
Abstract
The cell adhesion molecule L1 and the extracellular matrix protein Reelin play crucial roles in the developing nervous system. Reelin is known to activate signalling cascades regulating neuronal migration by binding to lipoprotein receptors. However, the interaction of Reelin with adhesion molecules, such as L1, has remained poorly explored. Here, we report that full-length Reelin and its N-terminal fragments N-R2 and N-R6 bind to L1 and that full-length Reelin and its N-terminal fragment N-R6 proteolytically cleave L1 to generate an L1 fragment with a molecular mass of 80 kDa (L1-80). Expression of N-R6 and generation of L1-80 coincide in time at early developmental stages of the cerebral cortex. Reelin-mediated generation of L1-80 is involved in neurite outgrowth and in stimulation of migration of cultured cortical and cerebellar neurons. Morphological abnormalities in layer formation of the cerebral cortex of L1-deficient mice partially overlap with those of Reelin-deficient reeler mice. In utero electroporation of L1-80 into reeler embryos normalised the migration of cortical neurons in reeler embryos. The combined results indicate that the direct interaction between L1 and Reelin as well as the Reelin-mediated generation of L1-80 contribute to brain development at early developmental stages.
Similar content being viewed by others


Introduction
During nervous system development, the cell adhesion molecule L11,2 plays crucial roles in proliferation, migration and survival of neural cells, and L1 participates in neuritogenesis and axonal outgrowth, guidance, pathfinding and fasciculation as well as in myelination and synaptogenesis3,4,5,6. L1-deficient mice show severe malformations and malfunctions of the nervous system7,8. In humans, mutations in L1 are associated with the L1 syndrome which comprises a spectrum of mild to severe congenital X-linked developmental disorders6,9. The L1 syndrome is characterised by developmental and mental retardation, intellectual deficits, hydrocephalus with stenosis of the aqueduct of Sylvius, corpus callosum agenesis, adducted thumbs, shuffling gait, aphasia and spastic paraplegia6,9.
The protein backbone of L1 consists of a C-terminal intracellular tail, a transmembrane domain and an extracellular part composed of 6 immunoglobulin-like (Ig) and 5 fibronectin type III (FN III) domains. Full-length L1 can undergo proteolytic ectodomain shedding to release soluble fragments and to generate transmembrane fragments10,11,12, which have been implicated in distinct L1 functions10,11,12,13,14,15,16,17,18,19,20. Cleavage of L1 within the third FN III domain by the serine proteases trypsin13, proprotein convertase PC5a10, and plasmin14,15,16 leads to promotion of neurite outgrowth and stimulates migration of neuronal cells.
The extracellular matrix protein Reelin controls the sequential lamination of the cerebral cortex, and the cortical layers are disorganised in the natural Reelin-deficient mutant reeler 21,22,23,24,25,26,27. Since Reelin was found to show serine protease activity cleaving extracellular matrix proteins28 and since Reelin-mediated cleavage of the cell surface glycoprotein Caspr had been reported to alter neurite outgrowth29, we investigated whether Reelin cleaves L1 and whether the serine protease activity of Reelin affects L1-dependent functions in the nervous system.
Here, we show that full-length Reelin and its fragments N-R2 and N-R6 bind to L1. We also show that full-length Reelin and its fragment N-R6 proteolytically cleave L1 within amino acids 840–845, and that serine1283 in Reelin is required for the cleavage of L1 by Reelin. Furthermore, we show that the protease activity of Reelin is required for L1-mediated neurite outgrowth and neuronal migration, and we provide evidence that cleavage of L1 by Reelin affects early stages of brain development.
Results
Reelin proteolytically cleaves cell adhesion molecule L1
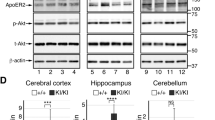
To determine if Reelin cleaves L1 we probed tissue from detergent-solubilised brain regions of postnatal reeler mice and wild-type littermates for L1-fragments using immunoblot analysis with antibody 172, which recognises the intracellular L1 domain. In the cerebellum, cerebral cortex and hippocampus of reeler mice, the expression of a proteolytic 80 kDa L1 fragment (L1–80) was decreased when compared to its wild-type levels (Fig. 1a,b). The protein expression of the close homolog of L1 (CHL1) and the neural cell adhesion molecule NCAM were similar in the cerebellum, cerebral cortex and hippocampus of wild-type and reeler mice (Fig. 1c and Supplementary Fig. S1a,b). Since double knock-out of the Reelin receptors ApoER2 and VLDLR or deficiency of Dab1, which is the downstream signal transducer of ApoER2 and VLDLR, lead to a reeler-like phenotype30,31,32,33,34, we next analysed the expression of L1 proteolytic fragments in brain tissue homogenates from Dab1-deficient and ApoER2 and VLDLR single and double knock-out mice. We found that the L1 expression in the cerebral cortex and cerebellum of these mice were similar to those in wild-type mice and heterozygous littermates (Fig. 1d and Supplementary Fig. S2), indicating that Reelin is involved in the proteolytic processing of L1 to generate L1-80 independently of the signalling cascade via ApoER2, VLDLR and Dab1.

L1-80 levels are decreased in reeler mice. (a) Immunoblot analysis of homogenates from cerebellum (cere), cerebral cortex (cortex) and hippocampus (hippo) of 6-day-old wild-type (WT) and reeler mice with an antibody against the intracellular L1domain (L1-ICD). L1-FL: full-length L1. (b) Quantification of L1-FL and L1-80 levels in homogenates from cerebellum, cerebral cortex and hippocampus of wild-type and reeler mice. Mean values + SEM from 6 independent experiments and differences between groups are shown (*p < 0.05, **p < 0.01, ***p < 0.005; two-tailed t-test). RI: relative intensity in arbitrary units (AU). (c) Unaltered NCAM and CHL1 expression levels in cerebellar homogenates from wild-type (WT) and reeler mice. (d) Unaltered L1-80 levels in cerebellar and cerebral cortex homogenates from wild-type (Dab1 +/+), heterozygous (Dab1 +/−), and Dab1 knock-out (Dab1 −/−) mice, and in homogenates from ApoER2-VLDLR double knock-out (A−/−V−/−), heterozygous (A+/+V−/−, A−/−V+/+) and wild-type (A+/+V+/+) littermates. (a,c,d) Representative immunoblots out of 6 independent experiments are shown and display all L1, CHL1 and NCAM forms. GAPDH antibody was used to control loading and only the regions of the blots with GAPDH bands are shown.
Since Reelin has been described to cleave fibronectin28 and since L1 contains five FN III domains, we tested whether Reelin can generate L1-80 and analysed which structural motifs in L1 are recognised by Reelin to generate L1-80. Freshly homogenized hippocampus from newborn reeler mice was incubated either with supernatants from HEK cells which were transfected to express and secrete Reelin or from mock-transfected HEK cells. L1-80 was detected in the homogenates treated with Reelin-containing supernatant, but not in the homogenates treated with mock-supernatant devoid of Reelin (Fig. 2a). L1-80 was also detected in embryonic reeler cerebral cortex homogenates after treatment with Reelin-containing supernatant, but not after treatment with Reelin-lacking mock-supernatant (Supplementary Fig. S3). In additional experiments, Reelin and L1 were expressed in L1-lacking HEK cells by co-transfection, and cell lysates were subjected to immunoblotting using L1 antibody 172. HEK cells transfected with L1 alone and non-transfected cells were used for comparison. L1-80 was detectable only in lysates from L1- and Reelin-expressing cells (Fig. 2b). These findings suggest that Reelin cleaves full-length L1 to generate L1-80.

