
Abstract
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) share overlapping genetic causes and disease symptoms, and are linked neuropathologically by the RNA binding protein TDP-43 (TAR DNA binding protein-43 kDa). TDP-43 regulates RNA metabolism, trafficking, and localization of thousands of target genes. However, the cellular and molecular mechanisms by which dysfunction of TDP-43 contributes to disease pathogenesis and progression remain unclear. Severe changes in the structure of neuronal dendritic arbors disrupt proper circuit connectivity, which in turn could contribute to neurodegenerative disease. Although aberrant dendritic morphology has been reported in non-TDP-43 mouse models of ALS and in human ALS patients, this phenotype is largely unexplored with regards to TDP-43. Here we have employed a primary rodent neuronal culture model to study the cellular effects of TDP-43 dysfunction in hippocampal and cortical neurons. We show that manipulation of TDP-43 expression levels causes significant defects in dendritic branching and outgrowth, without an immediate effect on cell viability. The effect on dendritic morphology is dependent on the RNA-binding ability of TDP-43. Thus, this model system will be useful in identifying pathways downstream of TDP-43 that mediate dendritic arborization, which may provide potential new avenues for therapeutic intervention in ALS/FTD.
Similar content being viewed by others

Introduction
Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) are clinically linked, rapidly progressing neurodegenerative diseases that affect both distinct and overlapping regions of the brain. Nearly 50% of ALS patients show some level of cognitive impairment while 25–30% of FTD patients exhibit motor neuron dysfunction1,2,3,4. Mutations in the TARDBP gene which encodes TAR DNA-binding protein of 43 kDa (TDP-43) have been linked to both ALS and FTD in humans5,6. The connection between ALS and FTD has been further confirmed at the molecular level by the identification of TDP-43 as the major component of ubiquitin–positive inclusions in both ALS and the most common pathological form of FTD7,8.
While aggregate-forming proteins are often thought to cause disease via toxic gain-of-function (GOF) mechanisms9,10,11,12, several lines of evidence suggest that pathologically altered TDP-43 causes neurodegeneration by both GOF and loss-of-function (LOF) mechanisms. First, in patients, cytoplasmic TDP-43 inclusions occur concomitantly with depletion of TDP-43 protein from the nucleus8. Second, TDP-43 has been shown to downregulate the expression of its own mRNA transcript13, and in transgenic mice, overexpression of human TDP-43 results in downregulation of endogenous TDP-4314, suggesting an impairment of TDP-43 function by an interplay between both LOF and GOF mechanisms. Third, TDP-43 LOF in animal and cellular models phenocopies many defects seen when overexpressing either wild-type or mutant forms of TDP-43 that are implicated in human disease15,16,17. Thus, LOF and GOF hypotheses are not mutually exclusive and it is possible that both mechanisms contribute to neurodegeneration18.
TDP-43 is a highly conserved, ubiquitously expressed, multifunctional nucleic acid-binding protein composed of two RNA recognition motifs (RRM), nuclear localization (NLS) and export signals (NES), and a carboxy-terminal glycine rich region. TDP-43 regulates transcription19,20, as well as miRNA processing21,22,23,24,25 and mRNA splicing26,27,28,29,30,31,32. Although it mostly resides in the nucleus, TDP-43 also shuttles to the cytoplasm33 where it is involved in stress granule formation34,35,36,37,38,39,40, axonal transport of target mRNAs41,42, and mRNA translation36,43,44,45,46,47. TDP-43 interacts with several proteins that are implicated in its RNA-processing functions36,48 including the heterogenous nuclear ribonucleoprotein complex which binds to the TDP-43 carboxy-terminal domain49. Despite the numerous, well-described functions and interactions of TDP-43, it is not well understood exactly which TDP-43-dependent cellular processes become defective in ALS/FTD and contribute to disease etiology.
Defects in dendritic arbor elaboration and maintenance have been associated with many neurodegenerative diseases50,51,52. In animal models of ALS, cortical neuronal degeneration along with regression of apical dendrites has been observed in presymptomatic SOD1G39A mice53,54. Cortico-spinal Motor Neurons (CSMNs; known as Betz cells in humans) also show apical dendrite regression in sporadic, familial, and FTD-ALS patients, indicating the loss of the dendritic arbor as an important marker of cellular pathology in ALS55. Given that TDP-43 is involved in the splicing, transport, and stability of several mRNAs which encode proteins that play important roles in neuronal arbor elaboration and synaptic function13,41,56,57, we asked whether TDP-43 dysfunction altered neuronal morphology.
To explore this question, we took the approach of overexpressing or knocking down TDP-43 in cultured mammalian neurons isolated from rat hippocampus or cortex and asked if manipulating TDP-43 protein levels interfered with dendritic complexity. Our results demonstrate that both overexpression and knockdown of TDP-43 result in reduced dendritic branching, without affecting neuronal viability. In addition, the effect of TDP-43 overexpression on dendritic loss is dependent on TDP-43 RNA binding. Our findings demonstrate that aberrant levels of TDP-43 disrupt neuronal morphology, an observation that has important implications for understanding disease pathology.
Results
Increased TDP-43 expression diminishes dendritic complexity
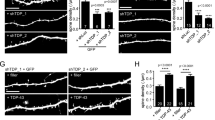
We investigated the effects of TDP-43 overexpression on neuronal morphology in hippocampal and cortical neurons, both of which are susceptible to neurodegeneration in ALS and FTD58,59,60,61,62. Our first experiments were performed using cultured prenatal rodent hippocampal neurons. We chose this primary neuronal culture system since it has been used extensively to investigate the cell biological mechanisms of dendritic growth and branching63,64,65,66,67 and because of its established use as a model to study TDP-43 biology68,69. We isolated hippocampal neurons from E18 rat pups and cultured them on a glial monolayer. Neurons were co-transfected at 2 days in vitro (DIV) with a plasmid expressing GFP to visualize neuronal morphology, and either a plasmid expressing the human TDP-43 gene under the control of a strong promoter (pCMV) at a concentration of 200 ng/well or 500 ng/well in a 24-well plate, or an empty vector control at 500 ng/well (Fig. 1a). At DIV 7, the cells were fixed and imaged, and dendritic complexity was quantified by Sholl Analysis70. Using this approach, we found that TDP-43 overexpression led to reduced dendritic branching (Fig. 1a,b). Increasing the concentration of the TDP-43-expressing plasmid during transfection further decreased the complexity of the dendritic arbor (Fig. 1a,b), indicating that TDP-43-mediated branching defects are dose-dependent.

Overexpression of TDP-43 leads to reduced dendritic complexity. Representative images showing DIV 7 (a) hippocampal neurons and (c) cortical neurons transfected at DIV 2 with a plasmid expressing GFP and an empty vector control, TDP-43 (200 ng/well), or TDP-43 (500 ng/well). Sholl analysis for indicated conditions in (b) hippocampal neurons (Control, N = 97; TDP-43 overexpression (200 ng/well), N = 100; TDP-43 overexpression (500 ng/well, N = 80) and (d) cortical neurons (N = 90 for each condition). Two-Way ANOVA with Tukey’s Test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The colour of the *represents the experimental condition being tested against control. For (b), statistical differences between TDP-43 200 ng/well and TDP-43 500 ng/well are as follows: *at 30 µm, **at 110 µm, ***at 40, 90, and 100 µm, and ****at 50, 60, 70, and 80 µm. For (d), statistical differences between TDP-43 200 ng/well and TDP-43 500 ng/well are as follows: **at 50, 70, 90, and 100 µm, ***at 60 and 80 µm, and ****at 110 µm.
