
Abstract
Salinity effects on microbial communities in saline soils is still unclear, and little is known about subsurface soil microbial communities especially in saline or hypersaline ecosystems. Here we presented the survey of the prokaryotic community in saline soils along a salinity gradient (17.3–148.3 dS/m) in surface (0–10 cm) and subsurface (15–30 cm) saline soils of Qarhan Salt Lake, China. Moreover, we compared them with three paired nonsaline normal soils. Using the high-throughput sequencing technology and several statistical methods, we observed no significant community difference between surface soils and subsurface soils. For environmental factors, we found that TOC was the primary driver of the prokaryotic community distribution in surface saline soils, so was pH in subsurface saline soils. Salinity had more effects on the prokaryotic community in subsurface saline soils than in surface saline soils and played a less important role in saline soils than in saline waters or saline sediments. Our research provided references for the prokaryotic community distribution along a salinity gradient in both surface and subsurface saline soils of arid playa areas.
Similar content being viewed by others


Introduction
Recently, people began to realize the importance of halophilic microorganisms in the industrial biotechnology because of their advantages, such as less fresh water consumption, low energy, continuous production, and low fixed capital investment1. And many halophilic hydrolases secreted by halophiles, such as amylases, lipases, and cellulases, are promising for industrial applications under diverse conditions due to their polyextremophilicity2,3. Hypersaline ecosystems, which represent a wide variety of ecosystem types, such as salt lakes, salt flats, playas, solar salterns, and ancient salt deposits, are globally distributed4,5,6. Halophilic and halotolerant microorganisms thrive broadly in these ecosystems; therefore, it is essential and valuable to make in-depth investigations on the microbial communities of these ecosystems.
A previous comprehensive analysis based on sequencing data from many researches of diverse physical environments had proved that salinity, rather than extremes of temperature, pH, or other environmental factors, mainly determined microbial communities7. However, the comprehensive analysis contained only saline waters or sediments in aquatic environments and no saline soils were included. Moreover, while the salinity has been characterized as one of primary factors of the microbial community distribution in aquatic environments, the extent of salinity effects on microbial communities in saline soils were still unclear8. And, few studies have focused on the correlation between salinity and the microbial community distribution in saline soils8,9,10. Therefore, more studies need to be done to clarify the extent of salinity effects on microbial communities in saline soils.
Present studies of microbial communities in soils focused exclusively on surface (0–10 cm) soils, and several studies11,12,13,14,15 have focused on microbial communities in both surface and subsurface nonsaline soil layers. Though microorganisms in surface soils were more dense and active collectively12, those in subsurface saline soils can also play an important role on nutrition cycling, ecosystem function, and soil respiration16,17. And, for microbial communities in saline or hypersaline soils, most previous studies9,10,18,19 just focused on the microbial community in surface soil layers (0–10 cm). No previous reports of microbial communities in subsurface saline soil layers (15–30 cm) seems to exist.
The Qaidam Basin is located in the northeast of Qinghai–Tibetan Plateau, China. There are twenty seven salt lakes in the basin; the playas and salt lakes take up about a quarter of the whole basin area20. Among all 27 lakes, Qarhan Salt Lake is the largest playa, where 70% of the total thickness of the upper salt-bearing deposits is composed of the halite layers20. It is the largest industrial base for potassium fertilizer in China, and it is also rich in Mg, Li, and many other salt resources. Its unique significances in scientific research, resource exploitation, and environmental protection are increasingly being recognized21. Many previous studies have focused on the prokaryotic communities in waters22,23,24,25 or sediments26,27 of the lakes in Qinghai-Tibetan Plateau, and they all observed that salinity was the primary driver of the microbial community distribution. However, the prokaryotic community distribution in saline soils and its correlations with environmental factors, especially the salinity, are still uncharacterized in this area.
The purposes of this research are: 1) to clarify the prokaryotic community distribution along several environmental gradients, especially the salinity gradient, in both surface and subsurface saline soils; 2) to characterize the prokaryotic community composition in saline soils and compare with that in nonsaline normal soils; 3) to ascertain the microbial resources for further exploitations.
Results
Physical and chemical properties of soils
The 23 saline soils from Qarhan Salt Lake showed a wide salinity gradient from 17.3 dS/m to 148.3 dS/m, a pH gradient from 7.36 to 8.92, a TOC (total organic carbon) gradient from 4.19% to 21.48%, and a water content gradient from 1.1% to 32.3% (Table 1).The data indicated a wide environmental range, especially the salinity range. In eight paired surface-subsurface saline soils, surface soils had higher salinity (except SS1 and SD1, SS8 and SD8), lower water contents (except SS4 and SD4), and lower pH values (except SS7 and SD7, SS8 and SD8) than subsurface soils (Table 1). The six nonsaline normal soils encompassed few variations of environmental factors compared with saline soils. Spearman’s rank correlation was conducted between EC (electrical conductivity) and other environmental factors (Supplementary Table S2). EC had significant (P < 0.05) positive correlations with concentrations of Na, Ca, Mg and significant negative correlations with other factors.