Reelin cleaves L1. (a) Freshly homogenized reeler hippocampus was treated with supernatants from HEK cells expressing (HEKrln) or lacking (HEKØ) Reelin. (b) HEKrln and HEKØ cells were transfected with wild-type L1 (L1) or mock DNA (−). (c) HEKrln cells were transfected with wild-type (L1wt) or mutated (L1mut) L1. (d) HEK cells were co-transfected with wild-type L1 and wild-type Reelin (rlnwt) or mutated Reelin (rlnmut). (a–d) Representative immunoblots of cell lysates out of six independent experiments using an antibody against the intracellular L1 domain (L1-ICD) and the GAPDH antibody are shown. The cropped blots display all L1 forms or the GAPDH band. Mean values + SEM from 6 independent experiments are shown for the levels of full-length L1 (L1-FL) and differences between groups are shown (*p < 0.05, **p < 0.0001; One-way ANOVA with Tukey’s Multiple Comparison Test). RI: relative intensity in arbitrary units (AU). (e) Purified brain L1 was incubated with heparin-purified wild-type (RLNwt) or mutated (RLNmut) Reelin protein in the absence or presence of the serine protease inhibitor PMSF (+PMSF) and then subjected to immunoblot analysis with L1 antibody recognising the intracellular domain (L1-ICD). (f) Schematic representation of L1 structure and cleavage by Reelin. Shown are the extracellular Ig and FN III domains, the transmembrane and intracellular domain (ICD).
Since the third FN III domain of L1 is a substrate for several proteases generating L1-8010,15, we tested whether Reelin also cleaves L1 at the previously identified motif 840RKHSKR845 10. Therefore, Reelin-expressing HEK cells were transfected with wild-type L1 or L1 harbouring mutations in this motif by changing it to 840SKHSSS845 10. L1-80 was detected in lysates of HEK cells co-expressing Reelin and wild-type L1, but not in cells co-expressing Reelin and mutated L1 (Fig. 2c). These results indicate that 840RKHSKR845 represents the cleavage site for Reelin in L1. Given that murine Reelin contains a serine in its enzymatically active centre at position 1283 (GKS1283D)28, we examined whether exchanging serine against alanine at this position would affect proteolysis of L1 by Reelin. HEK cells expressing L1 were transfected either with wild-type Reelin or Reelin with the S/A1283 mutation and cell lysates were probed by immunoblot analysis with L1 antibody 172. L1-80 was found in lysates of HEK cells expressing wild-type, but not mutated Reelin (Fig. 2d). In further experiments, we incubated heparin-purified wild-type Reelin35 or heparin-purified Reelin harbouring the S/A1283 mutation (Supplementary Fig. S4) with L1 immunoaffinity-purified from mouse brain2,11. L1-80 was only generated in the presence of wild-type Reelin, but not in the presence of mutated Reelin or in the concomitant presence of wild-type Reelin and the serine protease inhibitor phenylmethylsulfonyl fluoride (Fig. 2e). The combined findings show that S1283 in Reelin is essential for Reelin’s serine protease activity and for the generation of L1-80 by Reelin (Fig. 2f).
Protease activity of Reelin is required for L1-mediated neurite outgrowth and neuronal migration in vitro
To investigate whether L1-mediated neuronal migration or neurite outgrowth depend on Reelin’s serine protease activity, wild-type, reeler, and L1-deficient dissociated cerebellar neurons or cerebellar explants were treated with purified wild-type Reelin alone or in the presence of the serine protease inhibitor aprotinin or the Reelin function-inhibiting antibody CR5028,36,37. Neurons and explants treated with Reelin carrying the S/A1283 mutation as well as mock-treated and untreated neurons and explants were investigated for comparison. Explants from reeler and L1-deficient mice showed reduced neuronal migration when compared to wild-type explants (Fig. 3a,b). Wild-type Reelin promoted neuronal migration from explants of reeler and wild-type mice, but not from explants of L1-deficient mice, when compared to explants treated with mutated, proteolytically inactive Reelin or to mock-treated or non-treated explants (Fig. 3a,b). In the presence of the function-inhibiting Reelin antibody CR50 or the serine protease inhibitor aprotinin, neuronal migration from explants of reeler and wild-type mice was not enhanced by wild-type Reelin. Application of aprotinin or of Reelin antibody CR50 had no effect on migration from explants of L1-deficient mice (Fig. 3a,b). Since it has been shown by βIII-tubulin immunostaining that the thin processes around the explant core are of neuronal origin12, we conclude that in this assay the neuronal migration takes place along neurites and not along radial glial fibers. Application of wild-type Reelin to reeler and wild-type neurons led to enhanced total neurite lengths, whereas application of wild-type Reelin to L1-deficient neurons did not stimulate the extension of neurites (Fig. 3c). In the presence of Reelin antibody CR50 or aprotinin, wild-type Reelin did not enhance of neurite outgrowth from reeler and wild-type neurons (Fig. 3c). Application of aprotinin and CR50 together with Reelin did not affect neurite outgrowth in L1-deficient neurons (Fig. 3c). These results indicate that L1-mediated neurite outgrowth and neuronal migration in the cerebellum depend on the serine protease activity of Reelin.

Reelin promotes L1-dependent neurite outgrowth and neuronal migration in vitro via its serine protease activity. Assessment of neuronal migration out of cerebellar explants from wild-type, reeler and L1-deficient (L1−/y) mice (a,b) and assessment of neurite lengths in dissociated wild-type, reeler and L1-deficient (L1−/y) cerebellar neurons (c) after treatment with purified wild-type Reelin (RLNwt) or with purified mutated Reelin (RLNmut) in the absence or presence of the serine protease inhibitor aprotinin or the Reelin-inhibiting antibody CR50. Untreated (control) and mock-treated neurons and explants were used as controls. (a) Representative images of explants after treatment are shown; scale bars: 100 µm. The dashed lines represent the outward borders of migration; e: explant. (b,c) Numbers of migrating cells and total neurite lengths were determined evaluating 30 equal-sized explants (a) or 100 neurons (b) per group from 3 independent experiments. Mean values + SEM are shown for numbers of migrating cells or total neurite lengths relative to values observed for untreated explants and cells from wild-type mice (set to 100%). Differences between groups are indicated (***p < 0.0001; One-way ANOVA with Tukey’s Multiple Comparison Test). (d,e) Cerebral cortex explants from wild-type, reeler and L1-deficient mice were treated with mutated Reelin (RLNmut) or wild-type Reelin (RLNwt) in the absence or presence of aprotinin or Reelin antibody CR50. Untreated (control) and mock-treated explants were used as controls. (d) Representative images of untreated and Reelin-treated wild-type explants stained for βIII-tubulin (green) are shown; scale bars: 50 µm. Nuclei are stained with DAPI (blue). (e) Numbers of migrating cells were determined evaluating 10 equal-sized explants per group from 3 independent experiments. Mean values + SEM are shown for numbers of migrating cells relative to values observed for untreated explants from wild-type mice (set to 100%). Differences between groups are indicated (*p < 0.05; One-way ANOVA with Holm-Sidak Multiple Comparison Test).
