
Abstract
Extracellular ATP is a central signaling molecule in inflammatory responses. Pannexin1 (Panx1) channels release ATP in a controlled manner and have been implicated in various inflammatory pathologies, but their role in atherogenesis remains elusive. Using atherosclerosis-susceptible mouse models with ubiquitous deletion of Panx1 (Panx1 −/− Apoe −/−) or with Cre recombinase-mediated deletion of Panx1 in endothelial cells and monocytes (Tie2-Cre Tg Panx1 fl/fl Apoe −/−; Panx1 del Apoe −/−), we identified a novel role for Panx1 in the lymphatic vasculature. Atherosclerotic lesion development in response to high-cholesterol diet was enhanced in Panx1 del Apoe −/− mice, pointing to an atheroprotective role for Panx1 in endothelial and/or monocytic cells. Unexpectedly, atherogenesis was not changed in mice with ubiquitous Panx1 deletion, but Panx1 −/− Apoe −/− mice displayed reduced body weight, serum cholesterol, triglycerides and free fatty acids, suggesting altered lipid metabolism in these Panx1-deficient mice. Mechanistically, Panx1 −/− Apoe −/− mice showed impairment of lymphatic vessel function with decreased drainage of interstitial fluids and reduced dietary fat absorption. Thus, the detrimental effect of Panx1 deletion in endothelial and/or monocytic cells during atherogenesis is counterbalanced by an opposite effect resulting from impaired lymphatic function in ubiquitous Panx1-deficient mice. Collectively, our findings unveil a pivotal role of Panx1 in linking lymphatic function to lipid metabolism and atherosclerotic plaque development.
Similar content being viewed by others

Introduction
Atherosclerosis, the leading cause of mortality worldwide, is a chronic immuno-inflammatory disease of large and medium-sized arteries1. The disease involves the formation of plaques in the intima of arteries that are characterized by a dysfunctional endothelium, recruitment of leukocytes, lipid accumulation, smooth muscle cell (SMC) migration and proliferation, cell death and fibrosis. The cellular composition of atherosclerotic lesions determines their stability; stable plaques exhibit a thick fibrous cap with many SMCs while the necrotic core size and the number of macrophages are limited2. The most severe clinical events, such as myocardial infarction, follow the rupture of an atherosclerotic lesion, which exposes the pro-thrombotic material in the plaque to the blood resulting in thrombus formation and arterial occlusion.
Purinergic signaling in atherosclerosis has recently gained attention. In general, it appears that activation of P2 receptors by adenosine triphosphate (ATP) or other nucleotides promotes atherosclerosis whereas ATP hydrolysis by ecto-nucleotidases to adenosine displays an atheroprotective function3. Pannexin1 (Panx1) is important in arterial physiology, mostly through its capacity to form membrane channels that release nucleotides including ATP4. As such, Panx1 contributes to the coordination of SMC contraction in resistance arteries and to the endothelium-dependent regulation of arterial tone in conduit arteries5,6. Panx1 is also involved in ATP-mediated inflammasome activation, in chemotaxis of neutrophils and in activation of T cells7. Finally, Panx1 has been identified as the conduit for ATP from apoptotic cells to release a “find me” signal to recruit phagocytes at the early stages of programmed cell death8. Whether Panx1 plays a role in atherosclerotic lesion development and plaque stability remains, however, to be investigated.
Results and Discussion
Panx1 deletion in endothelial and monocytic cells promotes atherosclerosis
We first investigated Panx1 expression in carotid arteries of atherosclerosis-susceptible Apolipoprotein E-deficient (Apoe −/−) mice fed with high-cholesterol diet (HCD) by immunofluorescent staining. As shown in Fig. 1, non-diseased straight vessel parts showed Panx1 expression exclusively in the endothelium (Fig. 1A and B). Interestingly, an additional strong expression of Panx1 was observed in the membrane of macrophage foam cells in atherosclerotic lesions (Fig. 1C and D). Of note, fluorescent staining was absent from the media of diseased and non-diseased arteries, indicating a lack of Panx1 expression in the SMC layer of large arteries (Fig. 1A–D ). Absence of Panx1 in SMCs of large arteries was further confirmed by Western blot using endothelium-denudated aortas from WT and Panx1 −/− mice (Figure S1A). To investigate a potential contribution of Panx1 in endothelial cells (ECs) and in cells of monocytic origin to atherosclerosis, we generated mice with a conditional deletion of Panx1. Thus, we first interbred Panx1 fl/fl mice with Apoe −/− mice to generate Panx1 fl/fl Apoe −/− mice. Further breeding of these mice with mice harboring the Cre recombinase coding sequence under the control of the 2.1 kb Tie2 promoter9 resulted in Tie2Cre Tg Panx1 fl/fl Apoe −/− mice (hereafter referred to as Panx1 del Apoe −/−). Absence of Panx1 in the endothelium of thoracic-abdominal aortas and in bone marrow-derived macrophages (BMDMs) of Panx1 del Apoe −/− mice was confirmed by real-time polymerase chain reaction (PCR) (Fig. 1E and F). Moreover, absence of Panx1 from ECs of carotid arteries in Panx1 del Apoe −/− mice (Figure S1B) but presence of this protein in the epidermis (Figure S1C) and liver (Figure S1A) further demonstrated the specificity of Panx1 deletion in our Panx1 del Apoe −/− mice. We induced atherosclerosis in control Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice by feeding them from the age of 10 weeks with a HCD for 10 weeks. Mice in both groups gained weight with HCD and no differences were observed between Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice (Fig. 1G). Moreover, total cholesterol and triglyceride (TG) concentration in the serum of mice after HCD did not differ between the two genotypes (Fig. 1H and I ). Thoracic-abdominal aortas were longitudinally opened and stained for lipids with Sudan-IV, which is an indicator for the extent of atherosclerosis. We observed increased lipid staining in the thoracic-abdominal aortas of Panx1 del Apoe −/− mice (Fig. 1J and K ). In contrast, there were no differences in Sudan-IV staining in aortic sinuses from Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice (Fig. 1L and M ). Probably due to a combined action of elevated serum cholesterol and hemodynamic factors, atherosclerotic lesions first appear in aortic sinuses of Apoe −/− mice10. Hence, differences in atherogenesis due to genes important in the early phases of the disease might be plateaued out in the more advanced plaques in the aortic sinus after 10 weeks of HCD. To test this hypothesis, Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice were placed on a HCD for 5 weeks. Lipid staining in thoracic-abdominal aortas confirmed increased atherosclerotic plaque development in Panx1 del Apoe −/− mice after this shorter period of HCD (Figure S2). Altogether, these results point to a protective role for Panx1 expression in the endothelium and/or monocytes/macrophages in atherosclerosis.

