 23, 150–156 (2014).
Jongejan, F. & Uilenberg, G. The global importance of ticks. Parasitology 129, S3–S14 (2004).
Jonsson, N. N. The productivity effects of cattle tick (Boophilus microplus) infestation on cattle, with particular reference to Bos indicus cattle and their crosses. Vet. Parasitol. 137, 1–10 (2006).
Guerrero, F. D., Lovis, L. & Martins, J. R. Acaricide resistance mechanisms in Rhipicephalus (Boophilus) microplus. Rev. Bras. Parasitol. Vet. 21, 1–6 (2012).
Klafke, G. et al. Multiple resistance to acaricides in field populations of Rhipicephalus microplus from Rio Grande do Sul state, Southern Brazil. Ticks Tick Borne Dis. 8, 73–80 (2017).
de la Fuente, J. et al. A ten-year review of commercial vaccine performance for control of tick infestations on cattle. Anim. Health Res. Rev. 8, 23–28 (2007).
Chmelař, J. et al. Sialomes and mialomes: a systems-biology view of tick tissues and tick–host interactions. Trends Parasitol. 32, 242–254 (2016).
Francischetti, I. M. B. The role of saliva in tick feeding. Front. Biosci. 14, 2051 (2009).
Gulia-Nuss, M. et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun. 7, 10507 (2016).
Guerrero, F. D. et al. The Pacific Biosciences de novo assembled genome dataset from a parthenogenetic New Zealand wild population of the longhorned tick, Haemaphysalis longicornis Neumann, 1901. Data Br. 27, 104602 (2019).
Jia, N. et al. Large-scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities. Cell 182, 1328-1340.e13 (2020).
Ullmann, A. J., Lima, C. M. R., Guerrero, F. D., Piesman, J. & Black, W. C. IV. Genome size and organization in the blacklegged tick, Ixodes scapularis and the Southern cattle tick, Boophilus microplus. Insect Mol. Biol. 14, 217–222 (2005).
Barrero, R. A. et al. Gene-enriched draft genome of the cattle tick Rhipicephalus microplus: assembly by the hybrid Pacific Biosciences/Illumina approach enabled analysis of the highly repetitive genome. Int. J. Parasitol. 47, 569–583 (2017).
Karim, S., Singh, P. & Ribeiro, J. M. C. A deep insight into the sialotranscriptome of the Gulf Coast tick, Amblyomma maculatum. PLoS ONE 6, e28525 (2011).
Maruyama, S. R. et al. Mining a differential sialotranscriptome of Rhipicephalus microplus guides antigen discovery to formulate a vaccine that reduces tick infestations. Parasites Vectors 10, 206 (2017).
Perner, J. et al. RNA-seq analyses of the midgut from blood- and serum-fed Ixodes ricinus ticks. Sci. Rep. 6, 36695 (2016).
Xavier, M. A. et al. Tick Gené’s organ engagement in lipid metabolism revealed by a combined transcriptomic and proteomic approach. Ticks Tick Borne Dis. 10, 787–797 (2019).
Heekin, A. M. et al. The ovarian transcriptome of the cattle tick, Rhipicephalus (Boophilus) microplus, feeding upon a bovine host infected with Babesia bovis. Parasites Vectors 6, 276 (2013).
Rachinsky, A., Guerrero, F. D. & Scoles, G. A. Differential protein expression in ovaries of uninfected and Babesia-infected southern cattle ticks, Rhipicephalus (Boophilus) microplus. Insect Biochem. Mol. Biol. 37, 1291–1308 (2007).
Tirloni, L. et al. Tick-host range adaptation: changes in protein profiles in unfed adult Ixodes scapularis and Amblyomma americanum saliva stimulated to feed on different hosts. Front. Cell. Infect. Microbiol. 7, 517 (2017).
Kotsarenko, K. et al. Newly identified DNA methyltransferases of Ixodes ricinus ticks. Ticks Tick Borne Dis. 11, 101348 (2020).
Garcia, G. R. et al. A transcriptome and proteome of the tick Rhipicephalus microplus shaped by the genetic composition of its hosts and developmental stage. Sci. Rep. 10, 12857 (2020).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245 (2019).