To examine whether neuronal migration along neurites in the cerebral cortex also depends on the serine protease activity of Reelin, cortical explants from wild-type, reeler and L1-deficient mice were treated as described above for treatment of cerebellum explants and were then immunostained for βIII-tubulin. In wild-type explants, Reelin promoted neuronal migration along βIII-tubulin-positive neurites (Fig. 3d). Similarly, Reelin promoted neuronal migration along neurites on reeler explants, but not on explants from L1-deficient mice (Fig. 3e). In comparison to untreated wild-type explants, untreated reeler and L1-deficient explants showed reduced neuronal migration (Fig. 3e). Mock treatment and application of mutated Reelin did not result in promotion of neuronal migration (Fig. 3e). Reelin antibody CR50 and aprotinin inhibited the Reelin-promoted neuronal migration from reeler and wild-type explants (Fig. 3e). Aprotinin and CR50 did not affect the migration from explants of L1-deficient mice (Fig. 3e). These results indicate that the serine protease activity of Reelin is required for L1-mediated neuronal migration in the cerebral cortex.
Reelin interacts with L1
Since the N-terminus of Reelin binds to RGD motifs38, which are present in L115, we investigated whether L1 interacts with Reelin or Reelin fragments39,40,41. ELISA was performed with purified recombinant Reelin or Reelin-containing HEK cell supernatants as coated substrates and with addition of L1-Fc or Fc. Coatings with supernatants lacking Reelin, mock supernatants or vehicle solution were used as negative controls. In turn, ELISA was also performed using substrate-coated L1-Fc or Fc and applying purified recombinant Reelin, Reelin-containing HEK cell supernatant, mock-purified cell culture supernatant or vehicle solution. L1-Fc showed pronounced binding to substrate-coated recombinant Reelin and to Reelin-containing cell supernatant, whereas Fc, mock-treated supernatant or vehicle solution did not show significant binding (Fig. 4a). Similarly, recombinant Reelin and Reelin-containing HEK cell supernatants bound to substrate-coated L1-Fc, but not to Fc, whereas the mock-purified cell culture supernatant or vehicle solution did not bind to L1-Fc or Fc (Fig. 4b). L1-Fc and Fc were also used for pull-down assays with supernatants of Reelin-transfected and non-transfected HEK cells. Immunoblot analysis with Reelin antibody G10 revealed that L1-Fc, but not Fc, precipitated full-length Reelin and the Reelin fragments N-R6 and N-R2 from Reelin-containing HEK cell supernatants, but not from Reelin-lacking HEK cell supernatants (Fig. 4c). The combined results indicate that L1 associates with Reelin and its fragments N-R6 and N-R2.

Full-length Reelin and Reelin fragment N-R6 interact with L1 and cleave L1 to accelerate migration in vitro. (a) ELISA using substrate-coated L1-Fc, Fc or vehicle solution (phosphate buffer saline, pH 7.3) and purified Reelin protein (RLNrec), mock-purified solution (mockrec) or cell culture supernatants from Reelin-expressing (HEKrln) or Reelin-lacking (HEKØ) HEK cells. (b) ELISA using substrate-coated purified Reelin protein (RLNrec), mock-purified solution (mockrec) or vehicle solution and L1-Fc or Fc. (a,b) Mean values + SEM for binding are shown (***p < 0.0001; One-way ANOVA with Tukey’s Multiple Comparison Test from 3 independent experiments with triplicates). (c) Pull-down with L1-Fc and Fc using supernatants from Reelin-expressing (HEKrln) and Reelin-lacking (HEKØ) HEK cells. Supernatant from Reelin-expressing HEK cells was used as input control and Reelin antibody G10 was used for detection. A representative immunoblot out of 3 independent experiments with Reelin-positive bands is shown and full-length Reelin (RN-450) and its fragments N-R6 and N-R2 are indicated. (d) Structure of Reelin and its proteolytic fragments after N- and C-terminal processing by ADAMTS-4. The enzymatically decisive serine residue in the GKS1283D sequence in murine Reelin is indicated by a red spot. (e) Lysates from co-transfected HEK cells expressing full-length Reelin (RLN-450) or Reelin fragments N-R6, N-R2, R3-6, R3-8 or R7-8 were subjected to immunoblot analysis with L1 antibody against the intracellular domain (L1-ICD) and GAPDH antibody to control loading. Representative immunoblots out of 6 independently experiment are shown and display all L1 forms and the GAPDH bands. Quantification of full-length L1 (L1-FL) levels (AU: arbitrary units) is shown in the lower panel. Mean values + SEM from 6 independent experiments are shown and differences between groups are indicated (**p < 0.001; One-way ANOVA with Tukey’s Multiple Comparison Test). (f) Images of scratches in monolayers of co-transfected HEK cells expressing L1 and full-length Reelin or one of the indicated Reelin fragments. Gap borders are highlighted with continuous white lines; scale bars: 50 µm. Mean values + SEM from 3 independent experiments with triplicates per group are shown for gap closure after scratching. Differences are indicated (**p < 0.005; One-way ANOVA with Tukey’s Multiple Comparison Test).
Reelin and its N-terminal fragment N-R6 are proteases in the embryonic, early developing cerebral cortex and generate L1-80
N- and/or C-terminal cleavage of Reelin by ADAMTS-4 (for sequences of Reelin fragments, see ref.40) yields two N-terminal fragments (N-R2 and N-R6), two fragments containing the central part of Reelin (R3-6 and R3-8), and one fragment containing the C-terminus (R7-8) (Fig. 4d). Thus, we analysed whether only full-length Reelin cleaves L1 or whether one of the Reelin fragments could also cleave L1 to generate L1-80. Full-length L1-expressing HEK cells were transfected with full-length Reelin or Reelin fragments N-R6, R3-8 or R3-6 which carry the GKS1283D motif. Cells expressing the Reelin fragments N-R2 or R7-8 which lack the GKS1283D motif were used as controls. L1-80 was detectable in lysates of HEK cells co-expressing full-length L1, full-length Reelin or the N-R6 Reelin fragment, but not in lysates of HEK cells co-expressing L1 and the Reelin fragments N-R2, R3-8, R3-6 or R7-8 (Fig. 4e), suggesting that only full-length Reelin and N-R6 generate L1-80. These results indicate that the N-terminal proteolytic processing of Reelin41 generates the fragments N-R2, which does not contain GKS1283D, and the proteolytically inactive R3-8 and R3-6.
We next studied which Reelin fragment affects L1-dependent cell migration in an in vitro scratch injury assay using transfected HEK cells. It was not possible to perform this assay with primary cultures of neurons, since these neurons do not form uniform monolayers and do not survive upon scratching. Moreover, the low transfection efficiency of neurons precludes their usage in this experimental setup. It was plausible to use HEK cells, because they use similar signalling pathways for migration as neurons do during development, i.e. they respond to Reelin with enhanced migration when expressing L1. Widths of gaps were measured 16 h after scratching of monolayers of HEK cells which were transfected to co-express L1 and full-length Reelin or Reelin fragments N-R6, N-R3, R3-8, R3-6 or R7-8. Only the N-R6 fragment and full-length Reelin, but not the N-R2, R3-8, R3-6 and R7-8 fragments, enhanced the gap closure (Fig. 4f), suggesting that the protease activities of N-R6 and full-length Reelin are required for L1-mediated cell migration.
Since full-length Reelin and the N-R6 fragment cleave L1, we investigated by immunoblot analysis, whether expression of full-length Reelin and/or its N-R6 fragment coincides with the generation of L1-80 at different developmental stages in wild-type cerebral cortex homogenates. Levels of N-R6 and L1-80 were increased between embryonic days 12 and 15. N-R6 levels were already detected at embryonic day 11, but levels of full-length Reelin were only detected at embryonic days 15 and 16 (Fig. 5a). These results suggest that during embryonic development of the cerebral cortex, Reelin is rapidly processed to N-R6 which generates L1-80 at early developmental stages. Moreover, the findings suggest that the N-terminal processing of Reelin abolishes its cleavage activity and limits the generation of L1-80 during embryonic development.