Statistics for 16S rDNA sequencing data
After quality filtering and chimera checking, 1,693,891 effective tags were obtained with an average length of 254 bp from all 29 soil samples, of which 1,335,650 tags belonged to 23 saline soil samples from Qarhan Salt Lake and 358,241 tags to 6 normal soil samples from Tianjin, China (Supplementary Table S1). We also summarized Q20, Q30, GC%, and the number of effective tags (Supplementary Table S1). The number of effective tags of each sample ranged from 47,504 to 67,321 among saline soils and from 51,102 to 64,426 among normal soils. The effective tags of each sample were subject to further analysis.
For OTU picking, we obtained 56,541 OTUs with a mean of 2,458 ± 904 (s.d.) OTUs from 23 saline soil samples and 33,877 OTUs with a mean of 5,646 ± 607 OTUs from six normal soil samples (Supplementary Table S1). The mean of OTUs in normal soils was more than twice as many as that in saline soils, which indicated low microbial diversity in saline soils. The sum of OTUs in saline soil samples (surface saline soils: 12075; subsurface saline soils: 11689) was higher than that in normal soils (surface normal soils: 10804; subsurface normal soils: 9683), and the sum of OTUs in surface soils was higher than in subsurface soils of both saline soils and normal soils (Fig. 1). Moreover, shared OTUs took up more than 70% between surface normal soils and subsurface normal soils (7612/10804), surface saline soils and subsurface saline soils (8591/12075). And shared OTUs took up about 50% between surface normal soils and surface saline soils (6295/12075), subsurface normal soils and subsurface saline soils (5329/11689) (Fig. 1). The number of OTUs shared by all four groups was 3550.

Venn diagrams of OTUs included in four groups of soils. We grouped all soil samples into NS, ND, SS, SD, representing shallow normal soil (surface normal soil), deep normal soil (subsurface normal soil), shallow saline soil (surface saline soil), deep saline soil (subsurface saline soil), respectively.
Prokaryotic community composition and alpha diversity analysis
Archaea and Bacteria accounted for an average percentage of 52.8% (12.3–86.7%) and 47.2% (13.3–87.7%) of all 1,335,650 effective tags respectively in saline soils, with a huge difference compared with 6.8% (2.9–12.1%), 93.2% (87.9–97.2%) in normal soils. Detailed relative abundance of each phylum/class were summarized in Supplementary Table S3. Saline soils showed low microbial complexity compared with normal soils, and there were high percentages of Euryarchaeota in most saline soil samples (Fig. 2).

Relative abundance of 19 most predominant phyla (including four classes in Proteobacteria) in all samples sorted from left to right with increasing salinity.
For Bacteria at phylum level, Proteobacteria (35.1% ± 23.8% in surface saline soils, 23.5% ± 22.9% in subsurface saline soils), Bacteroidetes (4.3% ± 2.9%, 5.6% ± 3.9%), and Gemmatimonadetes (2.3% ± 1.8%, 7.3% ± 3.1%) were the three most abundant phyla in saline soils (Supplementary Fig. S1). And within Proteobacteria, Gammaproteobacteria (28.5% ± 25.7% in surface saline soils, 18.3% ± 2.5% in subsurface saline soils) were the most abundant class, which accounted for more than half (68.7% ± 31.0%, 59.4% ± 29.2%) of Proteobacteria, followed by Alphaproteobacteria (4.0% ± 2.9%, 4.1% ± 3.1%), Deltaproteobacteria (1.5% ± 1.3%, 0.9% ± 0.5%), and Betaproteobacteria (0.6% ± 0.5%, 0.3% ± 0.3%). In normal soils, Proteobacteria (39.3% ± 0.8% in surface normal soils, 37.2% ± 0.7% in subsurface normal soils), Acidobacteria (17.8% ± 0.8%, 20.1% ± 2.7%), Actinobacteria (11.9% ± 2.5%, 11.6% ± 0.8%) were the three most abundant phyla. And within Proteobacteria in normal soils, Alphaproteobacteria (14.3% ± 1.3%, 12.7% ± 0.6%) were the most abundant phylum, followed by Gammaproteobacteria (13.5% ± 2.2%, 12.0% ± 1.3%), Deltaproteobacteria (6.3% ± 1.2%, 7.0% ± 0.8%), and Betaproteobacteria (4.7% ± 1.4%, 5.2% ± 0.8%) (Supplementary Fig. S1). Standard deviations of phyla/classes in saline soils were much higher than in normal soils, indicating wide variations of phyla/classes in saline soils.
For Archaea, almost all effective tags were assigned to Halobacteria of Euryarchaeota in saline soils. But in normal soils, about 25% of Archaea tags were assigned to Thaumarchaeota except Halobacteria of Euryarchaeota (Fig. 2).
ANOSIM results showed significant prokaryotic community differences between surface saline soils and surface normal soil (R = 0.37, P < 0.05), subsurface saline soils and subsurface normal soils (R = 0.36, P < 0.05). To further identify the phyla/classes which contributed primarily to the community variations, we conducted SIMPER analysis between surface normal soils and surface saline soils, subsurface normal soils and subsurface saline soils (Supplementary Table S4). The overall community dissimilarity (the sum of contribution) between surface normal soils and surface saline soils was 68.9%, and the value was 72.1% between subsurface normal soils and subsurface saline soils at the phylum/class level. Euryarchaeota made the greatest contribution to the community variations in both surface and subsurface soils between saline soils and normal soils. Gammaproteobacteria and Deltaproteobacteria played a more important role in differentiating the surface soil community, but Alphaproteobacteria contributed more in subsurface soil community (Supplementary Table S4).
