
Abstract
Photoreceptor death and retinal gliosis underlie the majority of vision threatening retinal diseases including retinal detachment (RD). Although the underlying pathobiology of vision limiting processes in RD is not fully understood, inflammation is known to play a critical role. We conducted an iTRAQ proteomic screen of up- and down-regulated proteins in a murine model of RD to identify potential targetable candidates. Macrophage migration inhibitory factor (MIF) was identified and evaluated for neurotoxic and pro-gliotic effects during RD. Systemic administration of the MIF inhibitor ISO-1 significantly blocked photoreceptor apoptosis, outer nuclear layer (ONL) thinning, and retinal gliosis. ISO-1 and MIF knockout (MIFKO) had greater accumulation of Müller glia pERK expression in the detached retina, suggesting that Müller survival pathways might underlie the neuroprotective response. Our data show the feasibility of the MIF-inhibitor ISO-1 to block pathological damage responses in retinal detachment and provide a rationale to explore MIF inhibition as a potential therapeutic option for RD.
Similar content being viewed by others
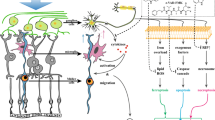


Introduction
Retinal detachment (RD) is a prevalent cause of visual loss, making a major impact on visual ability and quality of life1. Diverse retinal diseases may threaten sight because of RD formation including from ocular trauma, retinal tear, retinopathy of prematurity, diabetes, macular degeneration and intraocular tumors2. Surgical repair has a high rate of re-attaching the retina; however, permanent visual disability may occur despite prompt and successful surgical repair, with impaired reading vision in 40–60% of patients with macula-affecting RDs3,4. Different mechanisms, particularly photoreceptor degeneration and retinal gliosis result in vision loss following detachment. Thus, adjunctive neuroprotective and anti-gliotic strategies to reduce the neurodegeneration induced by RD could potentially benefit multiple diseases.
Given the known importance of inflammation on photoreceptor apoptosis5,6, retinal gliosis, and fibrosis7,8 after RD, we examined the inflammatory proteins identified in a proteomic screen of a murine model of RD. Macrophage migration inhibitory factor (MIF) was the only cytokine identified. It is pro-inflammatory, playing a key role in cell-mediated immunity9 and various autoimmune and central nervous system disorders10,11,12,13,14 and its role in RD is unclear. This study reports the results of a proteomic screen of murine experimental RD and evaluates the effects of MIF genetic depletion and pharmacologic inhibition.
Results
Proteomic analysis identifies up-regulation of MIF in experimental chronic RD
An iTRAQ proteomics discovery study was performed to evaluate up- and down-regulated proteins in an RD experimental model15 at early (2 week, n = 3) and later (4 week, n = 3) timepoints compared to controls (n = 2). 560 proteins with a false discovery rate < 0.05 were identified and relatively quantified (Fig. 1a; Table S1). The heatmap shows subtle differences in protein expression between control, 2 week and 4 week RD conditions.

Proteomic analysis of murine RD. (a) Heatmap of proteins with FDR < 0.05 identified on iTRAQ proteomic screen. Proteins extracted from individual murine retina plus vitreous at week 2 (n = 3) or week 4 (n = 3) post RD creation were labeled with iTRAQ probes in an 8-plex assay. Control murine retinas (n = 2) were sham-treated (label #119) or untreated (#121). Red represents strong protein expression and blue represents lack of expression. Validation of up-regulated proteins in homogenized retina plus vitreous was performed by ELISA at four timepoints post RD for MIF with n = 15 individual mice/group. MIF expression was significantly elevated in RD eyes at week 1 (b, *p = 0.0430).
Due to the known importance of proteins with inflammatory/immunologic function on RD pathology, this subset of proteins (23/560 proteins) was further analyzed (Table 1). No chemokines and only one cytokine were identified on the screen, narrowing the list of targetable, potentially-therapeutic candidates. Many of the strongly up-regulated proteins identified have important cytoprotective and anti-apoptotic functions which could be beneficial in RD. For example, prothymosin alpha, which was up-regulated in 2 week RD retina but not expressed in 4 week RD or controls, is neuroprotective in the retina and brain during periods of oxidative stress and ischemic injury16,17, factors which contribute significantly to photoreceptor death in RD.
Macrophage migration inhibitory factor (MIF) was the solitary cytokine identified on the screen and was up-regulated in 2 week > 4 week RDs (1.91 and 1.63 fold, respectively). It was attractive for further study since it is associated with worse disease severity for several inflammatory and fibrotic diseases conditions, making MIF inhibitors desirable for clinical development12,18. Given the known importance of various inflammatory mediators on photoreceptor apoptosis and scar formation after RD5,6 and the negative role of MIF in many diseases with an inflammatory component, the potential role of MIF inhibitors as adjunctive therapy in retinal disease was interesting to explore.
A commercially available inhibitor, ISO-1 is considered a canonical MIF inhibitor in preclinical studies and has beneficially impacted neurologic and inflammatory disease in several animal models13,19,20. Thus, subsequent studies evaluated the impact of MIF inhibition in RD including genetic depletion and pharmacologic inhibition.
Confirmation of MIF iTRAQ result
To confirm that MIF levels were up-regulated in RD, the RD retina + vitreous at day 1, day 3, week 1 and week 2 (n = 15/group) and fellow eye controls was evaluated with ELISA. MIF was expressed at high levels in control retina and increased significantly after RD at week 1 (136.73 ± 7.23 pg MIF/µg total protein in controls vs 158.58 ± 9.87 in RD, p = 0.0430, Fig. 1b).
Genetic depletion of MIF reduces photoreceptor apoptosis in experimental RD
RDs were created as previously described15,21. MIFKO mice with BALB/c background were used. They developed excessive periocular bleeding during the RD procedure (n = 6/10 MIFKO with large hemorrhage and none in background controls). A second study was conducted which used MIFKO mice in a C57BL/6 background and controls (n = 5/group) and the use of cautery to control surface bleeding; none developed hemorrhage.
The ONL contains the cell bodies of the photoreceptors which undergo cell death after RD and the ONL subsequently becomes thinner as photoreceptors die22. To assess the effect of genetic MIF depletion in RD, both TUNEL positive photoreceptors and ONL thickeness measurements were analyzed in both strains of MIFKO mice and their background controls (n = 5/group, Fig. 2a–d). There was a significant reduction in apoptosis by TUNEL assay at day 14 in both the BALB/c mice (1058.40 ± 125.11 cells/mm2 background controls vs 293.90 ± 103.22 MIFKO, p = 0.0008) and C57BL/6 mice (897.22 ± 232.97 cells/mm2 background controls vs 436.46 ± 65.05 MIFKO, p = 0.0466, Fig. 2c). Surprisingly, this significant reduction in TUNEL accumulation did not translate to a significant protection of the ONL thickness in both BALB/c and C57BL/6 MIFKO mice compared to background controls (p = 0.4169 and 0.2378, respectively, Fig. 2d).