Targeted deletion of Panx1 in endothelial and monocytic cells favors atherosclerotic plaque development. Representative images of Panx1 immunofluorescent staining in ECs of a mouse carotid artery (A and B) and in macrophage foam cells within a carotid atherosclerotic lesion (C and D). Nuclei were stained with DAPI (blue) and elastic laminae were visualized with Evans Blue (red). Scale bars represent 50 or 25 μm, respectively. Panx1 expression in ECs (E) and BMDMs (F) of Panx1 fl/fl Apoe −/− (white bars) and Panx1 del Apoe −/− (black bars) mice was assessed by real-time qPCR (n = 6). (G) Weights of Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice before and after 10 weeks of HCD (n = 10). Serum total cholesterol (H) and TG (I) levels in Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice after 10 weeks of HCD (n = 10). Sudan-IV staining (J) and quantification of atherosclerotic lesion extent (K) in the thoracic-abdominal aortas and the aortic roots (L and M) of Panx1 fl/fl Apoe −/− and Panx1 del Apoe −/− mice after 10 weeks of HCD (n = 10). Scale bar represents 200 μm.
Ubiquitous Panx1 deletion affects serum lipid levels
Panx1 is a widely expressed protein and its expression is up-regulated under acute inflammatory conditions of variable origin4. Moreover, general blockade of Panx1 channels with the Food and Drug Administration (FDA)-approved drugs probenecid or mefloquine has been shown effective in mice in the context of acute P. aeruginosa pneumonia11, in acute cerebral ischemia-reperfusion injury12 and in a rodent model of multiple sclerosis known as experimental autoimmune encephalomyelitis13. To investigate whether ubiquitous deletion of Panx1 affects atherosclerosis in mice, we placed 10 weeks-old Panx1 −/− Apoe −/− and Apoe −/− mice on a HCD for 10 weeks and analyzed atherosclerotic lesion development. Surprisingly, lesion size was not affected by genotype both at the level of the aortic sinus and the thoracic-abdominal aorta (Figure S3A–D). It has previously been reported that Panx1 −/− mice generated with the knockout (KO)-first strategy may show some remaining Panx1 mRNA expression14, although this did not alter the protection that these KO-first Panx1 −/− mice displayed against seizures as compared to a mouse Panx1 −/− line of another origin15. Similarly, we found about 30% remaining Panx1 mRNA or protein in samples extracted from Panx1 −/− Apoe −/− bladders or kidneys, organs known to express relatively high levels of Panx1 (Figure S4A–C ). However, Panx1 protein was absent in atherosclerotic lesions of Panx1 −/− Apoe −/− mice whereas a strong Panx1 signal was detected in the endothelium and macrophages in atherosclerotic lesions of control Apoe −/− mice (Figure S4D–G). Thus, the lack of effect on atherosclerosis in Panx1 −/− Apoe −/− mice is likely not caused by remaining Panx1 protein expression.
Reasoning that endothelial and monocytic Panx1 expression appeared protective early in atherosclerotic plaque development, we then examined atherosclerotic lesion development in Panx1 −/− Apoe −/− and Apoe −/− mice after only 5 weeks of HCD. As expected, only few and small plaques were observed in the thoracic-abdominal aortas and aortic sinuses at this early time point (Fig. 2A), however, we could still not observe any difference in atherogenesis between both genotypes (Fig. 2B and C). Although the weight gain after 5 weeks HCD was similar in Panx1 −/− Apoe −/− and Apoe −/− mice, Panx1 −/− Apoe −/− mice were significantly smaller than Apoe −/− controls both before and after HCD (Fig. 2D and E), suggesting metabolic differences between control mice and mice lacking Panx1. In keeping with this idea, magnetic resonance imaging revealed that Panx1 −/− Apoe −/− mice had an increased fat mass and a decreased lean mass compared to their Apoe −/− counterparts (Fig. 2F and G). Micro X-ray computed tomography on the same mice revealed that both the subcutaneous and the visceral adipose tissue were significantly increased in Panx1 −/− Apoe −/− mice (Fig. 2H and I and Movie S1 (Apoe −/−) and S2 (Panx1 −/− Apoe −/−)). Measurements of total serum cholesterol, TG and free fatty acids (FFA) revealed that the levels of these parameters were much lower in Panx1 −/− Apoe −/− mice (Fig. 2J–L) whereas LDL and HDL levels were not different from Apoe −/− controls (Fig. 2M and N). It is well known that lowering serum cholesterol and TG levels protects against atherosclerosis16, thus the lack of effect of ubiquitous deletion of Panx1 on the extent of atherosclerosis is most likely explained by simultaneous opposite effects of Panx1 on lipid metabolism and inflammation. As the extent of atherosclerosis is however not necessarily linked to the vulnerability of lesions for rupture17, it remains crucial to study the effects of Panx1 deletion on plaque stability.

Ubiquitous Panx1 deletion does not affect atherogenesis. Sudan-IV staining (A) and quantification of atherosclerotic lesion extent in the thoracic-abdominal aortas (B) and in the aortic roots (C) of Apoe −/− (white bars) and Panx1 −/− Apoe −/− (grey bars) mice after 5 weeks of HCD (n = 10). Scale bar represents 200 μm. (D) Representative image illustrating the difference in size between Apoe −/− and Panx1 −/− Apoe −/− mice at the age of 10 weeks (n = 10). (E) Weights of Apoe −/− and Panx1 −/− Apoe −/− mice before and after 5 weeks of HCD (n = 10). Fat mass (F) and lean mass (G) of Apoe −/− and Panx1 −/− Apoe −/− mice were measured by MRI (n = 5). Subcutaneous adipose tissue (SAT; H) and visceral adipose tissue (VAT; I) in Apoe −/− and Panx1 −/− Apoe −/− mice was determined by micro X-ray computed tomography (n = 10). Serum total cholesterol (J), TG (K), FFA (L), LDL (M) and HDL (N) levels in Apoe −/− and Panx1 −/− Apoe −/− mice were measured after 5 weeks of HCD (n = 10).