Atella, G. C. et al. Oogenesis and egg development in triatomines: a biochemical approach. An. Acad. Bras. Cienc. 77, 405–430 (2005).
Seixas, A. et al. Localization and function of Rhipicephalus (Boophilus) microplus vitellin-degrading cysteine endopeptidase. Parasitology 137, 1819–1831 (2010).
Coons, L. B., Lamoreaux, W. J., Rosell-Davis, R. & Tarnowski, B. I. Onset of vitellogenin production and vitellogenesis, and their relationship to changes in the midgut epithelium and oocytes in the tick Dermacentor variabilis. Exp. Appl. Acarol. 6, 291–305 (1989).
Logullo, C. et al. Isolation of an aspartic proteinase precursor from the egg of a hard tick, Boophilus microplus. Parasitology 116, 525–532 (1998).
Sorgine, M. H. F. et al. A heme-binding aspartic proteinase from the eggs of the hard tick Boophilus microplus. J. Biol. Chem. 275, 28659–28665 (2000).
Seixas, A., Friesen, K. J. & Kaufman, W. R. Effect of 20-hydroxyecdysone and haemolymph on oogenesis in the ixodid tick Amblyomma hebraeum. J. Insect Physiol. 54, 1175–1183 (2008).
Seixas, A. et al. A Boophilus microplus vitellin-degrading cysteine endopeptidase. Parasitology 126, 155–163 (2003).
Oldiges, D. P. et al. A Rhipicephalus (Boophilus) microplus cathepsin with dual peptidase and antimicrobial activity. Int. J. Parasitol. 42, 635–645 (2012).
Pohl, P. C. et al. An extraovarian aspartic protease accumulated in tick oocytes with vitellin-degradation activity. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 151, 392–399 (2008).
Bergamo Estrela, A., Seixas, A., de Oliveira NunesTeixeira, V., Pinto, A. F. M. & Termignoni, C. Vitellin- and hemoglobin-digesting enzymes in Rhipicephalus (Boophilus) microplus larvae and females. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 157, 326–335 (2010).
Lu, S., Parizi, L. F., Torquato, R. J. S., Vaz Junior, I. S. & Tanaka, A. S. Novel pseudo-aspartic peptidase from the midgut of the tick Rhipicephalus microplus. Sci. Rep. 9, 435 (2019).
Seixas, A. et al. Expression profile of Rhipicephalus microplus vitellogenin receptor during oogenesis. Ticks Tick Borne Dis. 9, 72–81 (2018).
Neese, P. A., Sonenshine, D. E., Kallapur, V. L., Apperson, C. S. & Roe, R. M. Absence of insect juvenile hormones in the American dog tick, Dermacentor variabilis (Say) (Acari: Ixodidae), and in Ornithodoros parkeri Cooley (Acari: Argasidae). J. Insect Physiol. 46, 477–490 (2000).
Dias, F. A. et al. Ovarian dual oxidase (duox) activity is essential for insect eggshell hardening and waterproofing. J. Biol. Chem. 288, 35058–35067 (2013).
Oliveira, J. H. M. et al. Blood meal-derived heme decreases ROS levels in the midgut of Aedes aegypti and allows proliferation of intestinal microbiota. PLoS Pathog. 7, e1001320 (2011).
Ha, E. M., Oh, C. T., Bae, Y. S. & Lee, W. J. A direct role for dual oxidase in Drosophila gut immunity. Science 310, 847–850 (2005).
Bonnet, S. I., Binetruy, F., Hernández-Jarguín, A. M. & Duron, O. The tick microbiome: why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 7, 236 (2017).
Duron, O. et al. Tick-bacteria mutualism depends on B vitamin synthesis pathways. Curr. Biol. 28, 1896-1902.e5 (2018).
Guizzo, M. G. et al. A Coxiella mutualist symbiont is essential to the development of Rhipicephalus microplus. Sci. Rep. 7, 17554 (2017).
Guizzo, M. G. et al. Poor unstable midgut microbiome of hard ticks contrasts with abundant and stable monospecific microbiome in ovaries. Front. Cell. Infect. Microbiol. https://doi.org/10.3389/fcimb.2020.00211 (2020).