MIF genetic depletion reduces apoptosis but not loss of ONL thickness in RD. MIFKO mice with BALB/c and C57BL/6 backgrounds were used with appropriate background controls (n = 5/group). Representative photographs shows TUNEL-positive cells in day 14 detached retina from BALB/c background controls (a) and MIFKO mice (b). Scale bar denotes 50 microns. TUNEL-positive cells were significantly reduced in the ONL of day 14 BALB/c and C57BL/6 MIFKO mice (c, **p = 0.0008 and *p = 0.0466, respectively). However, the loss of the ONL thickness in these day 14 RDs was not significantly reduced in either BALB/c or C57BL/6 MIFKO mice compared with background controls (d, p = 0.4169 and p = 0.2378, respectively). No significant changes were observed in cleaved caspase-3 positive cells (e, p = 0.5295). Cleaved PARP-positive cells were significantly reduced in day 14 MIFKO mice (f, *p = 0.0224). Confocal micrographs shows cleaved PARP positive cells in day 14 detached retina from C57BL/6 background controls (g) and MIFKO mice (h). Arrows denote positive cells. Scale bar denotes 25 microns.
To further explore the impact of genetic MIF depletion on photoreceptor cell death pathways in RD, we analyzed important apoptotic mediators. Activated caspase-3 plays a central role in executing the cleavage of DNA in apoptosis and can be activated by intrinsic and extrinsic apoptosis pathways. We found that the cleaved caspase-3 accumulation in the ONL in detached retina was not significantly different in MIFKO mice vs controls (Fig. 2e). Furthermore, we evaluated the effect of genetic depletion of MIF on accumulation of cleaved poly (ADP-ribose) polymerase (PARP) in detached photoreceptors (Fig. 2f–h). PARP is a multifunctional protein that plays a role in apoptosis, DNA repair, and cell death through caspase-dependent and -independent pathways. Similar to the reduction in TUNEL, MIFKO eyes had significantly reduced cleaved PARP accumulation compared to background controls at day 14 (1021.49 ± 321.37 cells/mm2 background control vs 193.59 ± 34.58 MIFKO, p = 0.0224, Fig. 2f). Taken together, these studies on photoreceptor apoptosis and cell death suggest that MIF plays a role in promoting photoreceptor apoptosis during RD, but despite significant reductions in TUNEL and cleaved PARP accumulation, global genetic depletion of MIF could not overcome permanent loss of ONL thickness. To better understand the role of MIF in RD, concurrent studies were undertaken to evaluate the impact of pharmacologic inhibition of MIF.
Pharmacologic inhibition of MIF protects photoreceptors after experimental RD
We investigated the ability of systemically administered ISO-1 to prevent apoptosis and photoreceptor death (n = 6, C57BL/6 mice/group, Fig. 3). ISO-1 significantly reduced TUNEL positive cells in the ONL at day 3, when maximal apoptosis is expected (4125.89 ± 849.5 cells/mm2 retina vehicle vs 588.68 ± 409.5 ISO-1, p = 0.0036, Fig. 3f). The significant reduction in apoptosis by systemic ISO-1 at day 3 was replicated in an independent experiment using 35 mg/kg instead of 40 mg/kg (n = 6/group, p = 0.008). The higher numbers of TUNEL positive cells in the ISO-1 vs MIFKO studies is best explained by time point; the numbers of TUNEL positive cells are maximal at day 1–3 RDs and decline significantly at later time points (day 14 RD was used in the MIFKO studies to allow evaluation of both TUNEL and ONL thickness in the same sample)23,24.

ISO-1 treatment blocks photoreceptor apoptosis and loss of ONL thickness. Experimental design (a): ISO-1(40 mg/kg/day) or vehicle treatment was initiated 1 day prior to RD creation and continued daily until sacrifice at day 3 or 14 (n = 6/group). Representative photographs show TUNEL positive cells (red) in the ONL in day 3 detached retina from vehicle (b) and ISO-1 treated mice (c). Scale bar denotes 50 microns. TUNEL positive cells were significantly reduced in ISO-1 treated mice at day 3 when apoptosis is maximal (f, *p = 0.0036), but not different at day 14 (not shown). A second independent study confirmed the significant reduction in TUNEL by ISO-1 (35 mg/kg/day, n = 6/group, p = 0.008, not shown). ONL thickness in ISO-1 treated eyes was significantly thicker than that of vehicle treated eyes at day 14 (g, *p = 0.0278). Confocal micrographs shows activated caspase-3 positive cells (green) in the ONL of day 3 background controls (d) and ISO-1 treated (e) mice (n = 10/group). Scale bar denotes 25 microns. Caspase-3 positive cells were not significantly reduced in ISO-1 treated animals compared to controls in day 3 and 14 (h, n = 10/group, p = 0.2593 and p = 0.1367, respectively). Cleaved PARP protein expression in ISO-1 treated eyes was not significantly different from vehicle treated eyes at either timepoints (i, n = 6, p = 0.3269 at day3 and n = 5, p = 0.3163 at day14). AIF protein expression in ISO-1 treated eyes was not significantly increased from vehicle treated eyes at day 3 (j, n = 6/group, p = 0.2891).
To determine whether ISO-1 could preserve photoreceptors in detached retina over time, the ONL thickness in day 14 RDs was quantitated (n = 6/group). The ONL thickness in the vehicle group dropped significantly to 83.70% ± 5.50 of fellow-eye control while in the ISO-1 group ONL thickness was preserved at 99.28% ± 4.63 (p = 0.0278, Fig. 3g).
ISO-1 impact on caspase-dependent and -independent apoptosis pathways in RD
To evaluate whether systemic ISO-1 inhibited caspase-3-mediated apoptosis of photoreceptor cells in RD, confocal analysis was performed at day 3 and day 14 (n = 10/group). ISO-1 treatment did not significantly decrease activated caspase-3 positive cells in the ONL, although there was a trend toward a decrease at day 14 (62.68 ± 32.38 cells/mm2 retina vehicle vs 23.88 ± 8.51 ISO-1, p = 0.1367, Fig. 3h).
PARP is another component of the cell-death machinery that we hypothesized could be affected by ISO-1. However, confocal analysis of cleaved PARP immunostaining in day 3 and 14 RDs showed no difference in accumulation of cleaved PARP in the ONL in ISO-1 treated eyes compared with controls (606.26 ± 287.78 cells/mm2 retina vehicle vs 836.59 ± 404.57 cells/mm2 retina ISO-1, p = 0.3269, n = 6 at day3 and 690.53 ± 421.20 cells/mm2 retina vehicle vs 424.65 ± 324.84 cells/mm2 retina ISO-1, p = 0.3163, n = 5 at day14, Fig. 3i). Caspase-independent mechanisms of apoptosis, such as up-regulation of apoptosis inducing factor (AIF), have been reported to account for a significant portion of the photoreceptor apoptosis in experimental RD25,26. To evaluate the potential effect of ISO-1 treatment on AIF expression in RD, we assayed AIF levels on western blot and IHC (n = 6/group). Western immunoblotting quatification showed that ISO-1 did not significantly alter AIF levels compared to vehicle (p = 0.2133, not shown). Similarly, confocal analysis showed AIF accumulation in the ONL was not significantly altered in ISO-1 treated retina compared to vehicle (397.47 ± 66.54 cells/mm2 retina vehicle vs 533.18 ± 226.59 ISO-1, p = 0.2891, Fig. 3j).