Panx1 deletion affects atherosclerotic lesion phenotype
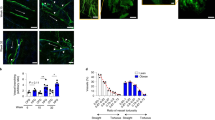
Rupture of atherosclerotic lesions may result from a loss of their mechanical stability, i.e. reduced tensile strength of the collagen cap covering the lesion and lipid core accumulation18. Indeed, histopathological analysis of plaques that have provoked fatal myocardial infarction show a large lipid core and reduced thickness of the fibrous cap, an accumulation of activated macrophages that produce enzymes digesting fibrillar collagen such as matrix metalloproteinases and a depletion of SMCs that synthesize arterial extracellular matrix18. Thus, we assessed whether Panx1 affects the phenotype of atherosclerotic lesions by exploring the plaque components in Panx1 −/− Apoe −/− and Apoe −/− mice after 5 weeks of HCD. As shown in Fig. 3A and E, immunostainings for macrophages revealed that the CD68+ area was significantly larger in atherosclerotic lesions of Panx1 −/− Apoe −/− mice compared to controls, but no differences were found in lipid/necrotic core size (Fig. 3B and F), collagen content (Fig. 3C and G) and SMC content (Fig. 3D and H) in the lesions of the two groups of mice. To investigate the effects of Panx1 deletion on plaque phenotype in more detail, we used shear stress-modifying casts known to induce atherosclerotic plaques with a stable (induced by oscillatory shear stress (OSS)) and vulnerable (induced by low laminar shear stress (LLSS)) phenotype in common carotid arteries of Apoe −/− mice19,20. As expected, the CD68+ area was increased in plaques induced by LLSS as compared to lesions induced by OSS in Apoe −/− mice, whereas an inverted pattern was observed for SMC content of these plaques (Figure S5A–D). Interestingly, such differences in plaque vulnerability markers were not found between LLSS- and OSS-induced atherosclerotic lesions in Panx1 −/− Apoe −/− mice (Figure S5A–D), suggesting that Panx1 deletion abrogated the development of a stable plaque phenotype under OSS. This effect seemed mostly due to reduced presence of SMCs in these lesions (Figure S5C,D), although the CD68+ area tended to be augmented as well (Figure S5A,B). Activated macrophages in atherosclerotic lesions express high levels of CD68 although this marker is also present in dendritic cells (DCs)21 and SMC-derived macrophages22. Therefore, we performed additional immunostainings using F4/80, a more specific marker for mature macrophages22, and observed a larger F4/80+ area in OSS-induced plaques of Panx1 −/− Apoe −/− mice compared with Apoe −/− mice (Figure S5E). Finally, LLSS-induced plaques in Panx1 −/− Apoe −/− mice displayed a larger necrotic core area than the lesions in Apoe −/− mice (Figure S5F) and no significant differences were found in the plaque collagen content between both genotypes (Figure S5G). Altogether, these results suggest that Panx1 contributes to the development of a more stable plaque phenotype through a reduction in the size of the necrotic core and in the macrophage content as well as an increase in the SMC content.

Reduced atherosclerotic plaque stability in mice lacking Panx1. Representative images and quantification of (immuno-)stainings for CD68 (A,E; brown signal), necrotic core (B,F; yellow-marked acellular areas), collagen (C,G; red signal) and α-SMA (D,H; green signal) performed on aortic roots of Apoe −/− (white bars) and Panx1 −/− Apoe −/− (grey bars) mice after 5 weeks of HCD (n = 10). MCP-1-induced chemotaxis (I) and adhesion onto collagen-coated (J) or fibronectin-coated (K) surfaces of Panx1 fl/fl Apoe −/− (white bars) and Panx1 del Apoe −/− (black bars) BMDMs (n = 6). Number of apoptotic cells (L) using TUNEL staining or proliferating cells (M) using PCNA staining performed on aortic roots of Apoe −/− (white bars) and Panx1 −/− Apoe −/− (grey bars) mice after 5 weeks of HCD (n = 10). Scale bars represent 200 μm for A, or 100 μm for B,C, and D.
To evaluate whether Panx1 affects macrophage recruitment, we first performed chemotaxis experiments using BMDMs from Panx1 del Apoe −/− and Panx1 fl/fl Apoe −/− control mice. In agreement with earlier studies on Panx1 −/− macrophages23,24, deletion of Panx1 from BMDMs (Fig. 1F) did not alter the chemotactic response to monocyte chemoattractant protein (MCP)-1 in these cells (Fig. 3I). Then, we tested the ability of these BMDMs to adhere to collagen-coated or fibronectin-coated surfaces. Adhesion to collagen or fibronectin was similar in BMDMs from both genotypes (Fig. 3J and K), thus it is unlikely that increased macrophage recruitment to atherosclerotic lesions would account for the increased F4/80+ content observed in plaques of Panx1-deficient mice (Figure S5E). In addition, these experiments point to a role for endothelial Panx1 rather than Panx1 in monocytic cells in early atherosclerotic plaque development (Fig. 1K). Alternatively, Panx1 may play a role in proliferation, apoptosis or clearance of macrophages from atherosclerotic lesions. Indeed, apoptotic cells release a “find me” signal to recruit phagocytes at the early stages of programmed cell death and Panx1 was identified as the conduit for ATP release from such apoptotic cells8. We therefore counted the number of apoptotic cells and proliferating cells in atherosclerotic plaques of Panx1 −/− Apoe −/− and Apoe −/− mice using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and proliferating cell nuclear antigen (PCNA) assays, respectively. As shown in Fig. 3L and M, we detected only few apoptotic and proliferating cells in atherosclerotic lesions in the aortic roots of Panx1 −/− Apoe −/− and Apoe −/− mice. Moreover, no remarkable changes in the number of apoptotic and proliferating cells were detected in atherosclerotic lesions between both genotypes (Fig. 3L and M). Taken together, these results indicate that increased F4/80+ content, and likely the increased CD68+ content, in atherosclerotic plaques of Panx1 −/− Apoe −/− mice is not due to a raise in chemotactic, adhesive, apoptotic or proliferative capacities of macrophages. This leads to the hypothesis that Panx1 may negatively regulate the exit of CD68+-F4/80+ cells from atherosclerotic lesions resulting in a more vulnerable plaque phenotype. The importance of the lymphatic vessels in removing immune cells and cholesterol from atherosclerotic plaques is increasingly recognized25.