As it is shown in Fig. 3, microorganisms in surface soils are richer than those in subsurface soils for both saline soils and normal soils. Surface soils had more OTUs than their corresponding subsurface soils except SS1 and SD1, SS2 and SD2 (Supplementary Table S1). Though we observed more Gammaproteobacteria in surface saline soils than in subsurface saline soils, and Bacteroidetes and Gemmatimmonadetes in surface saline soils were less than those in subsurface saline soils by summarizing prokaryotic community compositions in all paired saline soils (Supplementary Fig. S1). ANOSIM results showed no significant (P = 0.8 for normal soils and P = 0.26 for paired saline soils) prokaryotic community difference between surface soils and subsurface soils, and Student’s t-test results of each phylum/class were also unsignificant (P > 0.05) between the two groups in both saline soils and normal soils.

Alpha diversity boxplot among four groups same as Fig. 1 but without seven unpaired samples to remove the influence of environmental factors. The values of Observed OTUs were achieved after normalization (47504 tags per sample). The three lines of the box from bottom to top represents the first quartile (Q1), the second quartile (the median), and the third quartile (Q3), respectively; the two ends of the whiskers represent the minimum and maximum of the data within a group, respectively. Outliers were the values beyond the range (Q1 − 2 IQR, Q3 + 2 IQR). IQR (interquartile range) = Q3 − Q1.
Rarefaction curves of most samples (except UD3, SS8, and SD8) did not reach an obvious asymptote, indicating that there were still many undetermined species (Supplementary Fig. S2). Chao1 estimators suggested that our sequencing efforts contained about 54.8% ± 7.8% of the estimated diversity for saline soils and about 64.8% ± 1.3% for normal soils (Supplementary Table S1), which indicated larger proportions of unknown species in saline soils than in normal soils. The curves of six normal soils were obviously above curves of 23 saline soils, demonstrating low microbial diversities in saline soils (Supplementary Fig. S2). Shannon diversity index (5.9 ± 1.5 in saline soils and 10.2 ± 0.3 in normal soils) also verified that (Supplementary Table S1).
Correlations between environmental factors and the prokaryotic community distribution
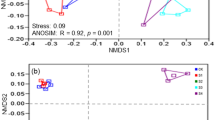
Two main axes explain 77.2% of the soil community variations, and soil samples can be clustered together by both the sizes and colors of points, indicating salinity, pH, and TOC can all affect the prokaryotic community distribution (Fig. 4). CCA biplots showed that pH, TOC, and EC were main factors that affected the prokaryotic community in saline soils: TOC was the strongest factor in surface saline soils (Fig. 5a), so was pH in subsurface saline soils (Fig. 5b).

Principal coordinate analyses (PCoA) based on the weighted Unifrace distance of all soil samples. The weighted Unifrace distance was calculated with the normalized OTU table (47504 sequences per sample). Soil samples were distinguished according to their salinity and pH (a), salinity and TOC (b). Four groups represented different saline level: Normal (six normal soils), Saline1 (EC 17–55 dS/m), Saline2 (EC 55–95 dS/m), Saline3 (> 95 dS/m).

Canonical correlation analysis (CCA) based on the normalized OTU table (47504 sequences per sample) of surface saline soils (a) and subsurface saline soils (b).
To further verify the main drivers of the prokaryotic community distribution, we conducted Mantel test between the prokaryotic community and each environment factor of saline soils in Qarhan Salt Lake (Table 2). Only EC, pH, and TOC had significant (P < 0.05) correlations with the prokaryotic community distribution in all saline soils. TOC and pH affected the prokaryotic communities in both surface saline soils and subsurface saline soils significantly, but EC only had significant effects on prokaryotic communities in subsurface saline soils (Table 2). Moreover, the r value of TOC (r = 0.54) were higher than that of pH (r = 0.49), suggesting TOC was the main driver of the prokaryotic community distribution in surface saline soils, so was pH in subsurface saline soils. In summary, both CCA and Mantel test proved that TOC was the main driver of the prokaryotic community distribution in surface saline soils, and pH was the main driver of that in subsurface saline soils of Qarhan Salt Lake, China.
Salinity effects on the prokaryotic community
Most samples can be clustered together according to their saline level (Fig. 4 and Supplementary Fig. S4). ANOSIM results further verified the significant prokaryotic community difference across different saline levels (R = 0.69, P < 0.01). Halobacteriaceae (belonging to Euryarchaeota of Archaea) and Moraxellaceae (belonging to Gammaproteobacteria of Bacteria) have deeper red grids than other families, suggesting they are two most abundant families in saline soils (Supplementary Fig. S4). Half of top 40 most abundant families belongs to Proteobacteria, indicating the widespread distribution of Proteobacteria in soils. Moreover, normal soils apparently had more complex communities and higher evenness than saline soils (Supplementary Fig. S4).