ISO-1 does not affect the number of F4/80 + cells after RD
Macrophages and microglia, identified by F4/80 + immunostaining, are recruited after experimental RD27. In a liver model, genetic depletion of MIF is associated with decreased F4/80 + cells28. Moreover, since macrophages can produce factors, like Monocyte chemoattractant protein-1 (MCP-1), that promote photoreceptor apoptosis in experimental RD5, the number of macrophages was quantitated (n = 6 mice/group, Fig. 4a). There was no statistically significant difference in F4/80 + cells between the treatment groups at day 3 (43.73 ± 15.33 cells/mm2 retina vehicle vs 29.85 ± 9.45 ISO-1, p = 0.2293) or day 14 (113.80 ± 33.60 cells/mm2 retina vehicle vs 117.84 ± 43.42 ISO-1, p = 0.4714, Fig. 4b). Similarly, the macrophage distribution (subretinal space, photoreceptor layer, and ganglion cell layer to ONL) was not significantly different (not shown).

Macrophage accumulation in RD is not affected by ISO-1. (a) Immunoperoxidase photomicrographs of F4/80+ cells (red) in the retina of C57BL/6 mice systemically treated with ISO-1 or vehicle demonstrates an accumulation of inflammation in detached retina compared with attached fellow-eye controls at day 3 and 14 (n = 6/group). (b) Quantification reveals the F4/80+ cell count was not affected by ISO-1 treatment compared with vehicle treatment at either timepoints (p = 0.2293, day3 and p = 0.4714, day14). Scale bar denotes 50 microns.
Expression of GFAP after RD is reduced by ISO-1 and MIFKO
Intermediate filaments like GFAP increase after RD29,30,31,32 and are a marker for gliotic changes in the retina important to the formation of excessive retinal scarring, a feature of proliferative vitreoretinopathy33,34. Moreover, mice with genetic depletion of the intermediate filaments GFAP and vimentin have decreased photoreceptor apoptosis and loss of ONL thickness after RD32. To determine the impact of ISO-1 systemic treatment on retinal intermediate filament expression, GFAP immunostaining was performed (n = 6/group, Fig. 5a–f). The normalized GFAP signal intensity was not significantly different in the ISO-1 treated mice at day 3, but was significantly decreased at day 14 (136.40 ± 12.48 mean intensity/mm2 retina vehicle vs 93.79 ± 14.69 ISO-1, p = 0.0256, Fig. 5k). Similarly, MIFKO retinas (n = 5/group) had significantly less GFAP reactivity at day 14 than background controls (83.69 ± 15.47 mean intensity/mm2 retina BALB/c control vs 43.44 ± 10.54 MIFKO, p = 0.0319, Fig. 5l).

ISO-1 treatment and MIF genetic depletion both reduce retinal gliosis. Immunofluorescence staining shows GFAP accumulation (green) after RD (b,c,e,f) compared with attached controls (a,d) in vehicle or ISO-1 treated animals at day 3 or day 14 (n = 6/group). Treatment with ISO-1 displayed significant reduction in gliosis in day 14 eyes (k, *p = 0.0256) while GFAP expression in day 3 detached retina was not significantly decreased (k, p = 0.1898). Detached retinas in day 14 BALB/c wild type controls (h) and MIFKO mice (j) displayed increased gliosis by GFAP accumulation over adjacent attached retina in controls (g,i). Detached retina in MIFKO mice had significantly reduced GFAP accumulation compared with background mice (l, n = 5/group, *p = 0.0319). Scale bar denotes 50 microns.
Pharmacologic inhibition of MIF increases retinal pERK accumulation after RD
Levels of phosphorylated ERK1/2 increase in retinal Müller cells and astrocytes after experimental RD32. Several MIF functions have been linked to intracellular signaling by ERK1/2 phosphorylation35,36 and it is unknown if ISO-1 could have any effect on retinal pERK accumulation. We hypothesized that pERK levels would be diminished by MIF inhibition during RD. To test this, we evaluated retinal pERK levels in vehicle- and ISO-1 treated murine RDs (n = 6/group, Fig. 6a–f). Immunofluorescence analysis showed that ISO-1 treatment was actually associated with an increase in retinal pERK levels in Müller glia compared with vehicle-treated RD retinas at day 14 (62.55 ± 9.16 mean intensity/mm2 retina vehicle vs 84.62 ± 5.97 ISO-1, p = 0.0356, Fig. 6k). The identity of Müller-glia progenitor cells was confirmed with Sox-2 immunofluorescence (not shown). Similarly, MIFKO mice at day 14 (n = 5/group) showed increased retinal pERK accumulation in RD (107.68 ± 27.60 mean intensity/mm2 retina BALB/c control vs 237.01 ± 48.54 MIFKO, p = 0.0246, Fig. 6l). Thus, ISO-1 treatment and MIF genetic depletion surprisingly had enhanced accumulation of pERK in the Müller glia during RD. Interestingly, several studies show that accumulation of retinal ERK1/2 phosphorylation in different models of retinal damage can be neuroprotective for photoreceptors37,38 and it is tempting to speculate that up-regulation of Müller survival pathways could be a mechanism underlying ISO-1 neuroprotection during RD.

Retinal pERK is up-regulated in ISO-1 and MIFKO RDs. Immunofluorescence staining shows pERK (green) accumulation in Müller glia after RD (b,c,e,f) compared with attached controls (a,d) in vehicle or ISO-1 treated animals at day 3 or day 14 (n = 6/group). Treatment with ISO-1 displayed significant elevation in pERK expression in day 14 eyes (k, *p = 0.0356) while pERK expression in day 3 detached retinas was not significantly elevated (k, p = 0.1948). Detached retinas in day 14 BALB/c wild type controls (h) and MIFKO mice (j) displayed increased pERK accumulation (red) compared to attached retina in controls (g,i). Detached retina in MIFKO mice had significantly elevated pERK accumulation compared with background mice (l, n = 5/group, *p = 0.0246). Scale bar denotes 50 microns.
ISO-1 does not significantly alter macrophage polarization in RD
Inacio et al. showed that ISO-1 was neuroprotective in a stroke model in rats, which correlated with a change in the quality rather than quantity of macrophages/microglia in the infarcted brain13. Macrophages and microglia have functional characterizations known as M1 polarization (producing pro-inflammatory products such as inducible nitric oxide synthase (iNOS)) or M2 polarization (characterized by a wound-healing function with products such as arginase-1 (Arg1))39. We investigated the expression of inflammatory mediators and M1 vs M2 gene expression in the retina + vitreous of vehicle or ISO-1-treated RD mice at day 3 post detachment (n = 9–11 mice/group, Fig. S1a ). We found no significant difference in M1 or M2 gene expression induced by systemic ISO-1 treatment in this RD model. Similarly, immunostaining for Arg1 in macrophage hot-spots in detached retina revealed no significant difference in the proportion of Arg1 positive macrophages between the groups (n = 12–13mice/group, Fig. S1b ).
Discussion
Proteomic iTRAQ screening identified several differentially expressed proteins in RD retina relevant to neuroprotection. Herein we focused on the proinflammatory cytokine MIF for its translational value; other interesting candidates will be explored in future studies.