As the cortex is primarily affected in FTD, we next sought to determine the effect of TDP-43 overexpression on dendritic arborization of dissociated cortical neurons from rodents. Similar to our result using hippocampal neurons, TDP-43 overexpression caused reduced dendritic branching in cultured cortical neurons in a dose-dependent manner (Fig. 1c,d). We traced individual neurites and found that TDP-43 overexpressing neurons also exhibit decreased total length for primary, secondary and tertiary neurites (Supplementary Fig. S1a). Further, neurons overexpressing TDP-43 display fewer secondary and tertiary branches (Supplementary Fig. S1b). By contrast, we found that control neurons and neurons overexpressing TDP-43 have the same number of primary neurites (Supplementary Fig. S1b), perhaps because these neurites formed prior to the time of transfection and TDP-43 overexpression71,72,73,74. Taken together, these data indicate that overexpression of TDP-43 reduces dendritic complexity in neurons isolated from two different brain regions that are also affected in human patients with ALS or FTD: hippocampus and cortex.
We were interested in understanding whether reduced dendritic branching was a general phenotype associated with overexpression of other ALS/FTD related genes. FUS (Fused in Sarcoma, also known as TLS) is an RNA-binding protein that is genetically linked to ALS and FTD and is structurally and functionally similar to TDP-4375; FUS is also ubiquitously expressed and predominantly localized to the nucleus, like TDP-4376. We tested the effect of FUS overexpression on dendritic morphology of cortical neurons using the same vector utilized previously for TDP-43 overexpression (pCMV). Neurons were co-transfected with GFP and either an empty vector control (500 ng/well), TDP-43 (500 ng/well), or FUS (500 ng/well). We analyzed dendritic complexity of the transfected neurons using Sholl analysis and found that FUS overexpression did not significantly alter dendritic branching compared to controls (Supplementary Fig. S2), indicating that decreased dendritic arborization is specific to manipulation of TDP-43.
To determine if reduced dendritic complexity could be attributed to poor cell health induced by TDP-43 overexpression, we asked if cortical neurons overexpressing TDP-43 displayed increased cell death relative to control transfected neurons. We assayed cell death using propidium iodide (PI), a membrane-impermeable nucleic acid stain that labels nuclei of dead cells. We quantified the percentage of transfected cells (co-labeled with GFP) that were positive for PI at DIV 4 or DIV 7 in control or TDP-43 overexpressing neurons that were transfected at DIV 2. We found no significant difference in the percentage of PI-positive neurons between the two conditions (Supplementary Fig. S3), indicating that reduced dendritic branching in neurons overexpressing TDP-43 is not a result of enhanced cell-death within this time-frame.
TDP-43 has been shown to regulate the expression of microtubule stabilizing proteins, such as MAP1B at Drosophila neuromuscular junctions45,46. Given that microtubule stabilization plays a major role in establishing and maintaining dendritic architecture, we asked whether there are similar changes in the expression of another microtubule regulator specifically found in neurons, Microtubule Associated Protein-2 (MAP2), which promotes microtubule stability in dendrites. We measured the levels of MAP2 in cortical neurons after overexpression of TDP-43 at 500 ng/well (Supplementary Fig. S4a). MAP2 protein levels in dendrites did not change significantly in the TDP-43 overexpression condition (Supplementary Fig. S4b), indicating that the reduction in dendritic branching does not result from a loss of MAP2 expression.
TDP-43 knockdown leads to reduced dendritic complexity
Given that depletion of nuclear TDP-43 is observed in ALS and FTD patient tissue8 and that TDP-43 LOF causes ALS-like phenotypes in mice77,78,79, we investigated whether TDP-43 knockdown affected neuronal morphology. We transiently transfected cortical neurons with GFP and a vector expressing previously validated shRNAs against TDP-4367 (33 ng/well or 100 ng/well), or an empty vector control. On DIV 7, we fixed these neurons, and confirmed depletion of TDP-43 by immunostaining with a polyclonal antibody recognizing TDP-43 (Fig. 2a). Relative levels of TDP-43 expression in both the nucleus and cytoplasm were calculated by normalizing intensity of TDP-43 staining within the soma to intensity of staining with an antibody that recognizes TFIIS, a ubiquitously expressed transcription elongation factor that is localized to the nucleus80 and is not expected to be affected by changes in TDP-43 expression levels (Fig. 2b). We then quantified neuronal morphology by Sholl analysis, and found that TDP-43 depletion, similar to TDP-43 overexpression, resulted in reduced dendritic complexity, consistent with previous reports67 (Fig. 2c,d). Both concentrations of the hairpin reduced the level of TDP-43 protein to a similar extent, indicating that increasing the dosage of hairpin doesn’t result in further reduction of TDP-43 protein or increased severity of the dendritic branching phenotype. Thus, both GOF and LOF of TDP-43 result in similar dendrite complexity phenotypes, suggesting that both decreased and increased TDP-43 expression causes misregulation of TDP-43 targets that control dendritic elaboration.

Knocking down TDP-43 expression results in reduced dendritic complexity. (a) Representative images of DIV 7 cortical neurons transfected on DIV 2 with GFP and either an empty vector or a short-hairpin targeting TDP-43 (shTDP; 33 ng/well). Neurons were stained using antibodies recognizing TDP-43 (magenta) and TFIIS (cyan) as a control. (b) Average fluorescence intensity of TDP-43 immunostaining in the soma (nucleus and cytoplasm) normalized to average fluorescence intensity of TFIIS in the nucleus (Control, N = 65; shTDP (33 ng/well), N = 58; shTDP (100 ng/well), N = 48; One-Way ANOVA with Tukey’s test, ****p < 0.0001. (c) Representative images showing DIV 7 cortical neurons transfected on DIV 2 with shRNA targeting TDP-43 (shTDP) at either 33ng/well or 100 ng/well or an empty vector control at 100 ng/well. (d) Sholl analysis comparing dendritic arbors of control neurons and shTDP neurons at the indicated concentrations (Control, N = 64; shTDP (33 ng/well), N = 50; shTDP (100 ng/well), N = 49). Two-Way ANOVA with Tukey’s test ****p < 0.0001. *represents comparisons between both shTDP conditions and the control condition.
Dendritic growth is suppressed by TDP-43 overexpression
Elaboration of the dendritic arbor is a dynamic process involving both growth and retraction of extending neurites81. Although TDP-43 overexpressing neurons display reduced dendritic branching, it remains unclear how this phenotype arises. We first quantified the expression levels of TDP-43 protein in neurons transfected with either an empty vector (500 ng/well) or TDP-43 (500 ng/well) over the time-course of our experiment, using a polyclonal TDP-43 antibody that recognizes both rat and human TDP-43, starting at 2 days post-transfection. Relative levels of TDP-43 expression were calculated by normalizing intensity of TDP-43 staining within the nucleus and cytoplasm to intensity of staining with a control antibody labeling TFIIS80. At DIV 4 (2 days post-transfection), we observed that neurons transfected with the TDP-43-expressing plasmid displayed approximately a 4-fold increase in TDP-43 protein levels compared to controls (Fig. 3a); this increased TDP-43 protein level remained constant for the remainder of the experiment (Fig. 3b,c). Given that TDP-43 predominantly localizes to the nucleus, we were curious whether TDP-43 protein levels were elevated in a specific subcellular compartment. To answer this question, we measured average fluorescence intensity for TDP-43 immunostaining within the Hoechst-labeled nuclear area or the GFP-filled cytoplasm in control and TDP-43 overexpressing neurons (Supplementary Fig. S5). We found that TDP-43 protein levels were increased in both the nucleus (Supplementary Fig. S5b) and the cytoplasm (Supplementary Fig. S5c). In addition, since TDP-43 has been reported to form pathological protein inclusions in post-mortem brain tissue, we asked if TDP-43 formed protein aggregates in our cell culture system by TDP-43 immunostaining. We did not observe TDP-43 inclusion formation by DIV 6 in our system. Instead, TDP-43 overexpression resulted in a diffuse localization pattern in both the nucleus and cytoplasm (Supplementary Fig. S5a).