Interaction between Reelin and L1 and functional roles of their fragments in the developing brain. (a,b) Immunoblot analysis of Reelin and L1 expression in wild-type and reeler cerebral cortex homogenates at different embryonic stages using the Reelin antibody (G10) or an antibody against the intracellular L1 domain (L1-ICD). GAPDH antibody was used to control loading. Representative immunoblots out of 6 independent experiments are shown and display all L1 and Reelin forms and the GAPDH bands. Quantifications of N-R6, full-length L1 (L1-FL) and L1-80 levels are given in arbitrary units (AU). Mean values ± SEM from 6 independent experiments and differences relative to the values at embryonic day 11 are shown (*p < 0.05; **p < 0.001; ***p < 0.0001; One-way ANOVA with Tukey’s Multiple Comparison Test). (c) L1-80 carrying a flag-tag was electroporated together with CAG-GFP into reeler embryos at embryonic day 13.5. Sixty hours after in utero electroporation, cerebral cortex was analysed for L1-80-expressing neurons using an antibody against the flag-tag (α-FT) and a fluorescently labelled secondary antibody (red). High numbers of GFP-positive (green) and L1-80-positive (red) neurons were found in the upper layers of the cerebral cortex from reeler mice after electroporation with L1-80 (white arrows) and in the cortex from wild-type mice. After electroporation with CAG-GFP coding for full-length L1 or with CAG-GFP alone, GFP-positive neurons were retained in the subventricular zone (svz). Inset: L1 promotes elongation of the leading processes of some electroporated neurons. The marginal zone (mz) in cortices of reeler mice contains more cells than observed in wild-type mice. White dashed lines indicate the border between the svz and intermediate zone (iz) and between iz and mz. Nuclear staining with DAPI (blue) is shown. Scale bars: 50 µm. (d) Schematic representation of migration behaviour of reeler neurons electroporated with L1-80 or full-length L1 in comparison to reeler and wild-type neurons. Assessment of numbers of electroporated neurons within the iz and mz area was performed. Mean values + SEM from 6 independent experiments are shown for the cell density in the iz-mz-area and differences between groups are indicated (ns: not significant; ***p < 0.0001; One-way ANOVA with Tukey’s Multiple Comparison Test).
It should be noted that L1 is cleaved by other serine proteases10,11,12, which, similarly to Reelin, cleave L1 at amino acids 840-845. To examine whether Reelin and/or its N-R6 fragment are responsible for the generation of L1-80 in the early cortical development, we measured L1-80 expression levels in reeler cerebral cortex at different embryonic stages. L1-80 was not detectable during embryonic corticogenesis in reeler mice and became detectable at low levels only at later stages of corticogenesis (Fig. 5b). These results implicate Reelin as serine protease in the embryonic cerebral cortex acting on L1 to produce L1-80.
To investigate whether L1-80 would normalise migration of reeler neurons during embryonic development of the reeler cerebrum, we electroporated L1-80 into the cerebral cortex of reeler embryos in utero at embryonic day 13.5 using a flag-tagged L1-80 plasmid in combination with a CAG-GFP plasmid for visualisation of the electroporated neurons. Reeler and wild-type embryos electroporated with CAG-GFP alone were used for comparison. Sixty hours after electroporation, L1-80-expressing neurons were identified as GFP-positive cells which were stained with the antibody recognising the flag-tag of L1-80 (Fig. 5c). Many double-labelled neurons had reached the reeler marginal zone as similarly observed for GFP-electroporated wild-type neurons (Fig. 5c,d). GFP-electroporated reeler neurons were misoriented and retained in the subventricular zone (Fig. 5c,d). In contrast to L1-80, full-length L1 did not promote migration of reeler neurons (Fig. 5c,d). Of note, some L1-electroporated neurons had elongated leading processes (Fig. 5c,d). These results indicate that L1-80 is important for the migration of cortical neurons and that the aberrant migration of reeler neurons can be partially rescued by the introduction of L1-80.
Displaced cortical neurons in the L1-deficient cerebral cortex
We next investigated whether L1-deficient cerebral cortices show deficits in positioning of cortical neurons and whether these deficits correlate at least partially with those in reeler mice. To this aim, sections of wild-type, L1-deficient and reeler cerebral cortices from 6-day-old mice were Nissl-stained and analysed by light microscopy. In contrast to reeler cerebral cortices, the marginal zones in wild-type and L1-deficient cerebral cortices appeared to harbour fewer neurons (Fig. 6a). Since Nissl staining does not reveal information on neuronal processes (Fig. 6a), we combined Nissl staining with Golgi impregnation. We found Golgi-impregnated pyramidal neurons uniformly oriented towards the marginal zone in the wild-type cerebral cortex, whereas pyramidal neurons of the L1-deficient cerebral cortex occasionally displayed aberrant apical dendrites (Fig. 6b). In the reeler cerebral cortex, we noticed many Golgi-positive neurons with misoriented apical dendrites, often pointing towards the subventricular zone (Fig. 6b). Furthermore, we immunostained sections of wild-type, L1-deficient and reeler cerebral cortices for the layer-specific markers Brn2 (upper layers) and Foxp2 (deep layers). In contrast to wild-type cerebral cortices, many Brn2-positive cells were abnormally found in deep layers, and many Foxp2-positive cells were found in upper layers of L1-deficient cortices (Fig. 6c). By contrast, in reeler mice Brn2-positive and Foxp2-positive cells were far more dislocated (Fig. 6c). Of note, immunostaining for Reelin revealed a decreased number of Reelin-expressing Cajal-Retzius cells in the marginal zone of cortices from L1-deficient mice in comparison to the marginal zone of cortices from wild-type mice (Fig. 6d,e). Additional Nissl staining of embryonic brain sections at an early developmental stage pointed to a less compact cortical plate in L1-deficient mice when compared to the dense cortical plate of wild-type littermates (Fig. 7a). Layer-specific immunostainings with Brn2, Cutl1 and Trb1 (superficial/upper layers) as well as a BrdU pulse chase labelling revealed misplaced neurons in the cerebral cortex of L1-deficient mice (Fig. 7a–c). Our combined analysis of pre- and postnatal brain development demonstrates that L1-deficient cortical neurons are delayed in their migration and have less uniformly orientated apical dendrites. However, these abnormalities were not as pronounced as those in reeler mice.

L1-deficient mice show deficits in positioning of cortical neurons and reduced number of Cajal-Retzius cells. (a) Nissl-staining of cerebral cortices from 6-day-old wild-type and L1-deficient (L1 −/y) littermates shows a cell-poor marginal zone (mz) followed by an intermediate zone (iz) and subventricular zone (svz). In contrast, the mz in the cerebral cortex of reeler mice contains by more neurons; scale bars: 100 µm. (b) Nissl staining combined with Golgi impregnation on cerebral cortical sections from 24-day-old wild-type and L1 −/y littermates in comparison to reeler mice of the same age (low magnification images are shown in the upper panels, scale bars: 50 µm; magnified areas are indicated by rectangles and presented in the lower panels, scale bars: 10 µm). (c) The left panels show cerebral cortical sections from wild-type, L1-deficient (L1 −/y) and reeler mice after immunostaining for Brn2 or Foxp2 (green) in combination with nuclear staining (DAPI, violet). White arrowheads show misplaced neurons; scale bars: 50 µm. The right panels show mean values + SEM for the numbers of Brn2- and Foxp2-positive cells in 16 cortical bins along the mz-svz axis analysing 12 sections of the same area from 4 animals per condition (***p < 0.0001; One-way ANOVA with Tukey’s Multiple Comparison Test; violet asterisks: differences between wild-type and reeler neurons; orange asterisks; differences between L1-deficient and wild-type neurons; blue asterisks: differences between reeler and L1-deficient neurons). (d) Immunostaining for Reelin (green) using antibody G10 revealed a reduced number of Cajal-Retzius cells in the mz of L1-deficient cortex when compared to wild-type littermates. Nuclear staining (DAPI, violet); scale bars: 100 µm. Mean values + SEM are shown for the numbers of Cajal-Retzius cells in the mz analysing 12 sections of the same area from 4 animals per condition (***p < 0.001; Student’s t-test).