MIF had a strong rationale for further exploration as a potential therapeutic target in RD. Its expression is associated with worse disease severity for several inflammatory and fibrotic diseases12,18. We demonstrated findings relevant to possible clinical translation: that systemic administration of the MIF-inhibitor ISO-1 blocked photoreceptor apoptosis and ONL thinning in detached retina. These results suggest a neuroprotective effect of the drug and identify MIF as a targetable molecule to potentially impact visual loss from RD.
Matsuda et al. has also shown that MIF may play a role in the regulation of ocular immunity and inflammation. In rat, they showed that MIF is expressed in the nonpigmented ciliary and iris epithelium40. In the rat cornea, MIF is expressed in the basal epithelium and endothelium and it is rapidly upregulated in the cornea and aqueous humor after penetrating injury41. MIF was also identified in human corneal epithelium and endothelium, suggesting a potential role in corneal immunity and uveitis42. In fact elevated MIF levels were detected in serum of patients with uveitis, with highest levels during active phases43,44. Posteriorly, Matsuda et al. identified constitutive MIF expression in the rat retina, primarily in the astrocytes, Müller glia, and RPE45.
In accordance with our findings of MIF in experimental RD, similar effects of MIF inhibition have been shown in other CNS disease models, including experimental stroke13, experimental autoimmune encephalitis46, and NMDA-induced retinal damage47 but not in a model of optic nerve injury48 or some other models of stroke49,50. Notably, the present study utilized female mice for the ISO-1 experiments and found a neuro-protective effect. In contrast, the study by Turtzo et al. found that MIF genetic depletion was deleterious in female mice compared to males after stroke. These findings suggest that there are likely different tissue- and model-specific neuroprotective effects of MIF as well as possible sex differences. Future studies should further evaluate these biologically relevant variables on MIF retinal biology.
Inflammation is one of the main mechanisms that lead to photoreceptor death after RD5 and MIF may contribute to the inflammatory cascade by inhibiting apoptosis in monocytes/macrophages51. Moreover, MIF inhibition or genetic depletion blocks the accumulation of infiltrating macrophages in some disease models20. However, in our model, the accumulation of F4/80 + macrophages located in the detached retina was not significantly altered by ISO-1. A possible explanation is that since MIF exists pre-formed in monocytes/macrophages52 and its pro-inflammatory action cannot be sufficiently prevented, at least not in the early stages after the detachment. Other studies of MIF in the CNS showed that MIF depletion was neuroprotective in a cerebral infarct model without changing the quantity of accumulating macrophages but by altering their surface expression13. Although we predicted that the macrophage phenotype would be shifted to a more M2-predominant polarization by ISO-1, levels of select M1 and M2 retinal gene expression were not significantly altered by ISO-1 treatment, nor was the fraction of arginase-1 positive M2 macrophages in the detached retina. These results do not exclude the possibility that inflammatory effects of MIF play a role in inducing neurodegeneration in RD and perhaps a different time point would allow detection of an effect. Future work will further evaluate the impact of timing and microglia depletion on MIF inhibitor-mediated neuroprotection.
The mechanism of ISO-1 neuroprotection during RD is unknown. Interestingly we found that in the MIFKO experiments, TUNEL and cleaved PARP were significantly reduced in the knockout mice, while cleaved caspase-3 did not reach significance. In the ISO-1 studies, we found that TUNEL was significantly reduced and ONL thickness was significantly protected by ISO-1, while caspase-3, cleaved PARP, and AIF (a caspase-independent mediator of apoptosis) were not significantly different between groups. These findings show that MIF inhibition in experimental RD has neuroprotective effects, yet the exact mechanism is unclear and may be different between the pharmacologic and global MIF depletion conditions. In the ISO-1 treated mice, there is not total depletion of MIF, compared to the MIF knockout. The ISO-1 treated mice are likely more physiologically relevant to what we would expect in the human. We are continuing work to better evaluate the relationship of MIF with caspase-dependent and independent cell death pathways in the retina.
Elegant studies by White et al.53 may contribute a potential explanation for variable cytoprotective results in MIF depletion studies due to the importance of the “compartmentalization” of MIF. In their murine myocardial infarction model they explored the importance of MIF expressed by the stromal cell compartment of the damaged tissue (cardiac myocytes) versus the bone-marrow-derived cell compartment. Chimeric MIF knockouts (MIFKO) were created so that the resident cells of the body remained either MIF wild-type (WT) or KO, and received either MIF WT or KO bone-marrow-derived cells. After myocardial infarction, having MIF WT cardiac myocytes and MIFKO bone marrow was cardioprotective, even more than global MIFKO in both compartments. In contrast, mice with MIF WT bone marrow and MIF KO cardiac myocytes had an increased risk of cardiac rupture after myocardial infarction even more than wild type mice. Liehn et al. showed similar results with CXCR2 chimeras54. It is possible that different compartments of MIF expression could be important in the retina as well. Interestingly, there is high baseline retinal MIF expression, particularly in the astrocytes and Müller glia45 and the functional role of MIF in this compartment is unknown. It is possible that Müller MIF might have a similar cytoprotective role as seen in the cardiac myocytes and may help explain why global genetic depletion of MIF did not produce as robust neuroprotection as the MIF inhibitor ISO-1. Future studies with Müller-selective MIF depletion as well as depletion of microglia/macrophages should investigate this, particularly as MIF inhibition may be considered therapeutically.
Another candidate neuroprotective mechanism of MIF inhibition was the up-regulation of the MAPK Müller survival pathway, which is neuroprotective for photoreceptors in different damage models37,38,55,56. Both ISO-1 and MIF genetic depletion enhanced the accumulation of pERK in the Müller glia during RD, which may promote photoreceptor survival. Future studies must be conducted to further elucidate the underlying mechanism of protection.
Proliferative vitreoretinopathy (PVR) is a feared complication after RD with excessive gliosis, scar formation, and severe vision loss. The activation of Müller glia and up-regulation of intermediate filaments like GFAP are particularly significant in PVR formation as well as in RD, with the intensity of staining correlating with the severity of detachment34. In our experiments, treatment with ISO-1 and genetic depletion of MIF both significantly decreased GFAP signal intensity, suggesting that retinal gliosis could be mitigated by MIF inhibition. This result is similar to findings in a neurodegenerative Alzheimer’s murine model in which MIF genetic depletion or blockade by ISO-1 decreased CNS GFAP levels57. In contrast, GFAP levels were not decreased in MIF knockouts in an experimental stroke model and the authors concluded that MIF is part of the gliotic process but not essential for it58.
Our preclinical ISO-1 data have clinical relevance to PVR; MIF levels are significantly elevated in the vitreous and subretinal fluid of patients with RD developing PVR compared with those without PVR59,60. Thus, there is a rationale for future clinical trials with MIF inhibitors, particularly for high-risk detachments.
Other prevalent vision-threatening retinal diseases such as age-related macular degeneration and diabetic retinopathy could also benefit from inhibition of gliosis as well as photoreceptor apoptosis and such strategies are being considered in the development of novel therapeutics for these diseases61,62. Proliferative diabetic retinopathy in particular may benefit from MIF inhibition; ocular MIF levels are elevated in patients with diabetic retinopathy, with the highest level in proliferative disease with the greatest degree of fibrosis, suggesting that MIF may play a role in the fibrovascular proliferative process63,64.