Martins, L. A. et al. The distinct transcriptional response of the midgut of Amblyomma sculptum and Amblyomma aureolatum ticks to Rickettsia rickettsii correlates to their differences in susceptibility to infection. Front. Cell. Infect. Microbiol. 7, 476 (2017).
Pavanelo, D. B. et al. Comparative analysis of the midgut microbiota of two natural tick vectors of Rickettsia rickettsii. Dev. Comp. Immunol. 106, 103606 (2020).
Esteves, E. et al. Antimicrobial activity in the tick Rhipicephalus (Boophilus) microplus eggs: Cellular localization and temporal expression of microplusin during oogenesis and embryogenesis. Dev. Comp. Immunol. 33, 913–919 (2009).
Fogaça, A. C. et al. Cysteine-rich antimicrobial peptides of the cattle tick Boophilus microplus: Isolation, structural characterization and tissue expression profile. Dev. Comp. Immunol. 28, 191–200 (2004).
Martins, L. A. et al. The transcriptome of the salivary glands of Amblyomma aureolatum reveals the antimicrobial peptide microplusin as an important factor for the tick protection against Rickettsia rickettsii infection. Front. Physiol. https://doi.org/10.3389/fphys.2019.00529 (2019).
Velentzas, A. D. et al. Global proteomic profiling of Drosophila ovary: a high-resolution, unbiased, accurate and multifaceted analysis. Cancer Genomics Proteomics 12, 369–384 (2015).
Xavier, M. A. et al. A proteomic insight into vitellogenesis during tick ovary maturation. Sci. Rep. 8, 4698 (2018).
Malumbres, M. Cyclin-dependent kinases. Genome Biol. 15, 122 (2014).
Gomes, H. et al. Identification and structural-functional analysis of cyclin-dependent kinases of the cattle tick Rhipicephalus (Boophilus) microplus. PLoS ONE 8, e76128 (2013).
Gomes, H. et al. Vaccination with cyclin-dependent kinase tick antigen confers protection against Ixodes infestation. Vet. Parasitol. 211, 266–273 (2015).
Funato, Y. & Miki, H. Nucleoredoxin, a novel thioredoxin family member involved in cell growth and differentiation. Antioxid. Redox Signal. 9, 1035–1057 (2007).
Jacobs, C. G. C., Spaink, H. P. & van der Zee, M. The extraembryonic serosa is a frontier epithelium providing the insect egg with a full-range innate immune response. Elife https://doi.org/10.7554/eLife.04111 (2014).
Jacobs, C. G. C. & Van Der Zee, M. Immune competence in insect eggs depends on the extraembryonic serosa. Dev. Comp. Immunol. 41, 263–269 (2013).
Jacobs, C. G. C. et al. Egg survival is reduced by grave-soil microbes in the carrion beetle, Nicrophorus vespilloides. BMC Evol. Biol. 14, 208 (2014).
Santos, V. T. et al. The embryogenesis of the Tick Rhipicephalus (Boophilus) microplus: the establishment of a new chelicerate model system. Genesis 51, 803–818 (2013).
Reck, J. et al. Systemic alterations of bovine hemostasis due to Rhipicephalus (Boophilus) microplus infestation. Res. Vet. Sci. 86, 56–62 (2009).
Tirloni, L. et al. Proteomic analysis of cattle tick Rhipicephalus (Boophilus) microplus saliva: a comparison between partially and fully engorged females. PLoS ONE 9, e94831 (2014).
Nuttall, P. A. & Labuda, M. Tick–host interactions: saliva-activated transmission. Parasitology 129, S177–S189 (2004).
Roberts, J. A. Resistance of cattle to the tick Boophilusmicroplus (Canestrini). II. Stages of the life cycle of the parasite against which resistance is manifest. J. Parasitol. 54, 667–673 (1968).
Schwarz, A. et al. De novo Ixodes ricinus salivary gland transcriptome analysis using two next-generation sequencing methodologies. FASEB J. 27, 4745–4756 (2013).
Kotsyfakis, M., Schwarz, A., Erhart, J. & Ribeiro, J. M. C. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci. Rep. 5, 9103 (2015).