Ubiquitous Panx1 deletion alters lymphatic function
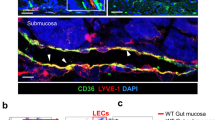
The lymphatic system forms a unidirectional transport pathway from the interstitial space to systemic veins regulating tissue fluid homeostasis, absorption of dietary fat and trafficking of antigen-presenting cells to draining lymph nodes (LNs)26. Macrophages express high levels of CD68 although this marker is also present in dendritic cells (DCs). DCs and to a lesser extent macrophages are triggered by inflammatory stimuli to exit from tissues to draining LNs where they present peptides on major histocompatibility complex molecules to initiate T cell responses27. Similar to ECs in blood vessels, lymphatic ECs (LECs) express Panx1 (Fig. 4A). Panx1 was also detected in a lymphatic endothelial cell line (LyEnd.528; data not shown). Lymphatic ECs are important in the trafficking of DCs to draining LNs by promoting DC entry into lymphatic vessels and by regulating their intra-lymphatic motility. To investigate whether Panx1 affects trafficking of DCs, we used a model of contact hypersensitivity (CH) to enhance migration of skin DCs to draining LNs29. Twenty-four hours after exposure of the skin in the right flank to acetone/dibutyl phthalate, cells were isolated from draining and non-draining LNs. T cells and B cells represented each about 40% (data not shown) and residential DCs less than 1% of the total cell population in draining and non-draining LNs of both Panx1 −/− and control wild-type mice (Fig. 4B). The percentage of migratory DCs arriving in CH draining LNs was also comparable in Panx1 −/− mice and control mice (Fig. 4B), suggesting that Panx1 is not required for regulatory DC – LEC interactions. In contrast, Panx1 channels are known to regulate leukocyte emigration through the venous endothelium during acute inflammation, a process that involves activation of endothelial type-1 tumor necrosis factor (TNF) receptors, recruitment of Src family kinases (SFK) and SFK-dependent phosphorylation of Panx1, ATP release and subsequent activation of P2Y receptors30.

Panx1 deficiency reduces lymphatic function. (A) Panx1 expression in aortic ECs and LECs from WT mice was assessed by real-time qPCR (n = 3–4). (B) Percentage of resident and migratory DCs in CH draining lymph nodes of control (white bars) and Panx1 −/− mice (grey bars) (n = 5). (C) Representative images of lymphatic drainage 1 and 15 min after injection of 5 μl of Evans Blue. Arrow points to lymphatic vessel and arrowhead to lymph node. Lymphatic function was measured by quantification of Evans Blue in the sera of Apoe −/− (white bar) and Panx1 −/− Apoe −/− (grey bar) mice (D; n = 4), and of Panx1 fl/fl Apoe −/− (white bar) and Panx1 del Apoe −/− (black bar) mice (E; n = 6). (F) Representative images of Hematoxylin/Eosin stained cryosections of tails (1 cm from top) from Apoe −/− and Panx1 −/− Apoe −/− mice. Asterisks denote regions rich in microvasculature. Scale bar represents 100 μm. Tail diameter quantification in Apoe −/− (white bars) and Panx1 −/− Apoe −/− (grey bars) mice was measured at (G) 1 cm from the basis and at (H) 4 cm from the tip of the tail (n = 16–19). (I) LYVE-1 immunostaining (red; arrows) in intestinal villi of Apoe −/− and Panx1 −/− Apoe −/− mice. Nuclei were stained with DAPI (blue). Scale bar represents 50 μm. TG (J) and FFA (K) concentration measured before and 3 hours after olive oil gavage of Apoe −/− (white bars) and Panx1 −/− Apoe −/− (grey bars) mice (n = 6).
To investigate whether Panx1 affects lymphatic flow, we compared drainage of interstitial fluids following injection of Evans Blue in the left footpad of Panx1 −/− Apoe −/− and Apoe −/− mice31. The dye progressively spreads throughout the lymphatic system to successive draining LNs (Fig. 4C). We collected sera 15 min after injection and the amount of dye was quantified. As illustrated in Fig. 4D, Evans Blue transport to the systemic circulation of Panx1 −/− Apoe −/− mice was considerably smaller than in control Apoe −/− mice, suggesting that lymphatic flow is impaired in mice with ubiquitous deletion of Panx1. Moreover, lymphatic vessel density was not different in the ears of Panx1 −/− Apoe −/− and Apoe −/− mice (Figure S6). Although mammalian lymphatic vasculature was originally thought to exclusively form by sprouting from embryonic veins, recent studies using Tie2 lineage tracking demonstrate a significant contribution of non-venous derived cells to the lymphatic vasculature32. In accordance, we did not find any difference in drainage of Evans Blue through lymphatics in Panx1 del Apoe −/− as compared to Panx1 fl/fl Apoe −/− control mice (Fig. 4E). As compared to wild-type mice, Apoe −/− mice exhibit tail swelling resulting from impaired interstitial fluids drainage33. To test whether Panx1 deficiency may exacerbate this effect, we measured tail diameters in Apoe −/− and Panx1 −/− Apoe −/− mice. Hematoxylin/Eosin stainings on cryosections of Panx1 −/− Apoe −/− tails suggested increased interstitial fluid content, in particular around the microcirculation, as compared to Apoe −/− tails (Fig. 4F). Indeed, diameters measured at 1 cm from the basis (Figs. 4G) or 4 cm from the tip of the tail (Fig. 4H) were increased in Panx1 −/− Apoe −/− mice, confirming impaired drainage of interstitial fluids in absence of Panx1. Together, these results suggest that Panx1 expression in lymphatic endothelium contributes to the drainage of interstitial fluid.
In view of these results, the reduced total cholesterol, TG and FFA serum values in Panx1 −/− Apoe −/− mice after HCD (Fig. 2J–L) might be caused by defective uptake or degradation of TG-rich remnants, such as chylomicrons. Lymphatic capillaries in the center of the villi in the small intestine, called lacteals, play a primary role in the absorption of dietary fat and fat-soluble vitamins26. As shown in Fig. 4I, lacteals were readily identified by LYVE-1 staining in the jejunum of both Panx1 −/− Apoe −/− and Apoe −/− mice. To study chylomicron absorption, we gavaged Panx1 −/− Apoe −/− and Apoe −/− mice with olive oil and measured accumulation of plasma TG and FFA 3 hours later. Panx1 −/− Apoe −/− mice had a blunted increase in plasma TG and a trend towards reduced FFA levels compared with control animals (Fig. 4J and K). As expected, total cholesterol levels remained unchanged in both genotypes (data not shown). Together, these results indicate that Panx1 in lacteals contributes to the uptake of dietary fat from the gut. Whether additional factors, such as feeding behavior, energy expenditure or lipoprotein breakdown, further contribute to the observed weight and TG differences between Panx1 −/− Apoe −/− and Apoe −/− mice remains to be investigated. Of note, selective ablation of Panx1 from adipocytes only modestly enhances parameters of insulin resistance, including glucose and insulin tolerance, after 12 weeks of diabetogenic diet without affecting weight, adiposity or FFA levels of the mice34.