Because surgical repair of RD alone is inadequate to prevent loss of photoreceprtors, additional treatment options aimed at the inflammatory and gliotic component of the neurodegenerative process may be valuable to reduce vision loss. To the best of our knowledge, this is the first study where ISO-1 was used as an inhibitor of MIF in an experimental model of RD. Our findings suggest a rationale to develop and trial MIF inhibitors as adjunctive therapeutics for patients with retinal detachment and other retinal diseases.
Methods
Reagents
ISO-1 ((S, R)-3-(4-hydroxyphenyl)-4, 5-dihydro-5-isoxazole acetic acid methyl ester) was obtained from EMD Millipore (Billerica, MA) and diluted in 10% DMSO for in vivo studies.
Animals and Retinal Detachment Surgery
This research adheres to the principles of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. It was conducted under a protocol approved by The Ohio States University Institutional Animal Care and Use Committee. Similar to our previous studies, adult female C57BL/6 mice (age 16–31weeks) were purchased from The Jackson Laboratory (Bar Harbor, ME)15. Better quality RDs are generated in mice aged 16 weeks or older (unpublished observation). For genetic depletion studies, BALB/c and C57BL/6 control mice and previously described Mif - knockout (MIFKO) mutants65 backcrossed > 10 generations to a BALB/c and C57BL/6 background (age 16–20 weeks) were used.
Retinal detachments (RD) were induced in anesthetized mice by subretinal injection of undiluted hyaluronic acid (HA, 10 mg/ml, Abbott Medical Optics, Abbott Park, IL) into left eyes as previously described21.
ISO-1 Studies
Starting 24 hours prior to RD induction, C57BL/6 mice were treated with daily intraperitoneal injections of freshly prepared ISO-1, (40 mg/kg diluted in saline with 10% DMSO) or vehicle until the day of euthanasia, similar to the protocol of Leng et al.20. A second study at 35 mg/kg was performed with similar results.
Enucleation, Fixation, and mRNA and Protein Isolation
Anesthetized mice were euthanized and eyes enucleated. Retinas, with vitreous, were dissected under an operating microscope using 0.12 forceps and Vannas scissors to cut the sclera and retina posterior to the limbus and remove the anterior cap of the eye. In the remaining posterior globe, the retina was gently separated from the underlying RPE and choroid and was cut near the insertion into the optic nerve. Retinas that were processed for RNA were dissected with instruments wiped with RNase-Zap and retinas were placed in TRIzol. For protein isolation retinas were placed in ice-cold 1x cell lysis buffer (#9803 S, Cell Signaling) freshly prepared with protease inhibitor cocktail set (#539134, Calbiochem) and 1 mM of phenylmethylsulfonyl fluoride (#AC215740010, Acros Organics)15.
iTRAQ labeling
Protein lysates from each retina were quantitated as previously described15. The retina + vitreous protein extracts (100 µg of protein per sample) were diluted with 50 mM TEAB, incubated with 2% SDS denaturant and 5 mM TCEP reducing agent. Fresh iodoacetamide (84 mM) was added and incubated before the addition of sequencing grade trypsin (Promega) for overnight incubation at 37 °C. The digests were dried in a vacuum centrifuge and iTRAQ labeling was carried out according to the instructions in the iTRAQ labeling kit (iTRAQ 8-plex One Assay Kit, Penn State University Proteomic Core).
2D –LC and Mass spectrometry
Liquid chromatography (LC) 2D separation and tandem mass spectrometry was performed at Penn State University Proteomic Core Facility. 2D-LC separations: SCX Separations were performed on a passivated Waters 600E HPLC system, using a 4.6 × 250 mm Polysulfoethyl aspartamide column (PolyLC, Columbia, MD) at a flow rate of 1 ml/min. Buffer A contained 10 mM ammonium formate, pH 2.7, in 20% acetonitrile/80% water. Buffer B contained 666 mM ammonium formate, pH 2.7, in 20% acetonitrile/80% water. The gradient was Buffer A at 100% (0–22 minutes following sample injection), 0% → 40% Buffer B (16–48 min), 40% → 100% Buffer B (48–49 min), then isocratic 100% Buffer B (49–56 min), then at 56 min switched back to 100% A to re-equilibrate for the next injection. The first 26 ml of eluant (containing all flow-through fractions) was combined into one fraction, then 14 additional 2-ml fractions were collected. All 15 of these SCX fractions were dried down completely to reduce volume and to remove the volatile ammonium formate salts, then resuspended in 9 µl of 2% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid and filtered prior to reverse phase C18 nanoflow-LC separation. For 2nd dimension separation by reverse phase nanoflow LC, each SCX fraction was autoinjected onto a Chromolith CapRod column (150 × 0.1 mm, Merck) using a 5 µl injector loop on a Tempo LC MALDI Spotting system (ABI-MDS/Sciex). Buffer C was 2% acetonitrile, 0.1% trifluoroacetic acid, and Buffer D was 98% acetonitrile, 0.1% trifluoroacetic acid.
The elution gradient was 95% C/ 5% D (2 µl per minute flowrate from 0–3 min, then 2.5 µl per minute from 3–8.1 min), 5% D → 38% D (8.1–40 min), 38% D → 80% D (41–44 min), 80% D → 5% D (44–49 min) (initial conditions). Flow rate was 2.5 µl/min during the gradient, and an equal flow of MALDI matrix solution was added post-column (7 mg/ml recrystallized CHCA (a-cyano-hydroxycinnamic acid), 2 mg/ml ammonium phosphate, 0.1% trifluoroacetic acid, 80% acetonitrile). (Gradient used prior to 2008 was 100% C (0–4 min), 0 → 10% D (4–10 min), 10% → 25% D (10–30 min), 25% → 40% D (30–35 min), 40% → 80% D (35–38 min), 80% D (38–42 min), 80% → 0% D (42–43 min), 0% D (43–50 min), with a flow rate of 3.0 µl per minute with an equal flow of MALDI matrix solution.) The combined eluant was automatically spotted onto a stainless steel MALDI target plate every 6 seconds (0.6 µl per spot), for a total of 370 spots per original SCX fraction.
Mass Spectrometry
After sample spot drying, thirteen calibrant spots (ABI 4700 Mix) were added to each plate manually. MALDI target plates (15 per experiment) were analyzed in a data-dependent manner on an ABI 5800 MALDI TOF-TOF. As each plate was entered into the instrument, a plate calibration/ MS Default calibration update was performed, and then the MS/MS default calibration was updated. MS Spectra were taken from 5500 MALDI Spots, averaging 500 laser shots per spot at laser power 2600. A plate-wide interpretation was then automatically performed, choosing the highest peak of each observed m/z value for subsequent MS/MS analysis. A total of 8118 MS/MS spectra were taken with up to 2500 laser shots per spectrum at Laser Power 3100, with CID gas Air at 1.2 to 1.3 × 10–6 Torr.