Histological analysis of embryonic wild-type, L1-deficient and reeler brain cortex sections and BrdU pulse chase labelling. (a) Nissl staining and layer-specific immunostaining of cortical sections of reeler, L1-deficient (L1 −/y) and wild-type (L1 +/y) mice at embryonic day 15; dashed lines indicate the area of the cortical plate (cp). Note that the L1 −/y cp appeared less compactly developed in comparison to the L1 +/y cp. The distribution of Tbr1-positive (red), Brn2-positive (red) and Cutl1-positive (green) cells in the L1 −/y cortex was slightly altered in comparison to the L1 +/y cerebral cortex, but these abnormalities were not as extensive as the misplacement of cells seen in the reeler cortex. Nuclear staining with DAPI (blue); mz, iz, svz: marginal, intermediate and subventricular zones; scale bars: 50 µm. (b,c) Altered distribution of BrdU-positive cells in the cerebral cortex of L1 −/y mice. A single BrdU dose (150 mg/kg) was injected intraperitoneally into timed-pregnant females carrying 13.5-day-old L1+/y and L1−/y embryos. Immunohistology for BrdU (red) was performed at birth (P0). The cerebral cortex was divided in 9 equidistant bins and the number of BrdU+ cells per mm2 was estimated for each bin (right); scale bars: 50 µm. Mean values ± SEM are shown for the numbers of BrdU-positive cells per mm2 analysing five sections from three animals per condition (*p < 0.05; Mann-Whitney U-Test).
Discussion
In this study, we show that Reelin and its here newly discovered binding partner and substrate L1 are functionally linked to each other. Both L1 and Reelin are required for neuronal migration and neurite outgrowth. These functions require Reelin’s proteolytic activity, which generates the C-terminal L1-80 fragment by a process that is independent of Reelin’s signalling via ApoER2, VLDLR and Dab1. Concomitantly generated N-terminal extracellular L1 fragment(s) could not be detected in these assays, implying a rapid degradation after generation of such fragment(s). Although the in vitro experiments indicate that proteolysis of L1 by Reelin takes place extracellularly, it is conceivable that Reelin cleaves L1 in the intracellular biosynthetic pathway. L1, but not its close homolog CHL1 or the neuronal cell adhesion molecule NCAM, is a substrate of Reelin. Reelin binds to L1 via N-terminal sequences present in full-length Reelin and the Reelin fragments N-R2 and N-R6. It remains to be shown whether other members of the L1 family or, more generally, the immunoglobulin superfamily, are also proteolytic substrate candidates for Reelin.
We have previously described a normal morphology of the cerebral cortex of L1-deficient mice based on our analysis of Nissl-stained sections42,43,44. In comparison to 6-day-old wild-type littermates, L1-deficient mice did not show obvious changes in cortical lamination following Nissl staining. However, Golgi impregnation and investigation of layer-specific markers in addition to BrdU pulse-chase labelling revealed abnormalities in the positioning and morphology of cortical neurons in L1-deficient brains. These novel findings call for a re-interpretation of previous results on neuronal lamination in the L1-deficient brain42,43,44 and underscore the importance of L1 for correct brain development. The morphological abnormalities observed in cortices in L1-deficient mice partially overlap with those seen in cortices of reeler mice, suggesting that the functional interplay between L1 and Reelin and cleavage of L1 by Reelin at early developmental stages controls cerebral cortex development. Indeed, we showed that reeler neurons in the cerebral cortex normalised their movement after in utero electroporation of L1-80. These findings further support the view that Reelin-mediated L1-cleavage contributes to cortical development. Moreover, our results are consistent with L1 being important for the migration of cerebellar granule cells1 and are in agreement with observations on the deficits in L1-deficient mice, which are characterized by misorientation of neuronal processes, impaired synaptogenesis and corticospinal tract malformation45,46. Notably, silencing of L1 in utero has been reported to result in delayed neuronal migration47. Migration defects of cortical neurons are known to contribute to mental diseases, such as the L1-syndrome5,48,49 or lissencephaly50. Thus, our results link proteolysis of L1 by Reelin to neurodevelopmental diseases, highlighting early migratory defects in the pathogenesis of these disorders.
Although some pathological features of the L1-deficient mice are also displayed by reeler mice, not all abnormalities overlap between both mutants. These findings are understandable under the premise that L1 and Reelin interact with multiple interaction partners that are distinct from each other, and thus are involved not only in overlapping functions, but also in varying pathways leading to diverse phenotypes.
It needs to be pointed out that L1 is cleaved by other proteases at amino acids 840–845 in late developing brain structures10,11,12,19, suggesting even more complex roles of L1 in the maintenance of cortical architecture and function. We here showed that full-length Reelin and its N-terminal fragment N-R6, but not other Reelin fragments, cleave full-length L1 and generate L1-80. Ontogenetic expression studies revealed a close correlation between the expression of N-R6 and L1-80 during the peak phase of neuronal migration, suggesting that both fragments contribute to migration.
Our results shed light on the importance of the N-terminal cleavage of Reelin by proteases, such as ADAMTS-4, to regulate Reelin’s functions. The N-terminal cleavage of Reelin abolishes the protease activity of N-R6, and the originating R3-6 and R3-8 fragments are unable to process L1. Interestingly, introduction of L1-80, but not of full-length L1, into the cerebral cortices of reeler mice by in utero electroporation partially normalises the migration of cortical neurons. Thus, our results indicate that Reelin’s N-terminus and its central part are required for Reelin’s serine protease activity to generate L1-80 and to promote L1-dependent functions via L1-80.
Since cleavage of the cell surface and transmembrane adhesion molecule L1 is crucial for cerebral cortex development and for directed neuronal motility, it will be important to analyse the molecular mechanisms underlying the directed migration of neurons from the subventricular zone to the marginal zone, when migrating neurons approach the upper cortical layers and detach from radial glial fibers51 in the terminal phase of the migratory process52. It would be also desirable to investigate whether the mechanisms of directed migration of neurons and their detachment from radial glial fibres depend solely on the L1–Reelin interaction, or whether other molecules determine the “go and stop” phenomena characteristic for neuronal migration in the cerebral cortex. It is conceivable that cytoskeletal elements at the leading edge of neurites extending along radial glial fibres are involved in regulation of migration. Moreover, cognate signal transduction mechanisms via the L1 fragment generated by Reelin could play a role in regulation of migration. Whether these downstream events would depend on the protease activity of Reelin or whether Reelin’s binding to its receptors at the cell surface would be sufficient for downstream signalling will be important to study in the future.
Methods
Animals
Reeler mice21, ApoER2 32 or VLDLR 53 single mutant mice, ApoER2 and VLDLR double mutant mice32, Dab1-deficient mice30 and L1-deficient mice7 were housed at 22 °C on a 12 h light/dark cycle with ad libitum access to food and water. All animal experiments were approved by the local authorities of the State of Hamburg (animal permit numbers ORG-679, ORG-604, ORG-578 and TVA 6/14) and conform to the guidelines set by the European Union.