TDP-43 overexpression reduces dendritic complexity by suppressing dendritic growth. (a) Average fluorescence intensity of TDP-43 immunostaining in the soma (nucleus and cytoplasm) normalized to average fluorescence intensity of TFIIS in the nucleus at DIV 4 (Control, N = 59, TDP-43 overexpression, N = 61), (b) DIV 5 (Control, N = 59, TDP-43 overexpression, N = 60), and (c) DIV 6 (Control, N = 60, TDP-43 overexpression, N = 60), respectively. ****p < 0.0001, unpaired t-test. (d) Representative images of an individual cortical neuron transfected with either an empty vector (control) or 500 ng/well TDP-43 (TDP-43 OE), repeatedly imaged at 8 hour intervals as indicated. (e) Average total dendrite length for each condition plotted as a function of time. *p < 0.05 for the comparison between TDP-43 overexpression and control condition using Unpaired t-test. Average total dendrite length for TDP-43 overexpression condition is not significantly different between 40 and 64 hours (Repeated Measures Two-Way ANOVA with Sidak’s multiple comparison). Control, N = 54; TDP-43 overexpression, N = 24. (f–h) Sholl analysis of hippocampal neurons comparing dendritic arbors between control and TDP-43 overexpressing neurons transfected on DIV 2 and fixed on (f) DIV 4 (Control, N = 52; TDP-43 overexpression, N = 54), (g) DIV 5 (Control, N = 62; TDP-43 overexpression, N = 61), and (h) DIV 6 (Control, N = 61; TDP-43 overexpression, N = 60). Two-way ANOVA with multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We reasoned that a TDP-43-dependent reduction in dendritic branching could be due to either a suppression of dendrite growth, or an increase in dendrite retraction. We used repeated imaging of individual cortical neurons over a period of 40 hours to track the effect of TDP-43 overexpression on dendrite extension. Cortical neurons were plated on separate 35 mm dishes and co-transfected 4 hours after plating with a plasmid expressing TdTomato to visualize neuronal morphology and either an empty vector (2 µg/dish) or a TDP-43-expressing plasmid (2 µg/dish). These vector concentrations scale proportionally with our previous transfections on 24-well plates at 500ng/well. The dendrites from each image were traced and measured for total dendrite length at each time point. We observed that dendrites from control neurons increased in length at each time point over the 40-hour imaging window (Fig. 3d,e). However, TDP-43-overexpressing neurons exhibited a reduced rate of dendritic growth compared to control neurons, and by 40 hours post-transfection halted their growth altogether (Fig. 3d,e), suggesting that TDP-43 overexpression suppresses dendritic outgrowth.
Similarly, hippocampal neurons overexpressing TDP-43 showed a consistent reduction in neurite outgrowth by Sholl analysis over time. We transfected hippocampal neurons with either a control plasmid (500 ng/well) or a TDP-43 plasmid (500 ng/well) and then fixed and imaged a subset of these neurons on each successive day starting at 2 days post-transfection (DIV4). We found that TDP-43 overexpressing neurons did not exhibit a statistically significant difference in dendritic complexity at DIV 4 (48 hours after transfection) (Fig. 3f). However, while the control transfected neurons increased their dendritic complexity from DIV4–DIV 6 by approximately 2-fold (e.g. the 50 µm circle radius), the TDP-43-overexpressing dendritic arbors did not change as drastically (Fig. 3g-h). Taken together, we conclude from these two experiments that decreased dendritic arborization in the TDP-43 overexpression condition is due to a suppression of dendritic growth starting approximately 40 hours after transfection which persists for at least 48 more hours.
Human TDP-43 mutations do not exacerbate the reduced branching phenotype
To date, over 50 nonsynonymous mutations in the human TDP-43 gene (TARDBP) have been identified in both familial and sporadic ALS patients82,83. Most of these mutations reside in the C-terminal glycine rich domain, which is essential in mediating protein-protein interactions49. A few of these mutations have been shown to impair the trafficking of TDP-43-containing RNP granules in axons41. In addition, mutant forms of TDP-43 have been reported to increase TDP-43 cytoplasmic localization in cultured cortical neurons68. Thus, we hypothesized that TDP-43 mutations might exacerbate the decreased dendritic branching phenotype observed upon overexpression of wild-type (WT) TDP-43. To test this hypothesis, we generated two TDP-43 constructs, each carrying a single point mutation previously described in human patients: TDP-43 A315T and TDP-43 M337V (Fig. 4a,b). We tested the effect of overexpressing these mutant forms of TDP-43 on dendritic branching by Sholl analysis at DIV 7, following 5 days of TDP-43 WT or mutant transfection. Interestingly, overexpression of either TDP-43 mutant caused a reduction in dendritic branching similar to the overexpression of WT TDP-43 (Fig. 4a,b). Our finding that the M337V and the A315T mutations do not cause a more severe dendritic growth phenotype than WT TDP-43 suggests that the TDP-43 dependent-signaling pathways which regulate dendritic branching are not additionally affected by these mutations.

Human TDP-43 mutations do not exacerbate the reduced branching phenotype (a) (top) Schematic of TDP-43 protein demonstrating location of A315T point mutation. NLS: Nuclear Localization Signal, RRM1, RRM2: RNA Recognition Motif, NES: Nuclear Export Signal, GRD: Glycine Rich Domain. (bottom) Sholl analysis for indicated conditions (Control, N = 60, TDP-43 overexpression, N = 60, TDP-43 A315T overexpression, N = 60). (b) (top) Schematic of TDP-43 protein demonstrating location of M337V point mutation; abbreviations as in a. (bottom) Sholl analysis for indicated conditions (Control, N = 60, TDP-43 overexpression, N = 60, TDP-43 M337V overexpression, N = 50). Two-Way ANOVA with Tukey’s test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The colour of the *represents the experimental condition being tested against control.