Ali, A. et al. Immunoprotective potential of a Rhipicephalus (Boophilus) microplus metalloprotease. Vet. Parasitol. 207, 107–114 (2015).
Kim, T. K. et al. Ixodes scapularis tick saliva proteins sequentially secreted every 24 h during blood feeding. PLoS Negl. Trop. Dis. 10, e0004323 (2016).
Narasimhan, S. et al. Immunity against Ixodes scapularis salivary proteins expressed within 24 hours of attachment thwarts tick feeding and impairs Borrelia transmission. PLoS ONE 2, e451 (2007).
Francischetti, I. M. B., Mather, T. N. & Ribeiro, J. M. C. Cloning of a salivary gland metalloprotease and characterization of gelatinase and fibrin(ogen)lytic activities in the saliva of the Lyme disease tick vector Ixodes scapularis. Biochem. Biophys. Res. Commun. 305, 869–875 (2003).
Markland, F. S. & Swenson, S. Snake venom metalloproteinases. Toxicon 62, 3–18 (2013).
Decrem, Y. et al. The impact of gene knock-down and vaccination against salivary metalloproteases on blood feeding and egg laying by Ixodes ricinus. Int. J. Parasitol. 38, 549–560 (2008).
Fló, M. et al. Functional diversity of secreted cestode Kunitz proteins: Inhibition of serine peptidases and blockade of cation channels. PLoS Pathog. 13, e1006169 (2017).
Nienaber, J., Gaspar, A. R. M. & Neitz, A. W. H. Savignin, a potent thrombin inhibitor isolated from the salivary glands of the tick Ornithodoros savignyi (Acari: Argasidae). Exp. Parasitol. 93, 82–91 (1999).
Macedo-Ribeiro, S. et al. Isolation, cloning and structural characterisation of boophilin, a multifunctional Kunitz-type proteinase inhibitor from the cattle tick. PLoS ONE 3, e1624 (2008).
Nazareth, R. A. et al. Antithrombotic properties of Ixolaris, a potent inhibitor of the extrinsic pathway of the coagulation cascade. Thromb. Haemost. 96, 7–13 (2006).
Kotsyfakis, M. et al. antiinflammatory and immunosuppressive activity of sialostatin L, a salivary cystatin from the tick Ixodes scapularis. J. Biol. Chem. 281, 26298–26307 (2006).
Mans, B. J. Evolution of vertebrate hemostatic and inflammatory control mechanisms in blood-feeding arthropods. J. Innate Immun. 3, 41–51 (2011).
Fogaça, A. C. et al. Ixodidin, a novel antimicrobial peptide from the hemocytes of the cattle tick Boophilus microplus with inhibitory activity against serine proteinases. Peptides 27, 667–674 (2006).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Mans, B. J., Ribeiro, J. M. C. & Andersen, J. F. Structure, function, and evolution of biogenic amine-binding proteins in soft ticks. J. Biol. Chem. 283, 18721–18733 (2008).
Sojka, D. et al. New insights into the machinery of blood digestion by ticks. Trends Parasitol. 29, 276–285 (2013).
Lara, F. A. Tracing heme in a living cell: hemoglobin degradation and heme traffic in digest cells of the cattle tick Boophilus microplus. J. Exp. Biol. 208, 3093–3101 (2005).
Landulfo, G. A. et al. Gut transcriptome analysis on females of Ornithodoros mimon (Acari: Argasidae) and phylogenetic inference of ticks. Rev. Bras. Parasitol. Vet. 26, 185–204 (2017).
Heekin, A. M. et al. Gut transcriptome of replete adult female cattle ticks, Rhipicephalus (Boophilus) microplus, feeding upon a Babesia bovis-infected bovine host. Parasitol. Res. 112, 3075–3090 (2013).
Anderson, J. M., Sonenshine, D. E. & Valenzuela, J. G. Exploring the mialome of ticks: an annotated catalogue of midgut transcripts from the hard tick, Dermacentor variabilis (Acari: Ixodidae). BMC Genomics 9, 552 (2008).