The role of the lymphatic system in the regulation of immune responses has been extensively studied, but research towards the role of this system in atherosclerotic disease is just at its infancy. Vuorio and colleagues reported increased plasma cholesterol levels and enhanced atherogenesis in independent mouse models with severe lymphatic insufficiency, which suggests a role for lymphatic vessels in the maintenance of serum lipoprotein balance and vascular homeostasis35. However, another recent study points to the lymphatic system as a critical player for removing cholesterol from atherosclerotic lesions by reverse cholesterol transport36. The reason for this discrepancy is not clear and remains to be investigated, one could for example hypothesize that the lymphatic system plays different roles in early and advanced stages of atherosclerosis. Interestingly, both in mice and human, lymphatic vessels are mostly present in the adventitial layer of arterial walls, and arteries with a dense lymphatic network seem naturally protected against atherosclerosis25, thus further supporting lymphatic transport function may improve cholesterol clearance in therapies aimed at reversing atherosclerosis. In the present study, we have identified Panx1 as a novel regulator of lymphatic function, thereby determining lipid metabolism as well as atherosclerotic plaque development and stability. Our knowledge on compounds precisely regulating the open probability of Panx1 channels, including already FDA-approved drugs, is rapidly increasing and holds promises for new therapeutic strategies in the prevention or treatment of atherosclerosis, a disease that is still the number one cause of mortality in industrialized countries.
Methods
Animals
All animal studies were performed after approval by the Swiss Federal Veterinary Office and were in accordance with the established Swiss guidelines and regulations. For atherosclerosis studies, we used 10 weeks-old male Panx1 −/− Apoe −/− , Apoe −/−, Tie2Cre Tg Panx1 fl/fl Apoe −/− (Panx1 del Apoe −/−) and Panx1 fl/fl Apoe −/− mice, on a C57BL/6 background, fed with HCD (1.25% cholesterol, 0% cholate; Research Diets Inc.) for 5 or 10 weeks. Mice were euthanized after general anesthesia induced by an intraperitoneal injection of ketarom (10 mg/kg xylazine mixed with 100 mg/kg ketamine). Blood was taken and mice were perfused with 0.9% NaCl. Aortic roots and pieces of flank skin were embedded in OCT compound (Tissue-Tek; Sakura) and snap-frozen. Carotid arteries and thoracic-abdominal aortas were fixed in 4% paraformaldehyde (PFA) for 2 hours. Carotids were then immerged overnight (ON) in 30% sucrose before being snap-frozen in OCT compound. Thoracic-abdominal aortas were rinsed with phosphate-buffered saline (PBS) and incubated overnight in Sudan-IV solution for lipid deposition analysis as described previously37. The vessels were opened longitudinally and the extent of atherosclerosis was determined by dividing Sudan-IV-positive area by the total surface of the thoracic-abdominal aorta. Atherosclerotic plaque formation was also assessed in aortic roots. For each root an average lesion area was quantified after Sudan-IV staining of 6 serial cross-sections (5 μm thickness, inter-distance 50 μm). Images were captured with Zeiss Axiophot microscope and quantification was performed by computer image analysis using Image J.
In vivo alteration of shear stress
A conical shear stress-modifying cast was used to induce defined changes in shear stress, as previously described19,20. In brief, 10 weeks-old male Panx1 −/− Apoe −/− and Apoe −/− mice were anesthetized with 5% isoflurane inhalation for induction, followed by 2% isoflurane for maintenance of anesthesia. The anterior cervical triangle was accessed by a sagittal anterior neck incision. Both halves of the vascular cast were placed around the right common carotid artery and fixed with a suture. Post-operative analgesia was performed with intraperitoneal injection of Buprenorphinum (0.05 mg/kg) for 3 days. Mice were fed a HCD and killed 10 weeks after cast placement following general anesthesia with ketamine 100 mg/kg and xylazine 10 mg/kg i.p. After perfusion with 0.9% NaCl, casted vessels were excised and the cast was removed. Samples were embedded in OCT compound and snap frozen. Five μm-thick serial cryosections were obtained from the 3 flow regions determined by the conical, progressively constrictive shape of the cast, i.e. upstream – LLSS, inside – high laminar shear stress (HLSS), downstream – OSS.
Histological analysis
Immunofluorescence: Cryosections (5 μm) of atherosclerotic lesions in carotids of 10 weeks-old male mice fed with a HCD were fixed 15 min in 4% PFA, permeabilized 15 min with 0.3% Triton X-100 and blocked 30 min in PBS containing 2% bovine serum albumin (BSA). Chicken anti-mouse Panx1414–425 antibodies38 (1:500) were then applied ON on the sections before detection with goat anti-chicken DyLight488 antibody (1:500; Jackson Laboratories) for 2 hours at RT and subsequent Evans Blue counterstaining. A similar protocol was applied to cryosections (5 μm) of flank skin. Moreover, 4% PFA-fixed jejunum cryosections (5 μm) were permeabilized 60 min with 0.2% Triton X-100 in PBS and blocked 30 min in PBS-BSA (2%). LYVE-1 antibody (1:50; Abcam) was incubated 1 hour. Secondary goat anti-rabbit Alexa 568 antibody (1:2000; Jackson Laboratories) was then incubated 1 hour at RT. All sections were counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI), mounted with Vectashield medium (Vector Laboratories) and analysed using a Zeiss Axioskop 2 microscope or a Zeiss LSM700 confocal microscope.
Immunostainings on cryosections of aortic roots or casted carotids. For macrophages: ON incubation at 4 °C of 100% acetone-fixed sections with CD68 antibody (1:100; Bio-Rad) followed by a 1 hour incubation at RT with corresponding horse radish peroxidase-conjugated antibody (1:500; Jackson Laboratories). Alternatively, macrophage staining was performed after a 2 hour incubation at RT of 4% PFA-fixed sections with F4/80 antibody (1/100; Caltag) followed by 1 hour incubation at RT with an Alexa 488 antibody (1:2000; Jackson Laboratories). For SMCs: α-SMA antibody39 (1:50) was incubated for 1 hour at RT on 4% PFA-fixed sections and detected by incubating with a goat anti-mouse IgG2a Alexa 488 antibody (1:100; Jackson Laboratories) for 30 min at RT. For proliferation: PCNA antibody (1:20; ThermoScientific) was incubated ON at 4 °C on sections fixed successively at RT with 4% PFA (15 min) and methanol (5 min), and detected with corresponding biotinylated secondary antibody (1:500; Jackson Laboratories). Collagen was detected by picrosirius red staining. Necrotic areas were quantified after Masson’s trichrome staining by measuring the surface of acellular and anuclear areas in the atherosclerotic lesions. Apoptosis was detected on 4% PFA-fixed sections using the DeadEnd Colorimetric TUNEL system (Promega). After counterstaining of nuclei with hemalun, images were captured using a Zeiss Axiophot microscope and quantifications were performed by computer image analysis using Image J. The percentage of positive cells was determined by dividing positively stained surface by total lesion surface area. Analysis of PCNA and TUNEL staining was performed by counting positive nuclei over lesion area.