Inhibition of dendritic branching by TDP-43 overexpression depends on TDP-43 N-terminus and RNA-binding
Initial reports from the literature indicate that protein inclusions found in cortical neurons are predominantly composed of TDP-43 C-terminal fragments (CTFs)7,8,84. Numerous reports have also indicated that these CTFs are toxic to neurons85,86,87,88,89,90. For example, overexpression of CTFs in cultured rat forebrain neural precursors impairs neurite formation during neuronal differentiation90. TDP-43 CTF lacks its native nuclear localization signal (NLS)33 and it has been shown to be enriched in the cytoplasmic compartment90. Furthermore, TDP-43 CTF also lacks its primary RNA-recognition motif (RRM1), so it cannot interact with its RNA targets26. We sought to determine if overexpression of the C-terminal domain could also suppress dendritic branching. To address this question, we co-transfected cultured cortical neurons on DIV 2 with a GFP-expressing plasmid along with an empty vector (500 ng/well), or TDP-43 (500 ng/well), or a plasmid expressing a truncated TDP-43 (500 ng/well) which only expresses the C-terminal region of TDP-43 composed of amino acid residues 170–414 (hereafter called TDP-43 CTF) (Fig. 5a). These neurons were fixed on DIV 7 and assayed for dendritic complexity by Sholl analysis. We found that overexpression of TDP-43-CTF had no effect on dendritic complexity (Fig. 5a). These results indicate that the C-terminal domain of TDP-43, consisting of residues 170–414, is not sufficient to suppress dendritic growth, suggesting that perhaps the N-terminus of TDP-43 is required for this function.

RNA binding ability of TDP-43 is important for regulating dendritic branching (a) (top) Schematic of TDP-43 protein demonstrating the C-terminal fragment (CTF, amino acid residues 170–414) being overexpressed. RRM2: RNA Recognition Motif, NES: Nuclear Export Signal, GRD: Glycine Rich Domain. (bottom) Sholl analysis for indicated conditions (N = 60 for all conditions). The colour of the *represents the experimental condition being tested against control. (b) (top) Schematic of TDP-43 protein demonstrating TDP-43 ∆N (amino acid residues 10–414). NLS: Nuclear Localization Signal, RRM1: RNA Recognition Motif. (bottom) Sholl analysis for indicated conditions (Control, N = 57, TDP-43 overexpression, N = 45, TDP-43 ∆N overexpression, N = 54). (c) (top) Schematic of TDP-43 protein demonstrating TDP-43 5FL (F147L/F149L/F194L/F229L/F231L). (bottom) Sholl analysis for indicated conditions (Control, N = 87, TDP-43 overexpression, N = 76, TDP-43 5FL overexpression, N = 90). The colour of the *represent the experimental condition being tested against control. (a–c) Grey vs pink = not significant. Two-Way ANOVA with Tukey’s test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Recently, the N-terminal domain of TDP-43 has received much attention. Several groups have demonstrated that the N-terminus of TDP-43 contributes to the formation of homodimers91,92,93,94 and enhances splicing activity93,94. Therefore, we investigated whether the overexpression of a TDP-43 construct containing amino acid residues 10–414 (hereafter referred to as TDP-43 ∆N) was sufficient to suppress dendritic branching. Cultured cortical neurons were co-transfected on DIV 2 with a GFP-expressing plasmid along with an empty vector (500 ng/well), or TDP-43 (500 ng/well), or TDP-43 ∆N (500 ng/well). On DIV 7, these neurons were fixed and assayed by Sholl analysis. We found that TDP-43 ∆N overexpression had no effect on dendritic branching (Fig. 5b), suggesting that homodimerization and/or splicing function may play a role in mediating reduced dendritic complexity.
TDP-43 is a RNA binding protein involved in several aspects of RNA metabolism including splicing, transport, and translation41,45,95. One model to explain our observations is that TDP-43 GOF or LOF causes misregulation of one or more of its target mRNAs, which regulate dendritic growth. Therefore, we asked if overexpression of a mutant form of TDP-43 that cannot bind RNA results in reduced dendritic complexity. We addressed this question by introducing five point mutations into TDP-43, F147/149 L in RRM1 and F194/229/231 L in RRM2, that have been shown to abolish its RNA-binding ability (hereafter called TDP-43 5FL)26. We co-transfected cortical neurons with a GFP-expressing plasmid along with an empty vector (500 ng/well), TDP-43 (500 ng/well), or a plasmid expressing TDP-43 5FL (500 ng/well) on DIV 2 and fixed these neurons on DIV 7. Overexpression of TDP-43 5FL had no effect on dendritic complexity (Fig. 5c), suggesting that the ability of TDP-43 to inhibit dendritic branching is due to its RNA-binding function.
Discussion
In this study, we demonstrate that increasing levels of TDP-43 in cultured mammalian neurons suppresses dendritic growth. This is in contrast to overexpression of the RNA binding protein and ALS gene, FUS, which had no effect on dendrite growth. Abnormal dendritic morphology is a common characteristic of several different neurological disorders96,97,98,99,100,101,102,103 and reduced dendritic complexity has been reported to precede neurodegeneration104. Importantly, TDP-43 overexpression also causes reduced dendritic complexity in both hippocampal and cortical neurons. The hippocampus and cortex are two regions of the brain that are prone to neurodegeneration in both ALS and FTD105. Cell morphological changes such as reduced dendritic complexity could interfere with circuit connectivity and disrupt cell to cell communication, ultimately affecting cognition and motor function.
In addition, we and others67 have demonstrated that RNAi-mediated TDP-43 knockdown in cortical neurons results in reduced dendritic branching. Indeed, TDP-43 knockdown and overexpression in several systems has resulted in similar cell morphological and behavioral phenotypes15,16,17. For example, both GOF or LOF of TDP-43 cause motor deficits in zebrafish15, defects in growth receptor signaling16, and affect expression of a set of proteins in a similar manner17. Taken together, these results suggest that both increased or decreased levels of TDP-43 negatively affects neuron function, perhaps via misregulation of a subset of critical TDP-43 targets13,106. Alternatively, TDP-43 LOF and GOF could affect dendritic growth through distinct mechanisms or targets.
We also asked if TDP-43 overexpression affected dendritic arborization by suppressing dendrite growth or promoting dendrite retraction. We distinguished between these mechanisms by performing a time-lapse analysis of dendrite elaboration in living cortical neurons overexpressing TDP-43, and an examination of neurite growth and branching in fixed neurons transfected with TDP-43 at 2 days, 3 days, and 4 days post-transfection. We found that dendritic growth is suppressed when TDP-43 protein levels are elevated, consistent with our live-imaging data in which TDP-43 overexpressing neurons stop dendritic growth at approximately 40 hours post-transfection. Thus, increased levels of TDP-43 alter dendritic complexity by suppressing dendritic growth, providing insight into possible cellular mechanisms by which TDP-43 regulates dendritic arborization.
Given that TDP-43 is an RNA-binding protein that regulates the splicing, transport, and stability of thousands of RNA transcripts, we asked if the RNA binding ability of TDP-43 was required to suppress dendritic branching. Interestingly, our data shows that reduction in dendritic branching due to TDP-43 overexpression is dependent on this function. This result is reminiscent of studies in C. elegans, D. melanogaster, and G. gallus models in which phenotypes associated with TDP-43 overexpression are abolished by disrupting TDP-43 RNA-binding ability107,108,109. Specifically, overexpression of wild-type TDP-43 led to a reduction of motor neuron number in G. gallus spinal cord while expression of an RNA-binding deficient TDP-43 did not cause motor neuron loss107. Thus, our results suggest that TDP-43 overexpression interferes with one or more RNA binding functions that is relevant to dendritic growth and branching.
At least one important function of TDP-43 that requires RNA-binding is splicing. Interestingly, the N-terminus of TDP-43 has been reported to play a vital role in splicing activity93,94. Therefore, we investigated whether overexpressing a TDP-43 construct lacking the first 9 amino acid residues could affect dendritic complexity. Consistent with previous reports, the extreme N-terminus of TDP-43 is required to suppress dendritic outgrowth93. TDP-43 ∆N still contains both RRMs and is presumably still capable of an RNA-binding interaction. Thus, our results indicate that, after RNA-binding, the splicing function of TDP-43 may also be necessary to suppress dendritic elaboration. However, we cannot rule out other possible functions that may become defective upon truncation of the TDP-43 N-terminus that lie downstream of RNA-binding such as RNA transport or translation repression.