Though TOC and pH were the main drivers, salinity can still strongly affect the prokaryotic community in saline soils. EC have significant correlations with the prokaryotic community distribution in subsurface saline soils but not with that in surface saline soils (Table 2), suggesting that salinity has more effects on subsurface saline soils. The prokaryotic community of different saline level had different compositions (Supplementary Fig. S3): medium saline soils (group S2 and D2) had higher percentages of Euryarchaeota and lower percentages of Proteobacteria, Bacteroidetes than low saline soils (group S1 and D1) or high saline soils (group S3 and D3) in both surface and subsurface soils. Within Proteobacteria, lower percentages of Gammaproteobacteria were in medium saline soils than in low saline soils or high saline soils for subsurface soils, and the same trend was observed between medium saline soils and high saline soils but not low saline soils for surface soils (Supplementary Fig. S3). That was probably because group S1 of surface soils contained only one soil sample (sample US2) with relatively high salinity (52.1 dS/m), suggesting that it was probably unrepresentative. We also observed significant correlations between salinity and percentages of several taxa across all saline soil samples (Supplementary Fig. S5). Salinity significantly (P < 0.05) affects the percentages of Euryarchaeota and Proteobacteria, though R 2 values were relatively low: percentage of Euryarchaeota peaks at about 85 dS/m; Proteobacteria reaches the lowest at also about 85 dS/m (Supplementary Fig. S5a). Gammaproteobacteria showed the same trend with Proteobacteria (Supplementary Fig. S5b).
Discussion
The extremely arid desert climate with high evaporation-precipitation ratio and strong sun exposure is observed in the area of Qarhan Salt Lake. These characters made the unique prokaryotic community in soils of Qarhan Salt Lake. Several reports showed that Bacteria were numerically dominant relative to Archaea in saline soils of the areas they explored10,28,29, and there were also reports showing that Archaea were dominant30,31. In the present study, we found that Archaea were dominant in saline soil samples with EC about 58 dS/m to 93 dS/m; Bacteria were dominant in soil samples with lower or higher salinity except three outliers (SD6, SD3, SS3) in Qarhan Salt Lake (Fig. 2). Almost all archaeal tags were assigned to Halobacteria of Euryarchaeota, which agreed with several previous microbial community studies of hypersaline waters22,32, saline soils or sediments10,26,33,34. Also, saline soil samples in which Archaea were dominant has low alkalinity (pH: 7.36–8.02) and low TOC contents (4.19–6.22%) except SS8 (Fig. 2 and Table 1). That agreed with the fact that most halophilic archaeal representatives (mainly Halobacteria) isolated from hypersaline lakes are aerobic, neutrophilic, and some are alkaliphilic35,36,37. And, specific membrane structures and catabolic pathways allows Archaea to out-compete Bacteria when faced with energy limitation, such as low nutrients in extreme conditions38.
For Bacteria in saline soils of the present study, Proteobacteria were the most abundant phylum, and within Proteobacteria, Gammaproteobacteria were the most abundant class, followed by Alphaproteobacteria, Deltaproteobacteria, and Betaproteobacteria, which was consistent with a previous meta-analysis of sequences in saline soils39. Except Proteobacteria, Bacteroidetes and Gemmatimonadetes were the other two most abundant phyla (Supplementary Fig. S1), agreed with previous reports10,28.
For Archaea in nonsaline soils from the eastern Tibetan Plateau, the Halobacteria were dominant only in dry soils; the most abundant phylum was Thaumarchaeota 40. Moreover, in nonsaline normal soils from Tianjin, China in the present study, Archaea only accounts for a small part, agreed with a previous report41. The most abundant archaeal phylum was Halobacteria, followed by Thaumarchaeota. Proteobacteria, Acidobacteria, and Actinobacteria were the three most abundant phyla of Bacteria (Supplementary Fig. S1), consistent with the bacterial community composition in Tibetan permafrost soils42. For Proteobacteria in normal soils, Alphaproteobacteria were the most abundant phylum, followed by Gammaproteobacteria, Deltaproteobacteria, and Betaproteobacteria.
Though with relatively high percentage of shared species (about 50%) (Fig. 1), significant community difference was observed, and Euryarchaeota contributed the most to the community variations in both surface and subsurface soils between saline soils and nonsaline soils. Because almost all archaeal tags were assigned to Halobacteria of Euryarchaeota, Archaea and Halobacteria had similar contributions to Euryarchaeota. Nonsaline normal soils had more complex community and higher evenness than saline soils (Fig. 2 and Supplementary Fig. S4). Nonsaline normal soils were also more diverse than saline soils (Supplementary Table S1 and Fig. 3). Keshri J. et al. has also found that the community in nonsaline soils was significantly different from that in saline soils and more diverse than saline soils18. Those also agreed with and supported the general belief that relatively low microbial diversity existed in “extreme” environments43. Within Proteobacteria, most saline samples (16/23) were dominant by Gammaproteobacteria, and Arit S. de León-Lorenzana et al.44 has observed a sharp relative abundance decrease of Gammaproteobacteria by flooding a saline soil, suggesting Gammaproteobacteria was more adaptive to saline environments than other proteobacterial classes. Moreover, it confirmed the widespread distribution of Halobacteria of Archaea, Proteobacteria of Bacteria in soils.