Magnetic resonance imaging and micro X-ray computed tomography
Whole-body composition was measured using an EchoMRI-700 quantitative nuclear magnetic resonance analyzer. Conscious mice were placed into instrument columns and total body fat, lean mass, body fluids and total body water were measured in a non-invasive manner by 1H-magnetic resonance spectroscopy. Values were expressed as percentage of total bodyweight. Mice were sacrificed by CO2 inhalation and transferred into the micro-CT Quantum GX (Perkin Elmer). Animals were scanned with a 60 mm field of view at 90 kV and 80 mA over 360 degrees using the whole body scan protocol: 3 scans of 8 seconds automatically stitched together to provide a continuous stack of whole body images. White adipose tissue analysis was performed by segmentation based on the low density (Hounsfield range of values between −50 and −200 HU) of the adipose tissue with Analyze 12.0 (AnalyzeDirect).
Oral lipid tolerance test and serum lipid analysis
The uptake of long-chain fatty acids was quantified as previously described40. In brief, Panx1 −/− Apoe −/− , Apoe −/−, Panx1 del Apoe −/− and Panx1 f/fl Apoe −/− mice were subjected to an ON fast and subsequently received olive oil (Sigma-Aldrich; 10 μl/g bodyweight) by oral gavage. Blood was obtained by submandibular puncture before (t = 0) and 3 hours after gavage. Total cholesterol, TG and FFA concentrations were measured in mouse sera after HCD or oral lipid tolerance test using a Cobas c111 analyser (Roche Diagnostics).
Lymphatic drainage, contact hypersensitivity and vessel density
Drainage of interstitial fluids was quantified using Evans Blue injections in the footpad of Panx1 −/− Apoe −/− , Apoe −/−, Panx1 del Apoe −/− and Panx1 f/fl Apoe −/− mice as described previously31. In brief, mice were anesthetized as described above. Then, the mice were injected with 5 μl 5% Evans Blue (dissolved in PBS) in the left footpad using a micro syringe. After 15 min, blood was collected by puncturing the left ventricle and centrifuged 15 min at 5000 rpm (4 °C). Formamide (Sigma) was added to each serum sample (500 μL formamide/200 μL serum) and the mix was incubated ON at 55 °C. Thereafter, presence of Evans Blue in the serum was quantified using a SpectraMax Paradigm Multi-Mode Microplate reader (excitation: 620 nm; emission 680 nm; Molecular Devices). In addition, tail diameters of Panx1 −/− Apoe −/− and Apoe −/− were determined using a digital caliper. Mice were briefly anesthetized with 5% isoflurane and the tail diameter was measured at 1 cm from the basis and at 4 cm from the tip. Tail diameters were normalized to mouse weight to correct for size differences between both genotypes.
CH assay was performed as described previously29. In brief, a mix of 1:1 acetone/dibutyl phthalate was applied on the skin in the right flank of the mice for 24 hours. Skin-draining LNs were carefully grinded and digested at 37 °C in RPMI containing 1 mg/ml Collagenase IV (Worthington Biochemical Corporation), 40 μg/ml DNase I (Roche) and 2% FBS for 40 min, gently mixing the samples every 20 min. The reaction was stopped by adding a 10% fetal calf serum solution containing 5 mM EDTA (FACS buffer). Samples were then filtered using a 70 μm cell strainer, centrifuged 5 min at 1300 rpm and resuspended in FACS buffer for flow cytometry staining.
To determine lymphatic vessel density, ears were fixed ON at 4 °C and subsequently washed with PBS. Then the inner and outer parts of the dermis of the ear were separated using forceps. After separation, in order to quantify the lymphatic area, whole mount samples were prepared and stained as described before41. In brief, samples were blocked ON at 4 °C in blocking buffer (0.5% BSA, 5% donkey serum, 0.3% Triton X-100, 0.1% Sodium Azide in PBS). Subsequently, samples were incubated ON at 4 °C with anti-LYVE-1 antibody (1:100, Abcam) in blocking buffer. Thereafter, samples were washed using washing buffer (0.3% Triton X-100 in PBS) and incubated ON with secondary antibodies (Alexa488, 1:500) in blocking buffer. Next, samples were washed using washing buffer and re-fixed for 48 hours at 4 °C using 4% PFA in PBS. Finally, samples were rinsed using PBS and mounted on a microscopy slide within the wall of three Secure-Seal spacers (Molecular Probes) in order to maintain the three-dimensional structure. Thereafter, LYVE-1 positive areas were visualized using a LSM700 confocal microscope (Zeiss). Three separate recordings were obtained. Z-stacks covering the layer of tissue in which lymphatics are present were obtained during these recordings in order to capture all the lymphatics. After imaging the Z-stacks were converted to a single plain using Fiji using the Z Project plugin. The LYVE-1 positive area was thereafter quantified manually by an observer unaware of the genotype of the mice and was subsequently normalized to the ear area.
Aortic and lymphatic EC isolation
Thoracic-abdominal aortas from Panx1 del Apoe −/− or Panx1 f/fl Apoe −/− mice were immerged in 100–150 μl PBS on a silicon support, longitudinally opened and pinned. The endothelial side was then delicately scraped with a scalpel blade. The samples were centrifuged 5 min at 1350 rpm and immediately processed for RNA extraction.
LN lymphatic ECs were obtained from skin LN isolated from 8–10 mice and digested in RPMI containing 1 mg/ml Collagenase IV (Worthington Biochemical Corporation), 40 µg/ml DNase I (Roche), and 2% fetal bovine serum gently mixing the samples every 10 min for a total incubation of 30 min. Undigested cells were further digested with 1 mg/ml Collagenase D, and 40 µg/ml DNase I (Roche) for not more than 20 min. The enzymatic reaction was stopped by addition of FACS buffer. Single cell suspensions were negatively selected using CD45 microbeads and magnetic bead column separation (Miltenyi Biotec).
Antibodies, flow cytometry, and cell sorting
Anti-gp38 (clone 8.1.1), anti-CD31 (clone 390), anti-CD11c (clone N418) and anti-IAb (AF6.120.1) were from BioLegend. Anti-CD16/32 FcyRIII (clone 2.4G2) and anti-CD45 (clone 30F11) were obtained from BD. For purification of lymphatic endothelial cells, enriched CD45-negative cells were FACS-sorted using a MoFlowAstrios (Beckman Coulter). Cells were acquired on a Gallios (Beckman Coulter) and analyzed using FlowJo software (Tree Star).