Similarly, CTFs are a pathological hallmark of ALS and FTD8,84, are enriched in the cytoplasm90 due to loss of the NLS, and are prone to aggregation88. It is hypothesized that CTFs may act as a sink and recruit wild-type TDP-43 to form pathological protein inclusions90,110,111,112. While a previous study demonstrated that overexpression of CTFs impairs neurite growth in NSC-34 cells90, we observed that overexpression of TDP-43 CTFs in mammalian cortical neurons does not alter dendrite morphology. The disparity in results between our two studies may lay in the fact that different cell types were employed for these experiments. Our results indicate that the pathological function of CTFs is independent of an effect on dendrite elaboration. Importantly, because the CTF lacks the TDP-43 RNA binding domain, these results support our conclusion that RNA binding ability is required for TDP-43 inhibition of dendrite growth.
In conclusion, our data indicate that either increasing or decreasing TDP-43 levels has negative consequences for dendrite elaboration in both cortical and hippocampal neurons, which could contribute to the pathology of diseases such as ALS and FTD. Although the molecular mechanisms behind TDP-43 mediated regulation of dendritic architecture remain to be elucidated, our data demonstrate that TDP-43 RNA-binding function plays a major role in altering neuronal morphology. In addition, our results further support the relevance of studying TDP-43 overexpression in neuronal cultures, as the phenotypic consequences of this genetic manipulation is dependent upon a well-established biological function of TDP-43. Thus, a careful examination of RNA misregulation in our culture system could yield insights into signaling pathways that are perturbed upon TDP-43 misexpression and are relevant to dendritic growth. This system can potentially provide a platform to test avenues for therapeutic intervention in ALS/FTD.
Methods
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. All animal procedures were approved by the Brandeis University Institutional Animal Care and Usage Committee and all experiments were performed in accordance with relevant guidelines and regulations.
Plasmids
Plasmids expressing full length human TDP-43 (Addgene plasmid # 28206) and C-terminal truncated human TDP-43 (hTDP-43170–414; Addgene plasmid # 28196) were a gift from Dr. Zuoshang Xu (University of Massachusetts Medical School, Worcester MA)90. Plasmids expressing full-length human FUS (Addgene plasmid # 29609) and TDP-43 5FL (Addgene plasmid # 84914) was a gift from Aaron Gitler (Stanford University School of Medicine, Stanford CA). pCSCMV:tdTomato was a gift from Gerhart Ryffel (Addgene plasmid # 30530). Full-length TDP-43, FUS, and TDP-43 5FL were cloned into pCMV-myc vector backbone using Gibson Assembly (NEB). Constructs containing various mutations of TDP-43 were generated from pCMV-myc hTDP-43 plasmid by site-directed mutagenesis using QuickChange Site Directed Mutagenesis Kit (Stratagene, San Diego CA). TDP-43 ∆N was generated by cloning from the pCMV-myc hTDP-43 plasmid. Primers used to generate indicated mutations are as follows:
M337V: GAGCAGTTGGGGTATGGTGGGCATGTTAGCC
A315T: TGGTGGGATGAACTTTGGTACGTTCAGCATTAATCC
∆N: GAGGCCCGAATTCGGGGACGATGGTGTGACTGC
Primary Neuronal Cultures and Transfection
Dissociated rat cortical or hippocampal neurons were cultured on a feeder layer of astrocytes as described previously113. Briefly, a layer of confluent astrocytes was generated by plating the cells onto 12 mm glass coverslips coated with poly-D-lysine (20 μg/ml) and laminin (3.4 μg/ml) in 24 well plates. Dissociated hippocampal or cortical neurons from E18 rat embryos were plated on this layer of astrocytes at the density of 80,000 cells/well, and grown in Neurobasal medium supplemented with B27 (Thermo Fisher) at 37 °C.
Neurons were transfected at the desired stage by using the calcium phosphate transfection method114, which yields a low transfection efficiency, allowing development of transfected neurons within a large population of unaffected neurons, as well as better visualization of neuronal morphology for individual transfected neurons. All experimental conditions were co-transfected with pCMV-GFP plasmid at 500 ng/well to visualize cellular morphology. In addition, all wells were co-transfected with either pCMV-myc plasmid for control empty-vector conditions, or other plasmid DNA as described.
Analysis of Neuronal Morphology
Transfected neurons were fixed at the desired time-point using 4% paraformaldehyde + 4% sucrose solution in PBS for 8 minutes at room temperature followed by 3 washes in 1X PBS. Images of individual transfected neurons were acquired on Olympus Fluoview 300 confocal microscope using a 20X oil objective (numerical aperture [NA] 0.85) in order to image the entire dendritic arbor (5–10 steps at 1 μm optical sectioning). All conditions were imaged at similar acquisition settings for laser power and detector gain in a blinded manner, and unblinded only after analysis.
Image Analysis
Maximum Intensity Projections (MIP) were generated from individual Z stacks using ImageJ. For Sholl analysis, a series of 11 concentric circles (10 μm intervals) centered at the soma, starting at 10 µm, was overlaid on the MIP using the ‘Concentric circles’ plugin. The number of dendrites crossing each circle was measured manually. For each experimental condition, a total of 15–30 neurons were analyzed from 2–3 coverslips with 5–15 neurons per covers lip. The reported results combine data from at least 2 independent biological replicates. For measuring total neurite length and branch tip number, neurites were traced using the NeuronJ plugin in ImageJ. The sum length of all the neurites was calculated for 10–15 neurons from 2–3 coverslips and the results were combined from 2 independent biological replicates.
To measure TDP-43 protein levels, sum projections were generated from individual Z stacks with ImageJ. In ImageJ, using GFP as a marker for transfected cells, a region of interest was drawn around the soma and fluorescence intensity was measured in that area in the TDP-43 channel. TFIIS fluorescence intensity was measured by drawing a region of interest around the Hoechst-33342-labeled nucleus, and fluorescence intensity was measured in the TFIIS channel. TDP-43 intensity was normalized to TFIIS intensity for every cell.
Nuclear vs cytoplasmic levels of TDP-43 staining were measured as follows. For nuclear TDP-43 levels, the region of interest was defined by the Hoechst-33342 labeling and TDP-43 intensity was measured in the TDP-43 immunostained channel. The TDP-43 levels in the cytoplasm were determined by drawing a region of interest around the GFP-filled soma, measuring TDP-43 intensity in the TDP-43 channel, and subtracting the previously measured nuclear TDP-43 intensity.
MAP2 levels were quantified by measuring fluorescence intensity of MAP2 immunostaining from empty vector or TDP-43-transfected cells (500 ng/well). Sum projections of each condition were generated using ImageJ, with GFP as a marker for transfected cells. Average intensity was calculated by taking the mean intensity from 2 separate dendrites of a transfected neuron followed by normalizing to the average intensity from 2 other dendrites from non-transfected cells.