We observed trends of higher salinity, lower water contents, and lower pH in surface soils than in subsurface soils according to physical and chemical properties of eight paired surface-subsurface saline soils (Table 1), which was probably caused by strong sun exposure and high evaporation in Qarhan Salt Lake. In addition, we needed to point out that trends of salinity and pH between surface soils and subsurface soils were unsignificant (t-test, P > 0.05), and the water contents in subsurface soil were significantly (P = 0.048) higher than those in surface soils. Though with variations of environmental factors, microorganisms in surface soils were richer than those in their corresponding subsurface soils (Fig. 3 and Table 1). According to Hollister E. B. et al., the water effect on microbial community was primarily due to its influence on soil oxygen concentrations10. And another reports suggested the decrease of biomass along the increasing depth was initially caused by redox state (depth of a few centimeters), and further caused by other variables, such as nutrients and stress (the anoxic zone)45. Therefore, we hypothesized that low oxygen concentrations, which might be affected by high water contents, caused low microbial richness in subsurface soils (15–30 cm). That also agreed with previous reports12,13,15 microorganisms in surface soils were richer than those in their corresponding subsurface soils.
Different prokaryotic diversities were observed, but we observed no significant community difference between surface soils and subsurface soils in both saline soils and normal soils according to the results of ANOSIM and Student’s t-test, contrary to a previous report11. Therefore, we hypothesized that the depth of 15–30 cm probably was not deep enough to generate significant community difference from the surface soil (0–10 cm).
In the present study, though saline soils had much fewer diversities than normal soils (Supplementary Table S1, Fig. 3), however we didn’t observe decreasing microbial richness (observed OTUs) with increasing salinity (r = 0.015, P = 0.947) in saline soils, just as previous studies have observed22,25,46. Moreover, salinity had more effects on subsurface saline soils than surface saline soils according to the result of Mantel test (Table 2). That was probably because compared with subsurface soils, surface soils were more easily subject to other environmental factors47, such as strong sun exposure, precipitation or oxygen. The prokaryotic community composition in soils with different saline level differed from each other (Supplementary Fig. S3): higher percentage of Euryarchaeota and lower percentage of Proteobacteria, Gammaproteobacteria existed in medium saline soils (group S2 and D2) than in low saline soils (group S1 and D1) or high saline soils (group S3 and D3) for both surface and subsurface soils. Nonlinear fitting curves also verified the same trend: Proteobacteria reaches lowest at about EC 85 dS/m contrary to the percentage of Euryarchaeota (Supplementary Fig. S5a); Gammaproteobacteria showed the same trend with Proteobacteria (Supplementary Fig. S5b), which was possibly caused by the large proportion of Gammaproteobacteria in Proteobacteria. That agreed with a previous study on soils from the former lake Texcoco, which showed more Gammaproteobacteria clones were in low (0.65 dS/m) and high (158 dS/m) saline soils than in medium (56 dS/m) saline soils48.
Soil samples were well clustered by pH, TOC, and salinity (Fig. 4); pH and TOC all had significant correlations with the prokaryotic community distribution in both surface saline soils and subsurface saline soils (Table 2). Therefore, we can infer that the prokaryotic community in both surface saline soils and subsurface saline soils were affected by similar environmental factors, agreed with a previous research on the bacterial community distribution in nonsaline soils of the western Tibetan Plateau11.
Most previous studies focused on the microbial community in saline waters and sediments, and salinity has been characterized as one of the primary factors of microbial community distribution in aquatic environments7,8,22. Many previous studies have observed that salinity had the strongest effect on the prokaryotic community distribution in waters22,23,25, sediments26,27,49. In the two previous reports on saline soils9,10, the former showed salinity had the strongest effect on the bacterial community structure, but the latter showed the microbial community distribution was correlated with other factors better, including organic carbon contents, water contents, pH, and phosphorus contents. Also, another report of archaeal communities in saline soils showed that archaeal community structures considering phylogenetic information were correlated well with pH34. In the present study, by summarizing the results of PCoA, CCA, and Mantel test, we found that TOC was the main driver of prokaryotic community distribution in surface saline soils; pH was main driver in subsurface saline soils of Qarhan Salt Lake. Therefore, we confirmed that salinity played a more important role on the microbial community distribution in saline waters or saline sediments than in saline soils; the microbial community in saline soils was more sensitive to other environmental factors, such as TOC, pH, or water contents.
Conclusion
We collected 23 saline soils from playa of Qarhan Salt Lake to clarify the prokaryotic community distribution along several environmental gradients in both surface (0–5 cm) and subsurface (15–30 cm) saline soils. We showed high-resolution differences between saline soils and nonsaline soils in both prokaryotic diversities and communities. And we found that TOC was the main driver of the prokaryotic community distribution in surface saline soils, so was pH in subsurface saline soils; salinity had more effects on subsurface saline soils than surface saline soils. Our finding can provide references for sampling strategy and prokaryotic community compositions in saline soils of arid areas. Also, the distribution patterns of the prokaryotic community and taxa, especially some halophilic taxa (mainly Halobacteria of Archaea), along the environmental gradients in saline soils provided references for further new gene exploitations, such as some halophilic or halotolerant enzyme genes. At last, further in-depth investigations on saline soil microbial communities in large scale or with greater depth were expected.