BMDM isolation
BMDMs were isolated from Panx1 del Apoe −/− or Panx1 f/fl Apoe −/− mice by flushing femurs and tibiae of posteriors members. Cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Gibco) containing Glutamax, 10% fetal bovine serum (Sigma), 100 U/l penicillin, 5 g/l streptomycin (Invitrogen) and 50 μM β-mercaptoethanol. Macrophage colony-stimulating factor (M-CSF) necessary for cell differentiation was provided by supplementing the medium with 30% L929-conditioned medium.
Real-time PCR
Total RNA from mouse BMDMs, aortic ECs or lymph node ECs was obtained using the NucleoSpin kit (Macherey-Naegel). Reverse transcription was performed using the Quantitect Reverse Transcription kit (Qiagen) and real-time RT-PCR was performed with the ABI Prism StepOnePlus Sequence Detection System (Applied Biosystems) using the TaqMan Fast Universal master mix (Applied Biosystem). Mouse Panx1 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers and probes were purchased from Applied Biosystems. Gene expression was normalized to GAPDH expression.
Western blot
Proteins were extracted from endothelium denudated aortas, kidneys or livers from C57BL/6 wild-type, Panx1 −/− 38,42, Panx1 −/− Apoe −/− 14,15,43 , Apoe −/− 43, Panx1 del Apoe −/− or Panx1 f/fl Apoe −/− mice and Western blots were performed as described previously44. The anti-Panx1 antibody (1:400, Alomone labs) was used ON. Loading controls were performed by probing for GAPDH (1:30000, Millipore).
Transmigration assay
BMDMs were starved for 14 hours in IMDM without serum and without M-CSF. Cells were washed with PBS and detached by incubating for 5–10 min and flushing with cold PBS containing 0.5 mM EDTA. After washing with IMDM, cells were resuspended in chemotaxis medium (IMDM containing 1% BSA; Sigma-Aldrich) and migration toward 50 ng/ml MCP-1 (CCL2; Peprotec) was determined using Transwells. Briefly, cells were allowed to migrate 90 min (at 37 °C in a humidified 5% CO2 atmosphere) through two adjacent compartment separated by a 5 μm polycarbonate filter (Sigma). Cells were added in the upper compartment while the lower compartment was filled with medium (basal migration) or medium containing the chemoattractant MCP-1 (chemotaxis). The percentage of migrated cells was assessed using a Cyan cell analyser (Beckman Coulter).
Adhesion assay
To assess adhesion of mouse BMDMs, 96-well plates coated with 2.5 μg type 1 collagen (from rat tail, Sigma) per well and blocked with PBS containing 0.5% BSA were used. Assays were performed by plating 5 × 104 CFDA-SE-labeled (Molecular Probes) BMDMs per well. After 30 min incubation, non-adherent cells were removed by washing with PBS and fluorescence emitted by adhering cells was measured (excitation: 492 nm; emission 517 nm) using a SpectraMax Paradigm Multi-Mode Microplate Detection reader (Molecular Devices).
Statistical analysis
Statistical analysis was performed with Graphpad Prism 6 (v6.0c) and results were expressed as mean ± SEM. Two-group comparisons were performed using Student’s t-test. Multiple group comparisons were performed using one-way ANOVA with Bonferroni’s post-test. Differences with a P < 0.05 were considered as significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
References
Libby, P., Lichtman, A. H. & Hansson, G. K. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity 38, 1092–1104, https://doi.org/10.1016/j.immuni.2013.06.009 (2013).
Yla-Herttuala, S. et al. Stabilization of atherosclerotic plaques: an update. European heart journal 34, 3251–3258, https://doi.org/10.1093/eurheartj/eht301 (2013).
Ferrari, D., Vitiello, L. & Idzko, M. et al. Purinergic signaling in atherosclerosis. Trends in molecular medicine 21, 184–192, https://doi.org/10.1016/j.molmed.2014.12.008 (2015).
Penuela, S., Harland, L. & Simek, J. et al. Pannexin channels and their links to human disease. The Biochemical journal 461, 371–381, https://doi.org/10.1042/BJ20140447 (2014).
Lohman, A. W., Billaud, M. & Isakson, B. E. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovascular research 95, 269–280, https://doi.org/10.1093/cvr/cvs187 (2012).
Meens, M. J., Kwak, B. R. & Duffy, H. S. Role of connexins and pannexins in cardiovascular physiology. Cellular and molecular life sciences: CMLS 72, 2779–2792, https://doi.org/10.1007/s00018-015-1959-2 (2015).
Adamson, S. E. & Leitinger, N. The role of pannexin1 in the induction and resolution of inflammation. FEBS letters 588, 1416–1422, https://doi.org/10.1016/j.febslet.2014.03.009 (2014).
Chekeni, F. B. et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467, 863–867, https://doi.org/10.1038/nature09413 (2010).
Schlaeger, T. M. et al. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 94, 3058–3063 (1997).
Veillard, N. R., Steffens, S. & Burger, F. et al. Differential expression patterns of proinflammatory and antiinflammatory mediators during atherogenesis in mice. Arteriosclerosis, thrombosis, and vascular biology 24, 2339–2344, https://doi.org/10.1161/01.ATV.0000146532.98235.e6 (2004).
Wonnenberg, B. et al. Probenecid reduces infection and inflammation in acute Pseudomonas aeruginosa pneumonia. International journal of medical microbiology: IJMM 304, 725–729, https://doi.org/10.1016/j.ijmm.2014.05.002 (2014).
Wei, R. et al. Probenecid protects against cerebral ischemia/reperfusion injury by inhibiting lysosomal and inflammatory damage in rats. Neuroscience 301, 168–177, https://doi.org/10.1016/j.neuroscience.2015.05.070 (2015).
Lutz, S. E. et al. Contribution of pannexin1 to experimental autoimmune encephalomyelitis. PloS one 8, e66657, https://doi.org/10.1371/journal.pone.0066657 (2013).
Hanstein, R. et al. Promises and pitfalls of a Pannexin1 transgenic mouse line. Frontiers in pharmacology 4, 61, https://doi.org/10.3389/fphar.2013.00061 (2013).
Santiago, M. F. et al. Targeting pannexin1 improves seizure outcome. PloS one 6, e25178, https://doi.org/10.1371/journal.pone.0025178 (2011).