Cell death Assay
Hippocampal neurons were cultured in 24-well plates as described and transfected with pCMV-GFP plasmid along with either empty pCMV-myc control plasmid at 500ng/well or full-length hTDP-43 at 500 ng/well. Neurons were incubated in 2 µg/ml Propidium Iodide solution (Life Technology) in HEPES buffer (117 mM NaCl, 5.3 mM KCl, 1.8 mM CaCl2, 0.814 mM MgSO4, 1 mM NaH2PO4, 20 mM HEPES, 50 mM D-glucose, 0.1% BSA, 320 mOsm, pH 7.2) for 1 hour at 37 °C followed by 1X PBS wash. Cultures were fixed with 4% PFA + 4% sucrose solution in PBS. GFP-positive neurons were counted manually using a 60x objective and an epiflourescence microscope, and the presence or absence of PI fluorescence was noted for each GFP-positive neuron by flipping between filter sets. For each experimental condition, 2 coverslips each from 2 independent experiments were analyzed.
Immunostaining
Following primary antibodies were used for immunostaining, α-TDP-43 (10782-2-AP) from ProteinTech (used at 1:500); α-TFIIS (611204) from BD Transduction Laboratories (used at 1:275); α-MAP2 (M4403) from Sigma-Aldrich (used at 1:1000). Secondary antibodies were conjugated to Cy-3 or Cy-5 (1:500, Jackson ImmunoResearch Laboratories). Coverslips containing transfected neurons were fixed as described above followed by 3 washes with 1X PBS. The coverslips were then incubated with primary antibodies) diluted in gelatin blocking buffer at 4 °C overnight in a humidified chamber (30 minutes at room temperature for MAP2). Coverslips were then washed 3 times with 1X PBS and incubated with secondary antibodies at room temperature for 2 hours. Coverslips were then washed 3 times with 1X PBS, submerged in MilliQ water and then mounted on glass microscope slides with Aquamount (Lerner Laboratories). Neurons were imaged using an Ni-E inverted microscope equipped with a Nikon C2 confocal head and a 60X oil (NA 1.4) objective, using similar acquisition settings for laser power, offset and detector gain across conditions in a blinded manner.
Live Imaging
An astrocyte feeder layer and cortical neurons were plated as described above on 35 mm glass-bottom dishes. 4 hours after plating, neurons were transfected via the calcium phosphate method with a plasmid expressing TdTomato to fill the cell and an empty vector control or a vector expressing TDP-43; our empirical observation was that TdTomato is less phototoxic than GFP for these live imaging experiments. The next day (~24 hours later) the neurons were placed in an OKO labs temperature controlled environmental chamber (5% CO2, 95% O2) and 20–30 neurons were randomly selected from each condition. Images of the selected neurons were acquired every 8 hours for 40 hours using an Ni-E inverted microscope equipped with a Nikon C2 confocal head and a 40X (NA 0.95) objective. Images were collected using Nikon Elements AR software. The experimenters were not blinded to the condition during the selection of neurons but were blinded to each condition during analysis. Images from each time point were analyzed by tracing individual dendrites using ImageJ.
References
Ringholz, G. M. et al. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590, https://doi.org/10.1212/01.wnl.0000172911.39167.b6 (2005).
Wheaton, M. W. et al. Cognitive impairment in familial ALS. Neurology 69, 1411–1417, https://doi.org/10.1212/01.wnl.0000277422.11236.2c (2007).
Lomen-Hoerth, C., Anderson, T. & Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079 (2002).
Burrell, J. R., Kiernan, M. C., Vucic, S. & Hodges, J. R. Motor neuron dysfunction in frontotemporal dementia. Brain 134, 2582–2594, https://doi.org/10.1093/brain/awr195 (2011).
Sreedharan, J. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672, https://doi.org/10.1126/science.1154584 (2008).
Kovacs, G. G. et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord 24, 1843–1847, https://doi.org/10.1002/mds.22697 (2009).
Arai, T. et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351, 602–611, https://doi.org/10.1016/j.bbrc.2006.10.093 (2006).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133, https://doi.org/10.1126/science.1134108 (2006).
Scheuner, D. et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2, 864–870 (1996).
Imarisio, S. et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J 412, 191–209, https://doi.org/10.1042/BJ20071619 (2008).
Oikawa, T. et al. alpha-Synuclein Fibrils Exhibit Gain of Toxic Function, Promoting Tau Aggregation and Inhibiting Microtubule Assembly. J Biol Chem 291, 15046–15056, https://doi.org/10.1074/jbc.M116.736355 (2016).
Avila, J. et al. Tau phosphorylation in hippocampus results in toxic gain-of-function. Biochem Soc Trans 38, 977–980, https://doi.org/10.1042/BST0380977 (2010).
Polymenidou, M. et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14, 459–468, https://doi.org/10.1038/nn.2779 (2011).
Igaz, L. M. et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest 121, 726–738, https://doi.org/10.1172/JCI44867 (2011).
Kabashi, E. et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet 19, 671–683, https://doi.org/10.1093/hmg/ddp534 (2010).
Deshpande, M. et al. Role of BMP receptor traffic in synaptic growth defects in an ALS model. Mol Biol Cell 27, 2898–2910, https://doi.org/10.1091/mbc.E16-07-0519 (2016).
Prpar Mihevc, S., Baralle, M., Buratti, E. & Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci Rep 6, 33996, https://doi.org/10.1038/srep33996 (2016).
Lee, E. B., Lee, V. M. & Trojanowski, J. Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci 13, 38–50, https://doi.org/10.1038/nrn3121 (2011).
Abhyankar, M. M., Urekar, C. & Reddi, P. P. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem 282, 36143–36154, https://doi.org/10.1074/jbc.M705811200 (2007).
Ou, S. H., Wu, F., Harrich, D., Garcia-Martinez, L. F. & Gaynor, R. B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69, 3584–3596 (1995).
Buratti, E. et al. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J 277, 2268–2281, https://doi.org/10.1111/j.1742-4658.2010.07643.x (2010).
Fan, Z., Chen, X. & Chen, R. Transcriptome-wide analysis of TDP-43 binding small RNAs identifies miR-NID1 (miR-8485), a novel miRNA that represses NRXN1 expression. Genomics 103, 76–82, https://doi.org/10.1016/j.ygeno.2013.06.006 (2014).
Kawahara, Y. & Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci USA 109, 3347–3352, https://doi.org/10.1073/pnas.1112427109 (2012).
King, I. N. et al. The RNA-binding protein TDP-43 selectively disrupts microRNA-1/206 incorporation into the RNA-induced silencing complex. J Biol Chem 289, 14263–14271, https://doi.org/10.1074/jbc.M114.561902 (2014).
Zhang, Z. et al. Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS One 8, e76055, https://doi.org/10.1371/journal.pone.0076055 (2013).
Buratti, E. & Baralle, F. E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem 276, 36337–36343, https://doi.org/10.1074/jbc.M104236200 (2001).
Mercado, P. A., Ayala, Y. M., Romano, M., Buratti, E. & Baralle, F. E. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic acids research 33, 6000–6010, https://doi.org/10.1093/nar/gki897 (2005).
Bose, J. K., Wang, I. F., Hung, L., Tarn, W. Y. & Shen, C. K. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem 283, 28852–28859, https://doi.org/10.1074/jbc.M805376200 (2008).
Fiesel, F. C., Weber, S. S., Supper, J., Zell, A. & Kahle, P. J. TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res 40, 2668–2682, https://doi.org/10.1093/nar/gkr1082 (2012).