Methods
Site climate description and the sample collection
Qarhan Salt Lake has a mean annual temperature of 5.33 °C, a mean annual precipitation of 24 mm, and a mean annual evaporation of 3,564 mm, which indicates an extremely arid desert climate. The climate data were achieved from Qarhan meteorologic station.
In summer 2016, 15 playa sites of Qarhan Salt Lake were selected with a minimum distance of 100 m as sampling sites. The sampling strategy was similar to a previous reports28: a) maximum reachable sites in the interior playa even without roads; b) different locations with various salinity levels. Most locations of the playa were bare zones dotted with scarce vegetations, including Lycium ruthenicum and Phragmites australis. For each site, surface soils (0–10 cm) and subsurface soils (15–30 cm) were collected using a metal auger with a 4-cm diameter. For each soil sample, four samples collected in the vertices of 1-meter side square were mixed into a representative sample. If present, stones, solid salt crust, and roots were removed before sampling. Three paired nonsaline normal soils were collected with the same sampling strategy in Water Park, Tianjin, China. The vegetation of the normal soil locations included Cynodon dactylon, Ophiopogon japonicas, Setaria leucopila, etc, and we selected the locations with few grass to avoid the influence of grassroots. Locations and altitudes were recorded using a GPS. Collected soils were placed into 50 ml sterile plastic centrifuge tubes and stored at 4 °C during transportation. For each sample, one portion was air dried, 2 mm sieved, then physically and chemically analyzed; the other portion was immediately stored at −80 °C for further DNA extraction.
Physical and chemical analysis
The pH was measured in a 1:2.5 (w/w) aqueous solution with a pH meter (PHS-3C; INESA, Shanghai, China). The electrical conductivity (EC) was measured in a 1:5 aqueous solution with a conductivity meter (FE-30; Mettler Toledo, Switzerland). The water content was detected by oven drying fresh soil to a constant weight at 105 °C. Potassium dichromate heating oxidation method50 was used to determine the total organic carbon content (TOC). The concentrations of Mg, K, Na, Ca were measured with an atomic absorption spectroscopy (TAS-990; PERSEE, Beijing, China).
DNA extraction and sequencing
PowerSoil DNA Extraction Kit (Mo Bio Laboratories, CA, USA) was used to extract DNA from soils (about 0.25 g) following the manufacturer’s instructions. But we failed to extract enough DNA from seven samples among all 15 paired saline soil samples due to the low biomass in saline soils. And for the seven failures, we have tested three different kinds of soil weight (0.15 g, 0.25 g, and 0.5 g), all resulting in no success. Primers 515 F and 806R41, fused with a barcode and Illumina adaptor, were used to amplify V4 regions of prokaryotic 16S rRNA genes. The PCR products were sequenced using Illumina HiSeq. 2500 platform51 (250 bp pair-end reads) by Novogene (Beijing, China).
Analysis of Illumina-sequencing data
Barcodes and primers of generated Illumina-sequencing reads were removed, and the reads were merged using FLASH software52, generating raw tags. Raw tags were qualified using QIIME (version 1.9.1)53 software package: a) raw tags, which had three or more consecutive low-quality bases (the threshold value was 19), will be truncated at the first low-quality base; b) some tags, whose consecutive high-quality base length is shorter than 75% of tags length, were further filtered. Then, chimeras were identified and filtered through blasting against the ChimeraSlayer reference database54 by UCHIME Algorithm55. The effective tags were finally obtained. The obtained effective tags were used for further OTU (operational taxonomic unit) picking and diversity analysis with QIIME. OTUs were picked using the preferred open-reference56 OTU picking strategy in QIIME, which includes both close-reference and de novo OTU picking strategy; USEARCH57 was used to cluster sequences with ≥ 97% similarity together forming an OTU. A representative sequence was picked out from each OTU, and the taxonomy was assigned to each of the representative sequences with Ribosomal Database Project (RDP) classifier58 against Greengenes59 at a confidence threshold of 0.8 in QIIME. Representative sequences were aligned against the Greengenes core set with PyNAST60, then a phylogenetic tree was generated with FastTree61, which was used to calculated the weighted UniFrac distance62.
Before diversity analysis, the sequences of each sample were normalized to 47504 tags based on the number of tags in the sample with the fewest tags. Several alpha diversity indices, which includes observed OTUs, Chao1 richness estimator, and Shannon diversity index, were calculated with QIIME. Principal Coordinate Analysis (PCoA) based on weighted Unifrac distance matrix was conducted with the function “cmdscale” of package Stats in R (version 3.3.1). Heat map was drawn using the package Pheatmap in R. Canonical correlation analysis (CCA), analysis of similarity (ANOSIM), similarity percentage (SIMPER) analysis63, and Mantel test were conducted with the package Vegan64 in R. We chose CCA according to detrended correspondence analysis (DCA) and removed environmental factors with variance inflation factors of more than ten from CCA to reduce effects of the collinearity. Mantel test was performed to analyze the correlation between the prokaryotic community composition (Bray-Curtis distance based on OTU-level communities) and each environmental factor (Euclidean distance). ANOSIM was used to evaluate significant differences of the prokaryotic community composition among different groups.
Data availability
The original sequences of this research were deposited at the NCBI Sequence Read Archive under the accession number SRP108198. The accession number of each sample was also listed (Supplementary Table S1).