Up to 2500 laser shots at laser power 3100 were accumulated for each MS/MS spectrum. After the MS and MS/MS spectra from all 15 plates in a sample set had been acquired, protein identification and quantitation were performed using the Paragon algorithm as implemented in Protein Pilot 3.0 software (ABI/MDS-Sciex). Protein Pilot Software was utilized for searches with the following search parameters: Cys Alkylation – Iodoacetamide; ID Focus – Biological Modifications; Search Effort – Thorough. The database searched was Jan 1st 2010 mouse NCBI database Sequences containing 256336 Protein Sequences, plus 156 common lab contaminants. For estimation of false discovery rate (FDR), a simultaneous search was performed of a concatenated Decoy database which is the exact reverse of each protein sequence in the database plus 156 common human and lab contaminants (ABSciex_ContaminantDB_20070711). Total protein sequences searched in the database plus contaminants plus concatenated reverse decoy database was 512,156. The protein identifications at 95% confidence level were retained. The preset “Thorough” (iTRAQ or Identification) search settings were used where identifications must have a ProteinPilot Unused Score > 1.3 ( >95% Confidence interval). In addition, the only protein IDs accepted had a local “FDR” estimation of no higher than 5%, as calculated from the slope of the accumulated Decoy database hits by the PSPEP (Proteomics System Performance Evaluation Pipeline) program66.
One-matrix heatmaps of the data were generated using CIM Miner (http://discover.nci.nih.gov/cimminer/) by plotting the area of each protein with FDR < 5% for each iTRAQ group. Clustering was performed using the Euclidean distance method with the Average Linkage algorithm. The column order was kept fixed for the analysis. Fold-change was determined by calculating the ratio of the protein area of the RD group divided by the area of the control group for proteins with FDR < 5%. A subset of proteins with immune function was identified by searching the protein function column with keywords “immune,” “inflam” “inflammation”, “cytokine”, “chemokine”, “macrophage”, “monocyte”, “T cell”, “neutrophil”, “TNF”, “nitric oxide”, “MHC”, “interferon”,” phagocytosis”, and “microglia.”
Enzyme-Linked Immunosorbent Assay
MIF protein levels were measured in retina-vitreous protein extracts by a colorimetric sandwich ELISA kit (Mouse MIF DuoSet ELISA, R&D Systems, sensitivity range, 125–8000 pg/ml) per the manufacturer’s instructions. 96 well-plates were coated over night with the capture antibody, washed, blocked with blocking solution and incubated at room temperature for 1.5 hours. The biotinylated detection antibody was applied. Protein concentration was calculated from a standard curve of recombinant MIF. Samples were run in duplicate per assay. Equal amounts of extracted proteins determined by BCA assay in lysis buffer were loaded with lysis buffer per well. Samples were run in duplicate. The final results were expressed as the ratio of MIF protein expression in picograms per total protein in microgram.
Immunohistochemistry (IHC)
Immunofluorescence was performed to detect markers as described15 using antibodies to glial fibrillary acidic protein (GFAP) (mouse monoclonal, Sigma-Aldrich), phospho-p44/42 MAPK(Erk1/2) (137F5, rabbit monoclonal, Cell Signaling), cleaved caspase-3 (Asp175, rabbit polyclonal, Cell Signaling), cleaved PARP (Asp214, mouse monoclonal, Cell Signaling), AIF (H-300, rabbit ployclonal, Santa Cruz Biotechnology), Sox-2 (Y-17, goat polyclonal, Santa Cruz Biotechnology), and arginase-1 (N-20, goat polyclonal, Santa Cruz Biotechnology). Slides were blocked with PBS containing 5% bovine serum albumin, and 1% Triton-X 100. Alexa Fluor 488 or 568 conjugated secondary antibodies (Invitrogen) were applied after 30 minute incubation with 100% normal goat serum. Omission of the primary antibody was used as a control for background staining; omission of primary and secondary antibodies was used as a control for autofluorescence. Cell nuclei were counterstained with DAPI (Invitrogen) and DRAQ5 (Thermo Scientific). Immunoperoxidase IHC was performed to evaluate macrophages and microglia using antibody to F4/80 (rat monoclonal, IgG2b, AbD Serotec), since autofluorescence was a confounding factor in the subretinal space. Goat IgG (Vector Laboratories) in blocking solution was applied to block Fc receptors. Secondary goat-anti-rat(Invitrogen) conjugated with horseradish peroxidase were applied. ImmPACT NovaRED kit (Vector Laboratories) was used to visualize the markers. Hematoxylin was used as a nuclear counterstain.
TUNEL assay
TUNEL assay was performed to detect apoptosis and cell death say with the In Situ Cell Death Kit (TMR red; 1215679910, Roche Applied Science) per the manufacturer’s instructions.
Western Immunoblotting Assay
Quantitated equal amount of total protein was run with a precast 4–20% Tris-glycine gel (Novex Wedge well, Invitrogen). Proteins on the gel were electroblotted onto a polyvinylidene fluoride membrane (Bio-Rad Laboratories). After blocking with 5% non fat dry milk, the membrane was immunoblotted for AIF expression. The blots were developed using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Scientific). The expression of α-tubulin (11H10, rabbit monoclonal, Cell Signaling) as a loading control was also evaluated after stripping off AIF antibody to normalize the data.
Quantitative real-time (qRT)-PCR
qRT-PCR was performed as previously described15. Briefly, total RNA was extracted from individual retinas using TRIzol (Invitrogen). cDNA was synthesized (SuperScript VILO cDNA Synthesis Kit (Invitrogen)). Gene expression of murine Nos2, Ccl2, Cxcl10, Arg1, Mrc1, Tnfa, Il1b, and Cd163 was quantified using catalogued Taqman probes and normalized to Gapdh (Applied Biosystems, ABI, Table S2).
Image analysis
Photomicrographs were obtained using a Leica DM5000B fluorescent microscope and Leica DC500 digital camera, or a Nikon Eclipse 50i microscope and Nikon Digital Sight DS-U1 camera with NIS-Elements software for immunoperoxidase images. Confocal images were obtained using a Zeiss LSM 510 at the Hunt-Curtis Imaging Facility. Sections from at least five different animals at the area of detachment were evaluated. Treatment groups were analyzed in a masked fashion. To minimize the variability of region-specific differences within the retina, the same region of retina was evaluated for the fellow-eye control and RD areas. Identical illumination, microscope, and camera settings were used. Fixed areas were randomly sampled across all retinal layers in areas of maximal detachment or similar control retina areas. Peripapillary retina and ora serrata were excluded from analysis. The retina was selected as the region of interest from the 200X field of view. Image analysis was performed with ImageJ64 (ImageJ64, National Institutes of Health, Bethesda, MD, USA) as previously described29. For outer nuclear layer (ONL) thickness measurements, the ratio of ONL thickness to total retinal thickness was determined to minimize sectioning artefacts and artefacts from undulations in the retina induced by the RD. GFAP and pERK levels were quantitated, dividing the mean gray value of the selected retina by the retinal area in mm2. Figure images were optimized for color, brightness, and contrast. For macrophage quantitation and phenotypic analysis, hot-spots of F4/80 + cells were imaged at 200X magnification and counted per mm2 retina. The ratio of arginase-1 + to F4/80 + macrophages was determined per 200X field of view. For TUNEL analysis, cell counting was performed by using Image-Pro 6.2 (Media Cybernetics, Bethesda, MD, USA). Cells with immunofluorescence signal above the threshold within fixed threshold-designated retinal regions (measuring 0.047 mm2) in the ONL were counted. Counting was also performed using ImageJ, Retina Analysis Toolkit high-threshold macro, on cropped images of the entire ONL from a 900 × 900 pixel image to identify TUNEL-positive cells with signal above the threshold within fixed, threshold-designated retinal regions. Multichannel thresholding was performed to quantitate TUNEL-positive cells that also exhibited DAPI positivity.