Sojka, D. et al. Profiling of proteolytic enzymes in the gut of the tick Ixodes ricinus reveals an evolutionarily conserved network of aspartic and cysteine peptidases. Parasites Vectors 1, 7 (2008).
Franta, Z. et al. Dynamics of digestive proteolytic system during blood feeding of the hard tick Ixodes ricinus. Parasites Vectors 3, 119 (2010).
Parizi, L. F. et al. Peptidase inhibitors in tick physiology. Med. Vet. Entomol. 32, 129–144 (2018).
Lu, S. et al. A novel type 1 cystatin involved in the regulation of Rhipicephalus microplus midgut cysteine proteases. Ticks Tick Borne Dis. 11, 101374 (2020).
Kanost, M. R. Serine proteinase inhibitors in arthropod immunity. Dev. Comp. Immunol. 23, 291–301 (1999).
Gracasouza, A. et al. Adaptations against heme toxicity in blood-feeding arthropods. Insect Biochem. Mol. Biol. 36, 322–335 (2006).
Ribeiro, J. M. C. et al. An insight into the transcriptome of the digestive tract of the bloodsucking bug, Rhodnius prolixus. PLoS Negl. Trop. Dis. 8, e2594 (2014).
Padrón, A. et al. In depth annotation of the Anopheles gambiae mosquito midgut transcriptome. BMC Genomics 15, 636 (2014).
Dreher-Lesnick, S. M., Mulenga, A., Simser, J. A. & Azad, A. F. Differential expression of two glutathione S-transferases identified from the American dog tick, Dermacentor variabilis. Insect Mol. Biol. 15, 445–453 (2006).
Hajdusek, O. et al. Knockdown of proteins involved in iron metabolism limits tick reproduction and development. Proc. Natl. Acad. Sci. U.S.A. 106, 1033–1038 (2009).
Braz, G. R. C., Coelho, H. S. L., Masuda, H. & Oliveira, P. L. A missing metabolic pathway in the cattle tick Boophilus microplus. Curr. Biol. 9, 703–706 (1999).
Perner, J. et al. Acquisition of exogenous haem is essential for tick reproduction. Elife 5, e12318 (2016).
Citelli, M., Lara, F. A., da Vaz, I. S. & Oliveira, P. L. Oxidative stress impairs heme detoxification in the midgut of the cattle tick, Rhipicephalus (Boophilus) microplus. Mol. Biochem. Parasitol. 151, 81–88 (2007).
Onofre, G. R., Lara, F. A., Oliveira, P. L. & Silva, L.-C.F. Heparan sulfate glycosaminoglycan expression in the intestinal tract and ovary of fully engorged adult females of the cattle tick Boophilus microplus and in their laid eggs. Mol. Biochem. Parasitol. 130, 163–166 (2003).
Pichu, S., Yalcin, E. B., Ribeiro, J. M., King, R. S. & Mather, T. N. Molecular characterization of novel sulfotransferases from the tick, Ixodes scapularis. BMC Biochem. 12, 32 (2011).
Klaassen, L. W. & Kammer, A. E. Octopamine enhances neuromuscular transmission in developing and adult moths, Manduca sexta. J. Neurobiol. 16, 227–243 (1985).
Lara, F. A. et al. ATP binding cassette transporter mediates both heme and pesticide detoxification in tick midgut cells. PLoS ONE 10, e0134779 (2015).
Ross, B. D. et al. Ixodes scapularis does not harbor a stable midgut microbiome. ISME J. 12, 2596–2607 (2018).
Maplesden, D. C., McSherry, B. J. & Stone, J. B. Blood sugar levels in normal cows before and after treatment with prednisolone and dexamethasone. Can. Vet. J. 1, 309–312 (1960).
Pastorino, J. G., Shulga, N. & Hoek, J. B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618 (2002).
Honig Mondekova, H. et al. Characterization of Ixodes ricinus fibrinogen-related proteins (ixoderins) discloses their function in the tick innate immunity. Front. Cell. Infect. Microbiol. 7, 509 (2017).
Dong, Y. & Dimopoulos, G. Anopheles fibrinogen-related proteins provide expanded pattern recognition capacity against bacteria and malaria parasites. J. Biol. Chem. 284, 9835–9844 (2009).