References
Yin, J., Chen, J. C., Wu, Q. & Chen, G. Q. Halophiles, coming stars for industrial biotechnology. Biotechnol. Adv. 33, 1433–1442 (2015).
Delgado-Garcia, M., Valdivia-Urdiales, B., Aguilar-Gonzalez, C. N., Contreras-Esquivel, J. C. & Rodriguez-Herrera, R. Halophilic hydrolases as a new tool for the biotechnological industries. J. Sci. Food Agric. 92, 2575–2580 (2012).
Mathabatha, E. S. Diversity and industrial potential of hydrolase-producing halophilic/halotolerant eubacteria. African Journal of Biotechnology 9, 1555–1560 (2010).
Ventosa, A., Mellado, E., Sanchez-Porro, C. & Marquez, M. C. Halophilic and Halotolerant Micro-Organisms from Soils. (Springer Berlin Heidelberg, 2008).
Oren, A. Halophilic Microorganisms and their Environments. (Kluwer Academic Publishe, 2002).
McGenity, T. J., Gemmell, R. T., Grant, W. D. & Stan-Lotter, H. Origins of halophilic microorganisms in ancient salt deposits. Environ. Microbiol. 2, 243–250 (2000).
Lozupone, C. A. & Knight, R. Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104, 11436–11440 (2007).
Rath, K. M. & Rousk, J. Salt effects on the soil microbial decomposer community and their role in organic carbon cycling: A review. Soil Biol. Biochem. 81, 108–123 (2015).
Canfora, L. et al. Salinity and bacterial diversity: to what extent does the concentration of salt affect the bacterial community in a saline soil? PLoS One 9, e106662 (2014).
Hollister, E. B. et al. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4, 829–838 (2010).
Chu, H. et al. Bacterial community dissimilarity between the surface and subsurface soils equals horizontal differences over several kilometers in the western Tibetan Plateau. Environ. Microbiol. 18, 1523–1533 (2016).
Fierer, N., Schimel, J. P. & Holden, P. A. Variations in microbial community composition through two soil depth profiles. Soil Biology & Biochemistry 35, 167–176 (2003).
LaMontagne, M. G., Schimel, J. P. & Holden, P. A. Comparison of subsurface and surface soil bacterial communities in california grassland as assessed by terminal restriction fragment length polymorphisms of PCR-amplified 16S rRNA genes. Microb Ecol 46, 216–227 (2003).
Li, C. H., Yan, K., Tang, L. S., Jia, Z. J. & Li, Y. Change in deep soil microbial communities due to long-term fertilization. Soil biology & biochemistry 75, 264–272 (2014).
Serkebaeva, Y. M., Kim, Y., Liesack, W. & Dedysh, S. N. Pyrosequencing-based assessment of the bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions. PLoS One 8, e63994 (2013).
Wardle, D. A. The influence of biotic interactions on soil biodiversity. Ecol Lett. 9, 870 (2006).
Wagg, C., Bender, S. F., Widmer, F. & Mg, V. D. H. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc Natl Acad Sci USA 111, 5266 (2014).
Keshri, J., Mody, K. & Jha, B. Bacterial Community Structure in a Semi-Arid Haloalkaline Soil Using Culture Independent Method. Geomicrobiol J. 30, 517–529 (2013).
Lu, H. et al. Changes in soil microbial community structure and enzyme activity with amendment of biochar-manure compost and pyroligneous solution in a saline soil from Central China. European Journal of Soil Biology 70, 67–76 (2015).
Kezao, C. & Bowler, J. M. Late pleistocene evolution of salt lakes in the Qaidam basin, Qinghai province, China. Palaeogeogr., Palaeoclimatol., Palaeoecol. 54, 87–104 (1986).
Fan, Q., Ma, H., Ma, Z., Wei, H. & Han, F. An assessment and comparison of 230Th and AMS 14C ages for lacustrine sediments from Qarhan Salt Lake area in arid western China. Environmental Earth Sciences 71, 1227–1237 (2013).
Zhong, Z. P. et al. Prokaryotic Community Structure Driven by Salinity and Ionic Concentrations in Plateau Lakes of the Tibetan Plateau. Appl. Environ. Microbiol. 82, 1846–1858 (2016).
Wu, Q. L., Zwart, G., Schauer, M., Kamst-van Agterveld, M. P. & Hahn, M. W. Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl Environ Microbiol. 72, 5478–5485 (2006).
Liu, Y. Q. et al. Salinity Impact on Bacterial Community Composition in Five High-Altitude Lakes from the Tibetan Plateau, Western China. Geomicrobiol J. 30, 462–469 (2013).
Wang, J. et al. Do patterns of bacterial diversity along salinity gradients differ from those observed for macroorganisms? PLoS One 6, e27597 (2011).
Liu, Y. et al. Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiol Ecol. 92 (2016).
Yang, J., Ma, L., Jiang, H., Wu, G. & Dong, H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci Rep 6, 25078 (2016).
Pandit, A. S. et al. A snapshot of microbial communities from the Kutch: one of the largest salt deserts in the World. Extremophiles 19, 973–987 (2015).
Keshri, J., Mishra, A. & Jha, B. Microbial population index and community structure in saline-alkaline soil using gene targeted metagenomics. Microbiol Res. 168, 165–173 (2013).