Statistics
Data were collected on standardized tables after analysis in a masked fashion. Error bars are listed with standard error of the mean. Student’s T-test was performed to evaluate the differences between two experimental groups, with p ≤ 0.05 considered statistically significant. In experiments with multiple groups, ANOVA was performed with Tukey post-hoc testing using JMP software.
Study approval
This research adheres to the principles of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. It was conducted under a protocol approved by our Institutional Animal Care and Use Committee.
References
Haimann, M. H., Burton, T. C. & Brown, C. K. Epidemiology of retinal detachment. Arch Ophthalmol 100, 289–292 (1982).
Ryan, S. J. Retina, (Elsevier/Mosby, Philadelphia, 2006).
Ross, W. H. Visual recovery after macula-off retinal detachment. Eye (Lond) 16, 440–6 (2002).
Stoffelns, B. M. & Richard, G. Is buckle surgery still the state of the art for retinal detachments due to retinal dialysis? J Pediatr Ophthalmol Strabismus 47, 281–7 (2010).
Nakazawa, T. et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci USA 104, 2425–30 (2007).
Nakazawa, T. et al. Tumor necrosis factor-alpha mediates photoreceptor death in a rodent model of retinal detachment. Invest Ophthalmol Vis Sci 52, 1384–91 (2011).
Pastor, J. C., de la Rua, E. R. & Martin, F. Proliferative vitreoretinopathy: risk factors and pathobiology. Prog Retin Eye Res 21, 127–44 (2002).
Yu, J., Peng, R., Chen, H., Cui, C. & Ba, J. Elucidation of the pathogenic mechanism of rhegmatogenous retinal detachment with proliferative vitreoretinopathy by proteomic analysis. Invest Ophthalmol Vis Sci 53, 8146–53 (2012).
Calandra, T. & Roger, T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol 3, 791–800 (2003).
Wu, S. P. et al. Macrophage migration inhibitory factor promoter polymorphisms and the clinical expression of scleroderma. Arthritis Rheum 54, 3661–9 (2006).
Baugh, J. A. et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun 3, 170–6 (2002).
Greven, D., Leng, L. & Bucala, R. Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets 14, 253–64 (2010).
Inacio, A. R., Ruscher, K., Leng, L., Bucala, R. & Deierborg, T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J Cereb Blood Flow Metab 31, 1093–106 (2011).
Niino, M., Ogata, A., Kikuchi, S., Tashiro, K. & Nishihira, J. Macrophage migration inhibitory factor in the cerebrospinal fluid of patients with conventional and optic-spinal forms of multiple sclerosis and neuro-Behcet’s disease. J Neurol Sci 179, 127–31 (2000).
Kim, B. et al. Retinal MMP-12, MMP-13, TIMP-1, and TIMP-2 expression in murine experimental retinal detachment. Invest Ophthalmol Vis Sci 55, 2031–40 (2014).
Halder, S. K., Matsunaga, H., Yamaguchi, H. & Ueda, H. Novel neuroprotective action of prothymosin alpha-derived peptide against retinal and brain ischemic damages. J Neurochem 125, 713–23 (2013).
Ueda, H., Matsunaga, H., Uchida, H. & Ueda, M. Prothymosin alpha as robustness molecule against ischemic stress to brain and retina. Ann N Y Acad Sci 1194, 20–6 (2010).
Zernecke, A., Bernhagen, J. & Weber, C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation 117, 1594–602 (2008).
Al-Abed, Y. et al. ISO-1 binding to the tautomerase active site of MIF inhibits its pro-inflammatory activity and increases survival in severe sepsis. J Biol Chem 280, 36541–4 (2005).
Leng, L. et al. A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. Journal of immunology 186, 527–38 (2011).
Cebulla, C. M., Ruggeri, M., Murray, T. G., Feuer, W. J. & Hernandez, E. Spectral domain optical coherence tomography in a murine retinal detachment model. Exp Eye Res 90, 521–7 (2010).
Mantopoulos, D. et al. Tauroursodeoxycholic acid (TUDCA) protects photoreceptors from cell death after experimental retinal detachment. PLoS One 6, e24245 (2011).
Matsumoto, H. et al. Strain difference in photoreceptor cell death after retinal detachment in mice. Invest Ophthalmol Vis Sci 55, 4165–74 (2014).
Cook, B., Lewis, G. P., Fisher, S. K. & Adler, R. Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci 36, 990–6 (1995).
Hisatomi, T. et al. Critical role of photoreceptor apoptosis in functional damage after retinal detachment. Curr Eye Res 24, 161–72 (2002).
Hisatomi, T. et al. Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am J Pathol 158, 1271–8 (2001).
Kaneko, H., Nishiguchi, K. M., Nakamura, M., Kachi, S. & Terasaki, H. Characteristics of bone marrow-derived microglia in the normal and injured retina. Invest Ophthalmol Vis Sci 49, 4162–8 (2008).
Barnes, M. A. et al. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology 57, 1980–91 (2013).
Cebulla, C. M. et al. A chick model of retinal detachment: cone rich and novel. PLoS One 7, e44257 (2012).
Luna, G., Lewis, G. P., Banna, C. D., Skalli, O. & Fisher, S. K. Expression profiles of nestin and synemin in reactive astrocytes and Muller cells following retinal injury: a comparison with glial fibrillar acidic protein and vimentin. Mol Vis 16, 2511–23 (2010).
Fisher, S. K. & Lewis, G. P. Muller cell and neuronal remodeling in retinal detachment and reattachment and their potential consequences for visual recovery: a review and reconsideration of recent data. Vision Res 43, 887–97 (2003).
Nakazawa, T. et al. Attenuated glial reactions and photoreceptor degeneration after retinal detachment in mice deficient in glial fibrillary acidic protein and vimentin. Invest Ophthalmol Vis Sci 48, 2760–8 (2007).
Lewis, G. P., Chapin, E. A., Luna, G., Linberg, K. A. & Fisher, S. K. The fate of Muller’s glia following experimental retinal detachment: nuclear migration, cell division, and subretinal glial scar formation. Mol Vis 16, 1361–72 (2010).
Sethi, C. S. et al. Glial remodeling and neural plasticity in human retinal detachment with proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci 46, 329–42 (2005).
Lue, H. et al. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal 18, 688–703 (2006).
Shi, X. et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25, 595–606 (2006).
Fischer, A. J., Scott, M. A., Ritchey, E. R. & Sherwood, P. Mitogen-activated protein kinase-signaling regulates the ability of Muller glia to proliferate and protect retinal neurons against excitotoxicity. Glia 57, 1538–52 (2009).