Schleicher, T. R. et al. A mosquito salivary gland protein partially inhibits Plasmodium sporozoite cell traversal and transmission. Nat. Commun. 9, 2908 (2018).
Kongton, K., McCall, K. & Phongdara, A. Identification of gamma-interferon-inducible lysosomal thiol reductase (GILT) homologues in the fruit fly Drosophila melanogaster. Dev. Comp. Immunol. 44, 389–396 (2014).
Balce, D. R., Allan, E. R. O., McKenna, N. & Yates, R. M. γ-Interferon-inducible lysosomal thiol reductase (GILT) maintains phagosomal proteolysis in alternatively activated macrophages. J. Biol. Chem. 289, 31891–31904 (2014).
Belmonte, R., Cruz, C. E., Pires, J. R. & Daffre, S. Purification and characterization of Hb 98–114: a novel hemoglobin-derived antimicrobial peptide from the midgut of Rhipicephalus (Boophilus) microplus. Peptides 37, 120–127 (2012).
Fogaça, A. C. et al. Antimicrobial activity of a bovine hemoglobin fragment in the tick Boophilus microplus. J. Biol. Chem. 274, 25330–25334 (1999).
Smith, A. A. & Pal, U. Immunity-related genes in Ixodes scapularis perspectives from genome information. Front. Cell. Infect. Microbiol. 4, 116 (2014).
Kotsyfakis, M., Kopáček, P., Franta, Z., Pedra, J. H. F. & Ribeiro, J. M. C. Deep sequencing analysis of the Ixodes ricinus haemocytome. PLoS Negl. Trop. Dis. 9, e0003754 (2015).
Rosa, R. D. et al. Exploring the immune signalling pathway-related genes of the cattle tick Rhipicephalus microplus: from molecular characterization to transcriptional profile upon microbial challenge. Dev. Comp. Immunol. 59, 1–14 (2016).
Shaw, D. K. et al. Infection-derived lipids elicit an immune deficiency circuit in arthropods. Nat. Commun. 8, 14401 (2017).
Capelli-Peixoto, J. et al. The transcription factor Relish controls Anaplasma marginale infection in the bovine tick Rhipicephalus microplus. Dev. Comp. Immunol. 74, 32–39 (2017).
Liu, L. et al. Ixodes scapularis JAK-STAT pathway regulates tick antimicrobial peptides, thereby controlling the agent of human granulocytic anaplasmosis. J. Infect. Dis. 206, 1233–1241 (2012).
Abraham, N. M. et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. U.S.A. 114, E781–E790 (2017).
Narasimhan, S. et al. Gut microbiota of the tick vector Ixodes scapularis modulate colonization of the Lyme disease spirochete. Cell Host Microbe 15, 58–71 (2014).
Smith, A. A. et al. Cross-species interferon signaling boosts microbicidal activity within the tick vector. Cell Host Microbe 20, 91–98 (2016).
Bulet, P., Stocklin, R. & Menin, L. Anti-microbial peptides: from invertebrates to vertebrates. Immunol. Rev. 198, 169–184 (2004).
Kopáček, P., Hajdušek, O., Burešová, V. & Daffre, S. Tick innate immunity. Adv. Exp. Med. Biol. 708, 137–162 (2010).
Hynes, W. L., Stokes, M. M., Hensley, S. M., Todd, S. M. & Sonenshine, D. E. Using RNA interference to determine the role of varisin in the innate immune system of the hard tick Dermacentor variabilis (Acari: Ixodidae). Exp. Appl. Acarol. 46, 7–15 (2008).
Kocan, K. M. et al. Silencing expression of the defensin, varisin, in male Dermacentor variabilis by RNA interference results in reduced Anaplasma marginale infections. Exp. Appl. Acarol. 46, 17–28 (2008).
Ceraul, S. M., Dreher-Lesnick, S. M., Gillespie, J. J., Rahman, M. S. & Azad, A. F. New tick defensin isoform and antimicrobial gene expression in response to Rickettsia montanensis challenge. Infect. Immun. 75, 1973–1983 (2007).