Yousuf, B., Sanadhya, P., Keshri, J. & Jha, B. Comparative molecular analysis of chemolithoautotrophic bacterial diversity and community structure from coastal saline soils, Gujarat, India. BMC Microbiol. 12 (2012).
Canfora, L. et al. Spatial microbial community structure and biodiversity analysis in “extreme” hypersaline soils of a semiarid Mediterranean area. Applied Soil Ecology 93, 120–129 (2015).
Jiang, H. et al. Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl Environ Microbiol. 72, 3832–3845 (2006).
Zhang, L. et al. Bacterial and archaeal communities in the deep-sea sediments of inactive hydrothermal vents in the Southwest India Ridge. Sci Rep 6, 25982 (2016).
Navarro-Noya, Y. E. et al. Archaeal Communities in a Heterogeneous Hypersaline-Alkaline Soil. Archaea 2015, 646820 (2015).
Oren, A. Halophilic archaea on Earth and in space: growth and survival under extreme conditions. Phil. Trans. R. Soc. A. 372 (2014).
Andrei, A.-Ş., Banciu, H. L. & Oren, A. Living with salt: metabolic and phylogenetic diversity of archaea inhabiting saline ecosystems. FEMS Microbiol Lett. 330, 1–9 (2012).
Oren, A. Ecology of Halophiles (ed. Horikoshi K) 334–361. (Springer Japan, 2011).
Valentine, D. L. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat. Rev. Microbiol. 5, 316–323 (2007).
Ma, B. & Gong, J. A meta-analysis of the publicly available bacterial and archaeal sequence diversity in saline soils. World J Microbiol Biotechnol 29, 2325–2334 (2013).
Shi, Y. et al. The biogeography of soil archaeal communities on the eastern Tibetan Plateau. Sci Rep 6, 38893 (2016).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 108, 4516-4522 (2011).
Zhang, X. et al. The soil carbon/nitrogen ratio and moisture affect microbial community structures in alkaline permafrost-affected soils with different vegetation types on the Tibetan plateau. Res Microbiol. 165, 128–139 (2014).
Smith, J. J., Tow, L. A., Stafford, W., Cary, C. & Cowan, D. A. Bacterial diversity in three different Antarctic Cold Desert mineral soils. Microb Ecol. 51, 413–421 (2006).
de Leon-Lorenzana, A. S. et al. Reducing Salinity by Flooding an Extremely Alkaline and Saline Soil Changes the Bacterial Community but Its Effect on the Archaeal Community Is Limited. Frontiers in microbiology 8, 466 (2017).
Dong, H. et al. Microbial diversity in sediments of saline Qinghai Lake, China: linking geochemical controls to microbial ecology. Microb. Ecol. 51, 65–82 (2006).
Casamayor, E. O. et al. Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 4, 338–348 (2002).
Brady, N. C. & Weil, R. R. The nature and properties of soils. 960–961 (Pearson Education, Inc, 2002).
Valenzuela-Encinas, C. et al. Changes in the bacterial populations of the highly alkaline saline soil of the former lake Texcoco (Mexico) following flooding. Extremophiles 13, 609–621 (2009).
Lv, X. et al. Bacterial community structure and function shift along a successional series of tidal flats in the Yellow River Delta. Sci Rep 6, 36550 (2016).
Schumacher, B. A. Methods for the Determination of Total OrganicCarbon (TOC) In Soils and Sediments. Ecological Risk Assessment Support Center (2002).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Magoč, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Haas, B. J. et al. Chimeric 16 S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21, 494–504 (2011).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Rideout, J. R. et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2, e545 (2014).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267 (2007).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072 (2006).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. Plos One 5, e9490 (2012).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 71, 8228–8235 (2005).
Clarke, K. R. Non-parametric multivariate analysis of change in community structure. Austral Ecol. 18, 117–143 (1993).
Dixon, P. VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927–930 (2003).
Acknowledgements
The study was partly supported by National Natural Science Foundation of China (NO. 31470967) and National High Technology Research and Development Program of China (NO. 2015AA020701).
Author information
Authors and Affiliations
Contributions
H.H., X.Z., and Y.D. designed the research. X.Z. and Y.D. collected the samples. J.L., X.Z., and Y.X. measured the physical and chemical parameters of soils. K.X., S.Z., W.Z., and Y.D. extracted DNA from soils. K.X. analyzed the data and wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, K., Deng, Y., Zhang, S. et al. Prokaryotic Community Distribution along an Ecological Gradient of Salinity in Surface and Subsurface Saline Soils. Sci Rep 7, 13332 (2017). https://doi.org/10.1038/s41598-017-13608-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13608-5
This article is cited by
-
Response of the metabolic activity and taxonomic composition of bacterial communities to mosaically varying soil salinity and alkalinity
Scientific Reports (2024)
-
Microbial diversity in polyextreme salt flats and their potential applications
Environmental Science and Pollution Research (2024)
-
Thrive or survive: prokaryotic life in hypersaline soils
Environmental Microbiome (2023)
-
Chemical Links Between Redox Conditions and Estimated Community Proteomes from 16S rRNA and Reference Protein Sequences
Microbial Ecology (2023)
-
Sediment microbial community structure, enzymatic activities and functional gene abundance in the coastal hypersaline habitats
Archives of Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