Antibodies and reagents
Mouse antibody G10 against the N-terminal part of Reelin was from Merck Millipore and mouse Reelin antibody CR50 was a kind gift of André Goffinet (Institute of Neuroscience, University of Louvain, Brussels, Belgium) and Tom Curran (Children’s Mercy Hospital, Kansas City, MO, USA). Goat Brn2 antibody (sc-6029) was from Santa Cruz Biotechnologies (Heidelberg, Germany). Mouse antibody 172 recognising the intracellular L1 domain (anti-CD171; clone 74-5H7; Cat #38101) was from Biolegend (San Diego, CA, USA) and the polyclonal rabbit L1 antibody Anti-L1CAM (ab123990) was from Abcam (Cambridge, UK). Mouse GAPDH antibody (ab9482) and rabbit antibodies against Foxp2 (ab16046), Tbr1 (ab31940), and Cutl1 (ab140042) were from Abcam (Cambridge, UK). Goat CHL1 antibody (AF2147) was from R&D Systems. Production of rabbit NCAM antibodies and of L1-Fc comprising the extracellular part of mouse L1 fused to Fc part of human IgG have been described54. Aprotinin, phenylmethylsulfonyl fluoride and the Flag M2 mouse antibody (F1804) were from Sigma-Aldrich (Taufkirchen, Germany). Secondary antibodies coupled to horseradish peroxidase, Cy3 or Cy2 and human Fc were from Dianova (Hamburg, Germany).
CAG-IRES-GFP vector was a kind gift from Melanie Richter and Froylan Calderon de Anda (Center for Molecular Neurobiology, Hamburg). Plasmids carrying full-length Reelin and Reelin fragments N-R2, N-R6, R3-6, R3-8 and R7-840 and purified Reelin55 used for ELISA were kind gifts from Tom Curran and André Goffinet. HEK293T cells stably transfected with wild-type Reelin and mock-transfected HEK293T cells37,56 were kindly provided by Eckart Förster (Ruhr-University Bochum, Department for Neuroanatomy and Molecular Brain Research, Bochum, Germany).
Preparation of brain homogenates and Western blot analysis
Brain areas were homogenised in RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Nonidet P-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1× cocktail of protease inhibitors (Roche Diagnostics, Mannheim, Germany)) at 4 °C. After centrifugation at 20,000 × g and 4 °C for 10 min, equal amounts of total protein from the supernatants were subjected to Western blot analysis12 with Reelin antibodies G10 (diluted 1:1,000), L1 antibody 172 (diluted 1:1,000); CHL1, NCAM or GAPDH antibodies (diluted 1:2,000). Secondary antibodies coupled to horseradish peroxidase were used at a dilution of 1:10,000.
Tissue preparation, immunohistology and imaging
For paraffin sectioning, fixed brains were incubated in 70% ethanol at 4 °C overnight. Brains were dehydrated in ascending grades of ethanol and cleared in pure xylene. Following immersion in liquid paraffin at 60 °C for 24 h, samples were embedded in paraffin at ambient room temperature. Sections of 10 µm were cut on a microtome (Leica, Wetzlar, Germany) and collected on glass slides. Sections were deparaffinised in pure xylene several times, rehydrated in descending grades of ethanol and immunostained with Reelin antibody G10 (diluted 1:500), Brn2, Foxp2, Tbr1, or Cutl1 antibodies (diluted 1:100). Blocking solution (PBS containing 10% horse serum and 0.01% Triton X-100) was used for dilution of antibodies. Cy3- and Cy2-conjugated secondary donkey antibodies were used at a dilution of 1:500 in PBS. Sections were mounted in Fluoromount containing DAPI (Sigma-Aldrich). Images of immunostained brain sections were taken on a confocal fluorescence microscope (F1000, Olympus, Hamburg, Germany) or a Keyence Fluorescent Microscope (BZ-9000, Keyence, Neu-Isenburg, Germany) and were processed by using the ImageJ software.
Plasmids and site-directed mutagenesis
Wild-type and mutated L110 were cloned into the CAG-IRES-GFP vector using the In Fusion cloning kit (TaKaRa/Clontech, Saint-Germain-en-Laye, France). For the mutation of Reelin, sequences between the SnaBI and AgeI sites were amplified using the HiFi amplification kit (TaKaRa/Clontech), the plasmid coding for full-length Reelin, the forward primer 5′-AAC CCC ACC TAC GTA CCG GGA CAG GAA TAC-3′ and the reverse primer 5′-GT TAC TGC AAA CCG GTC TCC ATC CGC CTT TCC AAA CAC CAT GGC TG-3′ carrying the mutation (underlined triplet). The plasmid coding for full-length Reelin was digested with SnaBI and AgeI and fused with the amplicon with the mutation using the In Fusion cloning kit.
To amplify sequences coding for L1-80 or for the L1 signal sequence, PCR were performed using murine L1-cDNA10 as template and the forward primer 5′‐GAATTC CAT ATC CAC AAA AGC CAC ATA‐3′ containing an EcoRI site and the reverse primer 5′-CTCGAG TTC TAG GGC TAC TGC AGG ATT‐3′ containing a XhoI site or the forward primer 5′‐GAAGCTTTAGCCACCATGGTC ATG GTC GTG ATG CTG CGG TAC‐3′ containing a HindIII site and the reverse primer 5′-GAATTC GAG CAG GCA GGG GCT GCA GAG‐3′ containing an EcoRI site. The amplicons were used for T/A-cloning into pGEM®-T-Easy vector (Promega, Mannheim, Germany). Using EcoRI and Xhol restriction sites, the L1-80 amplicon was cloned into pcDNA3 Flag-His A plasmid (Promega) in frame with the Flag-His tag. The signal sequence of L1 was cloned in frame with the L1-80 amplicon via HindIII and EcoRI restriction sites.
HEK cell transfection, scratch assay, assessment of gap closure and pull-down experiments
Culturing and transfection of HEK293T cells, scratch assay and assessment of gap closure have been described11. Serum-free supernatants from transfected HEK cells were dialysed and concentrated 5-fold using 100 kDa cut-off centrifugation columns (Merck Millipore, Darmstadt, Germany) at 4 °C. Pull-down with L1-Fc and Fc using Protein A beads was described57.
ELISA
ELISA (see ref.12) was performed as follows: 5 µg of L1-Fc and Fc, 5 µg of serum-free Reelin-containing HEK supernatants and Reelin-devoid HEK supernatants (mock) concentrated 5-fold and dialysed through 100 kDa cut-off Amicon columns, were substrate-coated in 384-well microtiter plates with high binding surface (Corning, Tewksbury, MA, USA) at 4 °C overnight. After washing with PBS, blocking with 1% w/v bovine serum albumin (essentially fatty acid free; PAA Laboratories, Cölbe, Germany) in PBS at ambient room temperature for 1 h, and washing with PBST (PBS with 0.05% Tween 20), 5 µg of L1-Fc or Fc were added to the substrate-coated Reelin and HEK cell supernatants, and 5 µg of purified Reelin were added to the substrate-coated L1-Fc or Fc for incubation at room temperature for 1 h. After washing with PBST, primary mouse monoclonal Reelin antibody G10 followed by an anti-mouse HRP-coupled secondary antibody (Dianova) as well as O-phenylenediamine dihydrochloride (ThermoFisher Scientific, Darmstadt, Germany) as horseradish peroxidase (HRP) substrate were used for detection of Reelin. For detection of L1-Fc and Fc, human anti-Fc-HRP (Dianova, diluted 1:2000) antibody was used. The reaction was terminated by addition of 2.5 M sulphuric acid. Absorbance was measured at 490 nm with the ELISA reader (BioTek, Bad Friedrichshall, Germany).