Lu, X. et al. A novel defensin-like peptide from salivary glands of the hard tick, Haemaphysalis longicornis. Protein Sci. 19, 392–397 (2010).
Kopáček, P., Vogt, R., Jindrák, L., Weise, C. & Šafařík, I. Purification and characterization of the lysozyme from the gut of the soft tick Ornithodoros moubata. Insect Biochem. Mol. Biol. 29, 989–997 (1999).
Grunclová, L., Fouquier, H., Hypša, V. & Kopáček, P. Lysozyme from the gut of the soft tick Ornithodoros moubata: the sequence, phylogeny and post-feeding regulation. Dev. Comp. Immunol. 27, 651–660 (2003).
Lai, R. et al. A new type of antimicrobial protein with multiple histidines from the hard tick, Amblyomma hebraeum. FASEB J. 18, 1447–1449 (2004).
Silva, F. D. et al. Structure and mode of action of microplusin, a copper ii-chelating antimicrobial peptide from the cattle tick Rhipicephalus (Boophilus) microplus. J. Biol. Chem. 284, 34735–34746 (2009).
Silva, F. D. et al. Effects of microplusin, a copper-chelating antimicrobial peptide, against Cryptococcus neoformans. FEMS Microbiol. Lett. 324, 64–72 (2011).
Niebylski, M. L., Peacock, M. G. & Schwan, T. G. Lethal effect of Rickettsia rickettsii on its tick vector (Dermacentor andersoni). Appl. Environ. Microbiol. 65, 773–778 (1999).
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Nielsen, H., Brunak, S. & Von Heijne, G. Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. 12, 3–9 (1999).
Harris, M. A. et al. The Gene Ontology project in 2008. Nucleic Acids Res. 36, D440–D444 (2008).
Tatusov, R. L. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 (2015).
Acknowledgements
This work was supported by Conselho Nacional de Desenvolvimento Tecnológico—CNPq, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES and INCT–Entomologia Molecular, Brazil. JMCR and LT were supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (Vector-Borne Diseases: Biology of Vector Host Relationship, Z01 AI000810-20). This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). The authors thank Brian Brown, NIH Library Writing Center, for manuscript editing assistance.
Author information
Authors and Affiliations
Contributions
G.B., P.L.O., I.S.V.J., J.M.C.R. conceived and designed the study; L.T., G.B., R.D.N., R.D.M., F.A.L., P.L.O., I.S.V.J., J.M.C.R. acquired and analyzed the data; L.T., G.B., R.D.N., A.C.P.G., L.R.V., T.C.A., G.A.S., R.M.S., M.G.G., J.A.M., E.P.C., D.S., H.F.G., J.M., M.B.S.M., R.D.M., M.S.L., P.H.A., F.A.L., A.S., R.N.F., A.C.F., C.L., A.S.T., S.D., P.L.O., I.S.V.J., J.M.C.R. interpreted the data; L.T., G.B., R.D.N., A.C.P.G., L.R.V., T.C.A., G.A.S., R.M.S., M.G.G., J.A.M., E.P.C., D.S., H.F.G., J.M., M.B.S.M.D., R.D.M., M.S.L., P.H.A., F.A.L., A.S., R.N.F., A.C.F., C.L., A.S.T., S.D., P.L.O., I.S.V.J., J.M.C.R. wrote the first draft; L.T., P.L.O., I.S.V.J., J.M.C.R. substantially revised subsequent drafts. All authors edited the manuscript, commented on the text and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tirloni, L., Braz, G., Nunes, R.D. et al. A physiologic overview of the organ-specific transcriptome of the cattle tick Rhipicephalus microplus. Sci Rep 10, 18296 (2020). https://doi.org/10.1038/s41598-020-75341-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75341-w
This article is cited by
-
Changes in saliva protein profile throughout Rhipicephalus microplus blood feeding
Parasites & Vectors (2024)
-
Mining gene expression data for rational identification of novel drug targets and vaccine candidates against the cattle tick, Rhipicephalus microplus
Experimental and Applied Acarology (2023)
-
De novo assembled salivary gland transcriptome and expression pattern analyses for Rhipicephalus evertsi evertsi Neuman, 1897 male and female ticks
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