Brinton, E. A. Management of Hypertriglyceridemia for Prevention of Atherosclerotic Cardiovascular Disease. Endocrinology and metabolism clinics of North America 45, 185–204, https://doi.org/10.1016/j.ecl.2015.09.012 (2016).
Falk, E., Shah, P. K. & Fuster, V. Coronary plaque disruption. Circulation 92, 657–671 (1995).
Kwak, B. R. et al. Biomechanical factors in atherosclerosis: mechanisms and clinical implications. European heart journal 35(3013–3020), 3020a–3020d, https://doi.org/10.1093/eurheartj/ehu353 (2014).
Cheng, C. et al. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113, 2744–2753, https://doi.org/10.1161/CIRCULATIONAHA.105.590018 (2006).
Pfenniger, A. et al. Shear stress-induced atherosclerotic plaque composition in ApoE(−/−) mice is modulated by connexin37. Atherosclerosis 243, 1–10, https://doi.org/10.1016/j.atherosclerosis.2015.08.029 (2015).
Jongstra-Bilen, J. et al. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. The Journal of experimental medicine 203, 2073–2083, https://doi.org/10.1084/jem.20060245 (2006).
Shankman, L. S. et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature medicine 21, 628–637, https://doi.org/10.1038/nm.3866 (2015).
Qu, Y. et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. Journal of immunology 186, 6553–6561, https://doi.org/10.4049/jimmunol.1100478 (2011).
Wang, H. et al. Pannexin-1 influences peritoneal cavity cell population but is not involved in NLRP3 inflammasome activation. Protein & cell 4, 259–265, https://doi.org/10.1007/s13238-013-2114-1 (2013).
Kutkut, I., Meens, M. J. & McKee, T. A. et al. Lymphatic vessels: an emerging actor in atherosclerotic plaque development. European journal of clinical investigation 45, 100–108, https://doi.org/10.1111/eci.12372 (2015).
Aspelund, A., Robciuc, M. R. & Karaman, S. et al. Lymphatic System in Cardiovascular Medicine. Circulation research 118, 515–530, https://doi.org/10.1161/CIRCRESAHA.115.306544 (2016).
Randolph, G. J. Mechanisms that regulate macrophage burden in atherosclerosis. Circulation research 114, 1757–1771, https://doi.org/10.1161/CIRCRESAHA.114.301174 (2014).
Frontera, V. et al. Cutting edge: JAM-C controls homeostatic chemokine secretion in lymph node fibroblastic reticular cells expressing thrombomodulin. Journal of immunology 187, 603–607, https://doi.org/10.4049/jimmunol.1003441 (2011).
Dubrot, J. et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4(+) T cell tolerance. The Journal of experimental medicine 211, 1153–1166, https://doi.org/10.1084/jem.20132000 (2014).
Lohman, A. W. et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nature communications 6, 7965, https://doi.org/10.1038/ncomms8965 (2015).
Tammela, T. et al. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nature medicine 13, 1458–1466, https://doi.org/10.1038/nm1689 (2007).
Martinez-Corral, I. et al. Nonvenous origin of dermal lymphatic vasculature. Circulation research 116, 1649–1654, https://doi.org/10.1161/CIRCRESAHA.116.306170 (2015).
Lim, H. Y. et al. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. Am J Pathol 175, 1328–1337, https://doi.org/10.2353/ajpath.2009.080963 (2009).
Adamson, S. E. et al. Pannexin 1 is required for full activation of insulin-stimulated glucose uptake in adipocytes. Molecular metabolism 4, 610–618, https://doi.org/10.1016/j.molmet.2015.06.009 (2015).
Vuorio, T. et al. Lymphatic vessel insufficiency in hypercholesterolemic mice alters lipoprotein levels and promotes atherogenesis. Arteriosclerosis, thrombosis, and vascular biology 34, 1162–1170, https://doi.org/10.1161/ATVBAHA.114.302528 (2014).
Martel, C. et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. The Journal of clinical investigation 123, 1571–1579, https://doi.org/10.1172/JCI63685 (2013).
Wong, C. W. et al. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nature medicine 12, 950–954, https://doi.org/10.1038/nm1441 (2006).
Molica, F. et al. Functional role of a polymorphism in the Pannexin1 gene in collagen-induced platelet aggregation. Thrombosis and haemostasis 114, 325–336, https://doi.org/10.1160/TH14-11-0981 (2015).
Skalli, O. et al. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. The Journal of cell biology 103, 2787–2796 (1986).
Bernier-Latmani, J. et al. DLL4 promotes continuous adult intestinal lacteal regeneration and dietary fat transport. The Journal of clinical investigation 125, 4572–4586, https://doi.org/10.1172/JCI82045 (2015).
Bernier-Latmani, J. & Petrova, T. V. High-resolution 3D analysis of mouse small-intestinal stroma. Nat Protoc 11, 1617–1629, https://doi.org/10.1038/nprot.2016.092 (2016).
Bargiotas, P. et al. Pannexins in ischemia-induced neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America 108, 20772–20777, https://doi.org/10.1073/pnas.1018262108 (2011).
Zhang, S. H., Reddick, R. L. & Piedrahita, J. A. et al. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258, 468–471 (1992).
Morel, S. et al. Endothelial Cx40 limits myocardial ischaemia/reperfusion injury in mice. Cardiovascular research 102, 329–337, https://doi.org/10.1093/cvr/cvu063 (2014).
Acknowledgements
This work was supported by grants from the Swiss National Science Foundation (310030_143343 and 310030_162579 to B.R. Kwak). We thank Viviane Rochemont and Bernard Foglia for technical assistance. We are grateful to Dr. Urban Deutsch and Dr. Marie-Luce Bochaton-Piallat for providing us with mice harboring the Cre recombinase coding sequence under the control of the 2.1 kb Tie2 promoter and α-SMA antibody, respectively.
Author information
Authors and Affiliations
Contributions
F.M. and B.R.K. designed research; F.M., M.J.M., J.D., A.E., C.L.R., S.M., G.P., L.V. and V.B. performed research; O.R., M.C., S.H., E.S. contributed new reagents/analytic tools; F.M., M.J.M., J.D., L.V., M.C., S.H. and B.R.K. analyzed data; and F.M. and B.R.K. wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Molica, F., Meens, M.J., Dubrot, J. et al. Pannexin1 links lymphatic function to lipid metabolism and atherosclerosis. Sci Rep 7, 13706 (2017). https://doi.org/10.1038/s41598-017-14130-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14130-4
This article is cited by
-
Pannexin 1 as a driver of inflammation and ischemia–reperfusion injury
Purinergic Signalling (2021)
-
Pannexin 1 regulates adipose stromal cell differentiation and fat accumulation
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