Primary cell cultures and explants, treatment with additives, and assessment of neurite length and neuronal migration
Isolation and culturing of murine cerebellar and cortical neurons and explants have been described11,12,58. Briefly, cortical and cerebellar neurons as well as explants were treated with 10 µl of HEK cell supernatants per well, 1 µM aprotinin or 2 µl of Reelin antibody CR50 (1 mg/ml) per well for 16 h at 37 °C in a humidified atmosphere containing 5% CO2. Neurons and explants were then fixed and stained as described11,12. Neurite outgrowth and neuronal cell migration were analysed and quantified by measuring the total neurite length per neuron, the number of cells migrating out of the explant cores, and migration area using a Zeiss Kontron microscope with AxioVision 4.7 software (Carl Zeiss, Jena, Germany).
Purification of wild-type and mutated Reelin, and L1, proteolytic assay and treatment of explants and hippocampal and cerebral cortex homogenates
Wild-type and mutated Reelin were purified using Heparin High-Trap Sepharose columns (GE Healthcare Life Sciences, Solingen, Germany)35. Elution was performed in two steps using 140 mM and 400 mM NaCl, and eluates containing Reelin were dialysed using 100 kDa cut-off centrifugation columns (Amicon). L1 was immunopurified from mouse brains as described2,11. For the proteolytic assay, 10 ng of wild-type or mutated Reelin were incubated at 37 °C for 16 h in the absence or presence of 1 µM phenylmethylsulfonyl fluoride with 10 ng of L1 immunoaffinity purified from mouse brain. For the treatment of homogenates, neonatal reeler hippocampus or embryonic (E17) reeler cerebral cortex was freshly homogenized in PBS and incubated with 100 µl of HEK cell supernatants at 37 °C for 16 h.
In utero electroporation and histological analysis of electroprorated brains
For in utero electroporation, timed-pregnant mice at E13.5 were anaesthetised, the abdominal cavity of the pregnant mice was opened and the uterine horns bearing the embryos were exposed. Thereafter, 2 µg of CAG-IRES-GFP vector coding for wild-type L1, vectors coding for L1-80, N-R2, N-R6, R3-6, R3-8 or R7-8 or empty CAG-IRES-GFP vector pre-mixed with 0.01% Fast Green were unilaterally injected into the ventricle of an embryonic brain. Five electrical square unipolar pulses (amplitude of 30 V, duration of 50 ms, intervals of 950 ms) powered by a BTX electroporation apparatus (model BTX ECM 830; Harvard Apparatus, ThermoFisher Scientific) were applied to the embryos. The uterine horns were placed back into the abdominal cavity. After 60 h, pregnant mice carrying electroporated embryos were sacrificed, the embryos were isolated in ice-cold PBS and killed by decapitation. GFP-positive electroporated brains were collected under a fluorescence microscope and fixed in 4% formaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, at 4 °C overnight, followed by an overnight immersion in a solution of 15% sucrose in 0.1 M sodium cacodylate buffer, pH 7.4, at 4 °C.
Electroporated brains were embedded in 5% agarose and sectioned on a vibratome (Leica VT1000 S) at 80 µm and immersed in blocking solution (5% normal goat serum and 0.2% Triton-X 100 in 0.1 M PBS), washed, and incubated with an antibody recognising flag-tag (dilution 1:500) at 4 °C overnight. Sections were washed in PBS, incubated with Cy3-conjugated secondary antibody (dilution 1:250) at 4 °C overnight, counterstained with DAPI to label nuclei, mounted in Fluorescence Mounting Medium (Dako, Hamburg, Germany) and subjected to fluorescence microscopy.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Lindner, J., Rathjen, F. G. & Schachner, M. L1 mono- and polyclonal antibodies modify cell migration in early postnatal mouse cerebellum. Nature 305, 427–430 (1983).
Rathjen, F. G. & Schachner, M. Immunocytological and biochemical characterization of a new neuronal cell surface component (L1 antigen) which is involved in cell adhesion. EMBO J. 3, 1–10 (1984).
Keilhauer, G., Faissner, A. & Schachner, M. Differential inhibition of neurone-neurone, neurone-astrocyte and astrocyte-astrocyte adhesion by L1, L2 and N-CAM antibodies. Nature 316, 728–730 (1985).
Hortsch, M. The L1 family of neural cell adhesion molecules: old proteins performing new tricks. Neuron 17, 587–593 (1996).
Kanemura, Y. et al. Molecular mechanisms and neuroimaging criteria for severe L1 syndrome with X-linked hydrocephalus. J. Neurosurg. 105, 403–412 (2006).
Maness, P. F. & Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 10, 19–26 (2007).
Dahme, M. et al. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat. Genet. 17, 346–349 (1997).
Fransen, E. et al. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum. Mol. Genet. 7, 999–1009 (1998).
Katidou, M., Vidaki, M., Strigini, M. & Karagogeos, D. The immunoglobulin superfamily of neuronal cell adhesion molecules: lessons from animal models and correlation with human disease. Biotechnol. J. 3, 1564–1580 (2008).
Kalus, I., Schnegelsberg, B., Seidah, N. G., Kleene, R. & Schachner, M. The proprotein convertase PC5A and a metalloprotease are involved in the proteolytic processing of the neural adhesion molecule L1. J. Biol. Chem. 278, 10381–10388 (2003).
Lutz, D. et al. Myelin basic protein cleaves cell adhesion molecule L1 and promotes neuritogenesis and cell survival. J. Biol. Chem. 289, 13503–13518 (2014a).
Lutz, D., Wolters-Eisfeld, G., Schachner, M. & Kleene, R. Cathepsin E generates a sumoylated intracellular fragment of the cell adhesion molecule L1 to promote neuronal and Schwann cell migration as well as myelination. J. Neurochem. 128, 713–724 (2014b).
Sadoul, K., Sadoul, R., Faissner, A. & Schachner, M. Biochemical characterization of different molecular forms of the neural cell adhesion molecule L1. J. Neurochem. 50, 510–521 (1988).
Nayeem, N. et al. A potential role for the plasmin(ogen) system in the posttranslational cleavage of the neural cell adhesion molecule L1. J. Cell Sci. 112, 4739–4749 (1999).
Silletti, S., Mei, F., Sheppard, D. & Montgomery, A. M. Plasmin-sensitive dibasic sequences in the third fibronectin-like domain of L1-cell adhesion molecule (CAM) facilitate homomultimerization and concomitant integrin recruitment. J. Cell Biol. 149, 1485–502 (2000).
Mechtersheimer, S. et al. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J. Cell Biol. 155, 661–673 (2001).
Maretzky, T. et al. L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol. Cell. Biol. 25, 9040–9053 (2005).
Riedle, S. et al. Nuclear translocation and signalling of L1-CAM in human carcinoma cells requires ADAM10 and presenilin/gamma-secretase activity. Biochem. J. 420, 391–402 (2009).
Kiefel, H. et al. L1CAM: a major driver for tumor cell invasion and motility. Cell Adh. Migr. 6, 374–384 (2012).
Lutz, D. et al. Generation and nuclear translocation of sumoylated transmembrane fragment of cell adhesion molecule L1. J. Biol. Chem. 287, 17161–17175 (2012).
Falconer, D. S. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 50, 192–201 (1951).
D’Arcangelo et al. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723 (1995).