Mohagheghi, F. et al. TDP-43 functions within a network of hnRNP proteins to inhibit the production of a truncated human SORT1 receptor. Hum Mol Genet 25, 534–545, https://doi.org/10.1093/hmg/ddv491 (2016).
Prudencio, M. et al. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci USA 109, 21510–21515, https://doi.org/10.1073/pnas.1211577110 (2012).
Shiga, A. et al. Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS One 7, e43120, https://doi.org/10.1371/journal.pone.0043120 (2012).
Winton, M. J. et al. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283, 13302–13309, https://doi.org/10.1074/jbc.M800342200 (2008).
Colombrita, C. et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem 111, 1051–1061, https://doi.org/10.1111/j.1471-4159.2009.06383.x (2009).
Dewey, C. M. et al. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol 31, 1098–1108, https://doi.org/10.1128/MCB.01279-10 (2011).
Freibaum, B. D., Chitta, R. K., High, A. A. & Taylor, J. P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res 9, 1104–1120, https://doi.org/10.1021/pr901076y (2010).
Liu-Yesucevitz, L. et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5, e13250, https://doi.org/10.1371/journal.pone.0013250 (2010).
McDonald, K. K. et al. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20, 1400–1410, https://doi.org/10.1093/hmg/ddr021 (2011).
Meyerowitz, J. et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol Neurodegener 6, 57, https://doi.org/10.1186/1750-1326-6-57 (2011).
Moisse, K. et al. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res 1249, 202–211, https://doi.org/10.1016/j.brainres.2008.10.021 (2009).
Alami, N. H. et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543, https://doi.org/10.1016/j.neuron.2013.12.018 (2014).
Fallini, C., Bassell, G. J. & Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet 21, 3703–3718, https://doi.org/10.1093/hmg/dds205 (2012).
Wang, I. F., Wu, L. S., Chang, H. Y. & Shen, C. K. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 105, 797–806, https://doi.org/10.1111/j.1471-4159.2007.05190.x (2008).
Godena, V. K. et al. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS One 6, e17808, https://doi.org/10.1371/journal.pone.0017808 (2011).
Coyne, A. N. et al. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci 34, 15962–15974, https://doi.org/10.1523/JNEUROSCI.2526-14.2014 (2014).
Romano, M., Feiguin, F. & Buratti, E. TBPH/TDP-43 modulates translation of Drosophila futsch mRNA through an UG-rich sequence within its 5’UTR. Brain Res 1647, 50–56, https://doi.org/10.1016/j.brainres.2016.02.022 (2016).
Higashi, S. et al. TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J Neurochem 126, 288–300, https://doi.org/10.1111/jnc.12194 (2013).
Ling, S. C. et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proceedings of the National Academy of Sciences of the United States of America 107, 13318–13323, https://doi.org/10.1073/pnas.1008227107 (2010).
Buratti, E. et al. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280, 37572–37584, https://doi.org/10.1074/jbc.M505557200 (2005).
Lee, S. B., Bagley, J. A., Lee, H. Y., Jan, L. Y. & Jan, Y. N. Pathogenic polyglutamine proteins cause dendrite defects associated with specific actin cytoskeletal alterations in Drosophila. Proc Natl Acad Sci USA 108, 16795–16800, https://doi.org/10.1073/pnas.1113573108 (2011).
Kweon, J. H., Kim, S. & Lee, S. B. The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep 50, 5–11 (2017).
Kulkarni, V. A. & Firestein, B. L. The dendritic tree and brain disorders. Mol Cell Neurosci 50, 10–20, https://doi.org/10.1016/j.mcn.2012.03.005 (2012).
Jara, J. H., Villa, S. R., Khan, N. A., Bohn, M. C. & Ozdinler, P. H. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis 47, 174–183, https://doi.org/10.1016/j.nbd.2012.03.036 (2012).
Fogarty, M. J., Noakes, P. G. & Bellingham, M. C. Motor cortex layer V pyramidal neurons exhibit dendritic regression, spine loss, and increased synaptic excitation in the presymptomatic hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Neurosci 35, 643–647, https://doi.org/10.1523/JNEUROSCI.3483-14.2015 (2015).
Genc, B. et al. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci Rep 7, 41765, https://doi.org/10.1038/srep41765 (2017).
Ayala, Y. M., Pagani, F. & Baralle, F. E. TDP43 depletion rescues aberrant CFTR exon 9 skipping. FEBS Lett 580, 1339–1344, https://doi.org/10.1016/j.febslet.2006.01.052 (2006).
Costessi, L., Porro, F., Iaconcig, A. & Muro, A. F. TDP-43 regulates beta-adducin (Add2) transcript stability. RNA Biol 11, 1280–1290, https://doi.org/10.1080/15476286.2014.996081 (2014).
Frisoni, G. B. et al. Hippocampal and entorhinal cortex atrophy in frontotemporal dementia and Alzheimer’s disease. Neurology 52, 91–100 (1999).
Laakso, M. P. et al. Hippocampus and entorhinal cortex in frontotemporal dementia and Alzheimer’s disease: a morphometric MRI study. Biol Psychiatry 47, 1056–1063 (2000).
Takeda, T., Uchihara, T., Arai, N., Mizutani, T. & Iwata, M. Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: distinction from Alzheimer disease. Acta Neuropathol 117, 35–44, https://doi.org/10.1007/s00401-008-0447-2 (2009).
Abdulla, S. et al. Hippocampal degeneration in patients with amyotrophic lateral sclerosis. Neurobiol Aging 35, 2639–2645, https://doi.org/10.1016/j.neurobiolaging.2014.05.035 (2014).
Raaphorst, J. et al. Prose memory impairment in amyotrophic lateral sclerosis patients is related to hippocampus volume. Eur J Neurol 22, 547–554, https://doi.org/10.1111/ene.12615 (2015).
Mattson, M. P., Dou, P. & Kater, S. B. Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J Neurosci 8, 2087–2100 (1988).
Kossel, A. H., Williams, C. V., Schweizer, M. & Kater, S. B. Afferent innervation influences the development of dendritic branches and spines via both activity-dependent and non-activity-dependent mechanisms. J Neurosci 17, 6314–6324 (1997).
Papa, M., Bundman, M. C., Greenberger, V. & Segal, M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J Neurosci 15, 1–11 (1995).
Ghiretti, A. E., Kenny, K., Marr, M. T. 2nd & Paradis, S. CaMKII-dependent phosphorylation of the GTPase Rem2 is required to restrict dendritic complexity. J Neurosci 33, 6504–6515, https://doi.org/10.1523/JNEUROSCI.3861-12.2013 (2013).
Schwenk, B. M. et al. TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J 35, 2350–2370, https://doi.org/10.15252/embj.201694221 (2016).
Barmada, S. J. et al. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30, 639–649, https://doi.org/10.1523/JNEUROSCI.4988-09.2010 (2010).
Barmada, S. J. et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10, 677–685, https://doi.org/10.1038/nchembio.1563 (2014).
Sholl, D. A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87, 387–406 (1953).
Bray, D. Branching patterns of individual sympathetic neurons in culture. J Cell Biol 56, 702–712 (1973).
Banker, G. A. & Cowan, W. M. Further observations on hippocampal neurons in dispersed cell culture. J Comp Neurol 187, 469–493, https://doi.org/10.1002/cne.901870302 (1979).
Bartlett, W. P. & Banker, G. A. An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. II. Synaptic relationships. J Neurosci 4, 1954–1965 (1984).