Kassen, S. C. et al. CNTF induces photoreceptor neuroprotection and Muller glial cell proliferation through two different signaling pathways in the adult zebrafish retina. Exp Eye Res 88, 1051–64 (2009).
Mantovani, A. et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25, 677–86 (2004).
Matsuda, A., Kotake, S., Tagawa, Y., Matsuda, H. & Nishihira, J. Detection and immunolocalization of macrophage migration inhibitory factor in rat iris and ciliary epithelium. Immunol Lett 53, 1–5 (1996).
Matsuda, A., Tagawa, Y., Matsuda, H. & Nishihira, J. Expression of macrophage migration inhibitory factor in corneal wound healing in rats. Invest Ophthalmol Vis Sci 38, 1555–62 (1997).
Matsuda, A., Tagawa, Y., Matsuda, H. & Nishihira, J. Identification and immunohistochemical localization of macrophage migration inhibitory factor in human cornea. FEBS Lett 385, 225–8 (1996).
Kitaichi, N. et al. Prominent increase of macrophage migration inhibitory factor in the sera of patients with uveitis. Invest Ophthalmol Vis Sci 40, 247–50 (1999).
Kitaichi, N. et al. Increase of macrophage migration inhibitory factor in sera of patients with iridocyclitis. Br J Ophthalmol 84, 1423–5 (2000).
Matsuda, A., Tagawa, Y., Yoshida, K., Matsuda, H. & Nishihira, J. Expression of macrophage migration inhibitory factor in rat retina and its immunohistochemical localization. J Neuroimmunol 77, 85–90 (1997).
Cox, G. M. et al. Macrophage migration inhibitory factor potentiates autoimmune-mediated neuroinflammation. J Immunol 191, 1043–54 (2013).
Naruoka, T. et al. ISO-1, a macrophage migration inhibitory factor antagonist, prevents N-methyl-D-aspartate-induced retinal damage. Eur J Pharmacol 718, 138–44 (2013).
Sobrado-Calvo, P., Vidal-Sanz, M. & Villegas-Perez, M. P. Rat retinal microglial cells under normal conditions, after optic nerve section, and after optic nerve section and intravitreal injection of trophic factors or macrophage inhibitory factor. J Comp Neurol 501, 866–78 (2007).
Turtzo, L. C. et al. Deletion of macrophage migration inhibitory factor worsens stroke outcome in female mice. Neurobiol Dis 54, 421–31 (2013).
Zhang, S. et al. Down-regulation of MIF by NFkappaB under hypoxia accelerated neuronal loss during stroke. FASEB J 28, 4394–407 (2014).
Mitchell, R. A. et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibitingp53: regulatory role in the innate immune response. Proceedings of the National Academy of Sciences of the United States of America 99, 345–50 (2002).
Calandra, T., Bernhagen, J., Mitchell, R. A. & Bucala, R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. The Journal of experimental medicine 179, 1895–902 (1994).
White, D. A. et al. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J Mol Cell Cardiol 69, 32–42 (2014).
Liehn, E. A. et al. Compartmentalized protective and detrimental effects of endogenous macrophage migration-inhibitory factor mediated by CXCR2 in a mouse model of myocardial ischemia/reperfusion. Arterioscler Thromb Vasc Biol 33, 2180–6 (2013).
Groeger, G., Doonan, F., Cotter, T. G. & Donovan, M. Reactive oxygen species regulate prosurvival ERK1/2 signaling and bFGF expression in gliosis within the retina. Invest Ophthalmol Vis Sci 53, 6645–54 (2012).
Bringmann, A. et al. Cellular signaling and factors involved in Muller cell gliosis: neuroprotective and detrimental effects. Prog Retin Eye Res 28, 423–51 (2009).
Li, S. Q. et al. Deficiency of macrophage migration inhibitory factor attenuates tau hyperphosphorylation in mouse models of Alzheimer’s disease. J Neuroinflammation 12, 177 (2015).
Inacio, A. R., Bucala, R. & Deierborg, T. Lack of macrophage migration inhibitory factor in mice does not affect hallmarks of the inflammatory/immune response during the first week after stroke. J Neuroinflammation 8, 75 (2011).
Ricker, L. J. et al. Chemokine levels in subretinal fluid obtained during scleral buckling surgery after rhegmatogenous retinal detachment. Invest Ophthalmol Vis Sci 51, 4143–50 (2010).
Mitamura, et al. Macrophage migration inhibitory factor levels in the vitreous of patients with proliferative vitreoretinopathy. Am J Ophthalmol. 128(6),763–5, PMID: 10612518 (1999).
Petrukhin, K. New therapeutic targets in atrophic age-related macular degeneration. Expert Opin Ther Targets 11, 625–39 (2007).
Bringmann, A. et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res 25, 397–424 (2006).
Tashimo, A. et al. Aqueous levels of macrophage migration inhibitory factor and monocyte chemotactic protein-1 in patients with diabetic retinopathy. Diabet Med 21, 1292–7 (2004).
Mitamura, Y. et al. Macrophage migration inhibitory factor levels in the vitreous of patients with proliferative diabetic retinopathy. Br J Ophthalmol 84, 636–9 (2000).
Bozza, M. et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med 189, 341–6 (1999).
Tang, W. H., Shilov, I. V. & Seymour, S. L. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J Proteome Res 7, 3661–7 (2008).
Acknowledgements
We thank Amanda Bingham, Steven Rasiah, Stanley Park, EmilyHonzel, Sumaya Hamadmad, Alana Reese, and Akhil Dhamija for excellent technical assistance and Jeffrey Parvin, MD, PhD for critical review of the manuscript. The authors declare no competing financial interests. Supported by NEI Award Number K08EY022672, NCATS Award Number KL2TR001068, and NIH-CCC award P30 CA016058 (Nucleic Acids Shared Resource). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institutions. Additional funds were provided by the Ohio Lions Eye Research Foundation, Fund #313310, and the Patti Blow Fund.
Author information
Authors and Affiliations
Contributions
Designing research studies (C.M.C., S.K.B., M.H.A.), conducting experiments (B.K., R.K., T.H.T., C.M.C.), acquiring data (B.K., R.K., T.H.T., D.M., C.M.C.), analyzing data (B.K., R.K., T.H.T., D.M., J.D., C.M.C., M.H.A., S.K.B.), providing reagents (A.R.S., M.H.A.), material support and critique (J.G.), and creating figures and writing the manuscript (C.M.C., B.K., R.K., T.H.T.).
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, B., Kusibati, R., Heisler-Taylor, T. et al. MIF Inhibitor ISO-1 Protects Photoreceptors and Reduces Gliosis in Experimental Retinal Detachment. Sci Rep 7, 14336 (2017). https://doi.org/10.1038/s41598-017-14298-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14298-9
This article is cited by
-
Targeting macrophage migration inhibitory factor (MIF): a promising therapy for inflammatory ocular diseases
Journal of Ophthalmic Inflammation and Infection (2023)
-
Rare variant analyses across multiethnic cohorts identify novel genes for refractive error
Communications Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