Curran, T. & D’Arcangelo, G. Role of reelin in the control of brain development. Brain Res. Brain Res. Rev. 26, 285–294 (1998).
Frotscher, M. Cajal-Retzius cells, Reelin, and the formation of layers. Curr. Opin. Neurobiol. 8, 570–575 (1998).
Rice, D. S. & Curran, T. Role of the reelin signaling pathway in central nervous system development. Annu. Rev. Neurosci. 24, 1005–1039 (2001).
Tissir, F. & Goffinet, A. M. Reelin and brain development. Nat. Rev. Neurosci. 4, 496–505 (2003).
Hevner, R. F., Daza, R. A., Englund, C., Kohtz, J. & Fink, A. Postnatal shifts of interneuron position in the neocortex of normal and reeler mice: evidence for inward radial migration. Neuroscience 124, 605–618 (2004).
Quattrocchi, C. C. et al. Reelin is a serine protease of the extracellular matrix. J. Biol. Chem. 277, 303–309 (2002).
Devanathan, V. et al. Cellular form of prion protein inhibits Reelin-mediated shedding of Caspr from the neuronal cell surface to potentiate Caspr-mediated inhibition of neurite outgrowth. J. Neurosci. 30, 9292–9305 (2010).
Howell, B. W., Gertler, F. B. & Cooper, J. A. Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 16, 121–132 (1997).
Hiesberger, T. et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24, 481–489 (1999).
Trommsdorff, M. et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701 (1999).
Sheldon, M. et al. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 389, 730–733 (1997).
Ware, M. L. et al. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron 19, 239–249 (1997).
Kohno, T. & Hattori, M. Re-evaluation of protease activity of reelin. Biol. Pharm. Bull. 33, 1047–1049 (2010).
Ogawa, M. et al. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 14, 899–912 (1995).
D’Arcangelo, G. et al. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J. Neurosci. 17, 23–31 (1997).
Senzaki, K., Ogawa, M. & Yagi, T. Proteins of the CNR family are multiple receptors for Reelin. Cell 99, 635–647 (1999).
Lambert de Rouvroit, C. et al. Reelin, the extracellular matrix protein deficient in reeler mutant mice, is processed by a metalloproteinase. Exp. Neurol. 156, 214–217 (1999).
Jossin, Y. et al. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J. Neurosci. 24, 514–521 (2004).
Hisanaga, A. et al. A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4) cleaves Reelin in an isoform-dependent manner. FEBS Lett. 586, 3349–3353 (2012).
Ohyama, K. et al. Neural cell adhesion molecule L1 is required for fasciculation and routing of thalamocortical fibres and corticothalamic fibres. Neurosci. Res. 48, 471–475 (2004).
Wiencken-Barger, A. E., Mavity-Hudson, J., Bartsch, U., Schachner, M. & Casagrande, V. A. The role of L1 in axon pathfinding and fasciculation. Cereb. Cortex 14, 121–131 (2004).
Demyanenko, G. P. et al. L1 and CHL1 cooperate in thalamocortical axon targeting. Cereb. Cortex 21, 401–412 (2011).
Itoh, K. et al. Brain development in mice lacking L1-L1 homophilic adhesion. J. Cell. Biol. 165, 145–154 (2004).
Jakeman, L. B., Chen, Y., Lucin, K. M. & McTigue, D. M. Mice lacking L1 cell adhesion molecule have deficits in locomotion and exhibit enhanced corticospinal tract sprouting following mild contusion injury to the spinal cord. Eur. J. Neurosci. 23, 1997–2011 (2006).
Tonosaki, M. et al. L1cam is crucial for cell locomotion and terminal translocation of the soma in radial migration during murine corticogenesis. PLoS One 9, e86186 (2014).
Jouet, M. & Kenwrick, S. Gene analysis of L1 neural cell adhesion molecule in prenatal diagnosis of hydrocephalus. Lancet 345, 161–162 (1995).
Schäfer, M. K. & Frotscher, M. Role of L1CAM for axon sprouting and branching. Cell. Tissue Res. 349, 39–48 (2012).
Hong, S. E. et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 26, 93–96 (2000).
Rakic, P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 145, 61–83 (1972).
Cooper, J. A. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 31, 113–119 (2008).
Frykman, P. K., Brown, M. S., Yamamoto, T., Goldstein, J. L. & Herz, J. Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 92, 8453–8457 (1995).
Chen, S., Mantei, N., Dong, L. & Schachner, M. Prevention of neuronal cell death by neural adhesion molecules L1 and CHL1. J. Neurobiol. 38, 428–439 (1999).
Benhayon, D., Magdaleno, S. & Curran, T. Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Brain Res. Mol. Brain Res. 112, 33–45 (2003).
Förster, E. et al. Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc. Natl. Acad. Sci. USA 99, 13178–13183 (2002).
Loers, G. et al. The interaction between cell adhesion molecule L1, matrix metalloproteinase 14, and adenine nucleotide translocator at the plasma membrane regulates L1-mediated neurite outgrowth of murine cerebellar neurons. J. Neurosci. 32, 3917–3930 (2012).
Lee, G. H. et al. Reelin induces Erk1/2 signaling in cortical neurons through a non-canonical pathway. J. Biol. Chem. 289, 20307–20317 (2014).
Acknowledgements
We thank E. Kronberg, S. Deutsch, U. Wolters, and S. Homann for technical support, A. Failla for the possibility to use the IMARIS software, and M. Richter and F. Calderon de Anda for providing the CAG-IRES-GFP vectors. We are very grateful to T. Curran and A. Goffinet for the Reelin antibodies and plasmids. G. D’Arcangelo and T. Pohlkamp contributed helpful comments, and G. Rune gave the opportunity to use the Keyence microscope. This study was supported by grants from the Deutsche Forschungsgemeinschaft (FR 620/12-2 and FR 620/13-1) and the Hertie Foundation to M.F. D.L. is supported by a FFM fellowship of the Medical Faculty, University Medical Center Hamburg-Eppendorf. M.S. is grateful to the Li Kashing Foundation for support.
Author information
Authors and Affiliations
Contributions
D.L. designed and performed experiments, analysed data and wrote the manuscript draft. A.S. designed and performed immunostaining experiments, Golgi silver impregnation and Nissl staining. A.S. and D.D. contributed to immunoblotting, preparations of homogenates and tissues and to perfusion of animals. H.K. contributed to the homogenate preparations, immunoblotting, cell cultures, genotyping, cloning and sequencing. G.W.-E. contributed to cloning and analysis of L1-80. B.B. performed immunostainings, helped with cell culture and mouse breeding, and contributed to the design of the initial experiments. R.K. provided the mutated L1 constructs, contributed to the design of the initial experiments and to writing of the manuscript. G.L. provided immunoaffinity purified L1 from mouse brain and contributed to writing of the manuscript. D.L., M.F. and M.S. conceived the idea on the relationship between L1 and Reelin and discussed all experiments. M.F. and M.S. contributed to manuscript writing and editing.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lutz, D., Sharaf, A., Drexler, D. et al. Proteolytic cleavage of transmembrane cell adhesion molecule L1 by extracellular matrix molecule Reelin is important for mouse brain development. Sci Rep 7, 15268 (2017). https://doi.org/10.1038/s41598-017-15311-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-15311-x
This article is cited by
-
SLPI is a critical mediator that controls PTH-induced bone formation
Nature Communications (2021)
-
Dynamics, nanomechanics and signal transduction in reelin repeats
Scientific Reports (2019)
-
L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing
Scientific Reports (2019)
-
Molecular basis of Mitomycin C enhanced corneal sensory nerve repair after debridement wounding
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