Kaech, S. & Banker, G. Culturing hippocampal neurons. Nat Protoc 1, 2406–2415, https://doi.org/10.1038/nprot.2006.356 (2006).
Mackenzie, I. R., Rademakers, R. & Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9, 995–1007, https://doi.org/10.1016/S1474-4422(10)70195-2 (2010).
Morohoshi, F. et al. Genomic structure of the human RBP56/hTAFII68 and FUS/TLS genes. Gene 221, 191–198 (1998).
Iguchi, Y. et al. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain 136, 1371–1382, https://doi.org/10.1093/brain/awt029 (2013).
Kraemer, B. C. et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 119, 409–419, https://doi.org/10.1007/s00401-010-0659-0 (2010).
Yang, C. et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 111, E1121–1129, https://doi.org/10.1073/pnas.1322641111 (2014).
Wind, M. & Reines, D. Transcription elongation factor SII. Bioessays 22, 327–336, https://doi.org/10.1002/(SICI)1521-1878(200004)22:4 <327::AID-BIES3> 3.0.CO;2-4 (2000).
Dailey, M. E. & Smith, S. J. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16, 2983–2994 (1996).
Scotter, E. L., Chen, H. J. & Shaw, C. E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 12, 352–363, https://doi.org/10.1007/s13311-015-0338-x (2015).
Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv Genet 91, 1–53, https://doi.org/10.1016/bs.adgen.2015.07.001 (2015).
Igaz, L. M. et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173, 182–194, https://doi.org/10.2353/ajpath.2008.080003 (2008).
Stallings, N. R., Puttaparthi, K., Luther, C. M., Burns, D. K. & Elliott, J. L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis 40, 404–414, https://doi.org/10.1016/j.nbd.2010.06.017 (2010).
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M. & Baloh, R. H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 106, 18809–18814, https://doi.org/10.1073/pnas.0908767106 (2009).
Wils, H. et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 107, 3858–3863, https://doi.org/10.1073/pnas.0912417107 (2010).
Zhang, Y. J. et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 106, 7607–7612, https://doi.org/10.1073/pnas.0900688106 (2009).
Igaz, L. M. et al. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J Biol Chem 284, 8516–8524, https://doi.org/10.1074/jbc.M809462200 (2009).
Yang, C. et al. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS One 5, e15878, https://doi.org/10.1371/journal.pone.0015878 (2010).
Chang, C. K. et al. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem Biophys Res Commun 425, 219–224, https://doi.org/10.1016/j.bbrc.2012.07.071 (2012).
Wang, Y. T. et al. The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J Biol Chem 288, 9049–9057, https://doi.org/10.1074/jbc.M112.438564 (2013).
Zhang, Y. J. et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum Mol Genet 22, 3112–3122, https://doi.org/10.1093/hmg/ddt166 (2013).
Jiang, L. L. et al. The N-terminal dimerization is required for TDP-43 splicing activity. Sci Rep 7, 6196, https://doi.org/10.1038/s41598-017-06263-3 (2017).
Buratti, E. et al. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20, 1774–1784, https://doi.org/10.1093/emboj/20.7.1774 (2001).
Raymond, G. V., Bauman, M. L. & Kemper, T. L. Hippocampus in autism: a Golgi analysis. Acta Neuropathol 91, 117–119 (1996).
Armstrong, D., Dunn, J. K., Antalffy, B. & Trivedi, R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol 54, 195–201 (1995).
Armstrong, D. D., Dunn, K. & Antalffy, B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol 57, 1013–1017 (1998).
Anderton, B. H. et al. Dendritic changes in Alzheimer’s disease and factors that may underlie these changes. Prog Neurobiol 55, 595–609 (1998).
Terry, R. D. et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580, https://doi.org/10.1002/ana.410300410 (1991).
Couch, B. A., DeMarco, G. J., Gourley, S. L. & Koleske, A. J. Increased dendrite branching in AbetaPP/PS1 mice and elongation of dendrite arbors by fasudil administration. J Alzheimers Dis 20, 1003–1008, https://doi.org/10.3233/JAD-2010-091114 (2010).
Becker, L., Mito, T., Takashima, S. & Onodera, K. Growth and development of the brain in Down syndrome. Prog Clin Biol Res 373, 133–152 (1991).
Takashima, S., Ieshima, A., Nakamura, H. & Becker, L. E. Dendrites, dementia and the Down syndrome. Brain Dev 11, 131–133 (1989).
Lopez-Domenech, G. et al. Loss of Dendritic Complexity Precedes Neurodegeneration in a Mouse Model with Disrupted Mitochondrial Distribution in Mature Dendrites. Cell Rep 17, 317–327, https://doi.org/10.1016/j.celrep.2016.09.004 (2016).
Ferrari, R., Kapogiannis, D., Huey, E. D. & Momeni, P. FTD and ALS: a tale of two diseases. Curr Alzheimer Res 8, 273–294 (2011).
Tollervey, J. R. et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14, 452–458, https://doi.org/10.1038/nn.2778 (2011).
Voigt, A. et al. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS One 5, e12247, https://doi.org/10.1371/journal.pone.0012247 (2010).
Ash, P. E. et al. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19, 3206–3218, https://doi.org/10.1093/hmg/ddq230 (2010).
Ihara, R. et al. RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum Mol Genet 22, 4474–4484, https://doi.org/10.1093/hmg/ddt296 (2013).
Che, M. X., Jiang, Y. J., Xie, Y. Y., Jiang, L. L. & Hu, H. Y. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J 25, 2344–2353, https://doi.org/10.1096/fj.10-174482 (2011).
Che, M. X., Jiang, L. L., Li, H. Y., Jiang, Y. J. & Hu, H. Y. TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett 589, 1920–1928, https://doi.org/10.1016/j.febslet.2015.06.009 (2015).
Nonaka, T., Kametani, F., Arai, T., Akiyama, H. & Hasegawa, M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet 18, 3353–3364, https://doi.org/10.1093/hmg/ddp275 (2009).
Ghiretti, A. E. & Paradis, S. The GTPase Rem2 regulates synapse development and dendritic morphology. Dev Neurobiol 71, 374–389, https://doi.org/10.1002/dneu.20868 (2011).
Xia, Z., Dudek, H., Miranti, C. K. & Greenberg, M. E. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci 16, 5425–5436 (1996).
Acknowledgements
This work was supported by a Blazeman Foundation for ALS Research Postdoctoral Fellowship to M.D. and R01NS065856 to S. P. We would like to thank Amanda K. Shilton and Paul Johnson for technical assistance.
Author information
Authors and Affiliations
Contributions
J.J.H., M.D., A.A.R, and S.P. designed the research while J.J.H., M.D., and L.S. performed experiments and analyzed the data. J.J.H. and S.P. wrote the manuscript with input from M.D. and A.A.R.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herzog, J.J., Deshpande, M., Shapiro, L. et al. TDP-43 misexpression causes defects in dendritic growth. Sci Rep 7, 15656 (2017). https://doi.org/10.1038/s41598-017-15914-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15914-4
This article is cited by
-
Mitigating a TDP-43 proteinopathy by targeting ataxin-2 using RNA-targeting CRISPR effector proteins
Nature Communications (2023)
-
Loss of TDP-43 function underlies hippocampal and cortical synaptic deficits in TDP-43 proteinopathies
Molecular Psychiatry (2023)
-
Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss
Acta Neuropathologica Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
