
Abstract
Studies on the halotolerance of bacteria are attractive to the fermentation industry. However, a lack of sufficient genomic information has precluded an investigation of the halotolerance of Halomonas beimenensis. Here, we describe the molecular mechanisms of saline adaptation in H. beimenensis based on high-throughput omics and Tn5 transposon mutagenesis. The H. beimenensis genome is 4.05 Mbp and contains 3,807 genes, which were sequenced using short and long reads obtained via deep sequencing. Sixteen Tn5 mutants with a loss of halotolerance were identified. Orthologs of the mutated genes, such as nqrA, trkA, atpC, nadA, and gdhB, have significant biological functions in sodium efflux, potassium uptake, hydrogen ion transport for energy conversion, and compatible solute synthesis, which are known to control halotolerance. Other genes, such as spoT, prkA, mtnN, rsbV, lon, smpB, rfbC, rfbP, tatB, acrR1, and lacA, function in cellular signaling, quorum sensing, transcription/translation, and cell motility also shown critical functions for promoting a halotolerance. In addition, KCl application increased halotolerance and potassium-dependent cell motility in a high-salinity environment. Our results demonstrated that a combination of omics and mutagenesis could be used to facilitate the mechanistic exploitation of saline adaptation in H. beimenensis, which can be applied for biotechnological purposes.
Similar content being viewed by others


Introduction
Halotolerant and halophilic bacteria have the ability to adapt to high salt concentrations. This trait can be exploited by the fermentation industry to conserve energy and water1. Due to their specific mechanisms of saline adaptation, halotolerant bacteria can tolerate high-salinity stress, whereas halophilic bacteria require a certain saline concentration to survive2. Several molecular approaches to saline adaptation have been discovered in bacteria, including the accumulation of compatible solutes, potassium uptake, and sodium effluxion3,4. Compatible solutes (e.g. betaine, ectoine, glutamate, trehalose, and proline) are low molecular weight compounds that are synthesized by bacteria to maintain cellular turgidity and electrolyte concentrations4. The role of compatible solutes overlaps with that of potassium uptake in the maintenance of osmotic equilibrium. When Escherichia coli (a non-halotolerant bacterium) is exposed to saline stress, potassium concentration first transiently increases and then rapidly decreases after the de novo synthesis of compatible solutes, indicating that the osmoprotective role of these compatible solutes supplants that of potassium3. By contrast, the potassium and glutamate concentrations of Halomonas elongata (a halotolerant bacterium) continue to increase even after ectoine is generated, suggesting that the halotolerant of E. coli and H. elongata differ3.
In Halomonas spp., potassium uptake and organic compatible solute synthesis controls respiratory chain and osmotic regulation under high-salinity conditions5. Trk, Ktr, and Kdp are three major systems of potassium uptake in bacteria3. The Trk system requires ATP and drives potassium uptake through the transmembrane electrochemical proton gradient3. TrkA of E. coli, which is involved in the Trk system, contains a nicotinamide adenine dinucleotide (NAD+)-binding domain that can bind NAD+ or NADH6. Moreover, a trkA mutant of H. elongata showed defects in potassium transportation3. The Ktr system is a Na+-dependent potassium uptake system, whereas the Kdp system is classified as a P-type K+-translocating ATPase that hydrolyzes ATP to generate the driving force for K+ uptake3,7. Therefore, sodium ion efflux and hydrogen and potassium ion uptake help maintain bacterial osmotic balance in high-salinity environments.
In general, bacteria maintain high intracellular potassium and low sodium concentrations for effective cellular enzyme activity, and some marine bacteria may maintain a sodium gradient for energy metabolism8,9. To prevent entry of sodium into the cell, the sodium-translocating NADH:quinone oxidoreductase (Na+-NQR) uses exergonic energy to pump hydrogen or sodium ions from the cytosol to the periplasm through symporters9. In Vibrio cholera, NqrA is a subunit of Na+-NQR that acts as a sodium pump and can bind quinone9,10. The operation of the respiratory chain generates an electrochemical sodium gradient to pump out sodium to maintain osmotic balance while NADH is oxidized by Na+-NQR9. Under high-salinity conditions, bacteria maintain a low internal sodium concentration via Na+/H+ antiporters9.
Cell motility and energy production also play critical roles in halotolerance11,12. Flagellum-related genes have been found to be down-regulated in highly saline environments, suggesting that decreased cell motility allows more energy to be available for osmoprotection11,12. Another explanation is that cell motility is correlated with sodium-driven motor activity11. Therefore, decreased motor activity can reduce sodium re-entry to maintain intracellular ion homeostasis11.
Currently, complete genomic sequence are available for approximately 6,892 of the 87,815 bacterial genomes in NCBI (2016), including 7 Halomonas spp. Microarray and next-generation sequencing (NGS) approaches have been used to examine transcriptome profiles related to saline adaptation in Bacillus sp., Desulfovibrio vulgaris, Staphylococcus sp., Mesorhizobium alhagi, and Jeotgalibacillus malaysiensis, and the alkaline response of Halomonas sp.1,12,13,14,15 Moreover, mutant lines have been generated by transposon mutagenesis to study halotolerance in several bacteria, including Caulobacter crescentus, Sinorhizobium fredii, Azospirillum brasilense, and Sinorhizobium meliloti 16,17,18,19. Moreover, Kindzierski et al. (2017) integrated metabolome and proteome analyses to study the osmoregulation of H. elongata and demonstrated that glycolysis- and tricarboxylic acid cycle-related enzymes are involved in compatible solute biosynthesis for osmoregulation20.
In this study, we isolated the NTU-111 strain of Halomonas beimenensis (hereafter referred to as H. beimenensis) and sequenced its entire genome using NGS. H. beimenensis gene expression profiles under various salt conditions were also analyzed by transcriptome analysis. Moreover, we identified 16 Tn5 insertion mutants that had lost the ability to tolerate 15% NaCl. Based on these studies, we propose a conceptual model for H. beimenensis adaptation to a high-salinity environment.
Results
H. beimenensis growth conditions and phylogenetic position
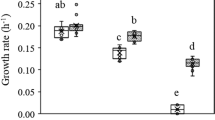
H. beimenensis requires at least 5% NaCl to survive, and no bacterial growth was observed in the presence of 0% NaCl condition (Fig. 1A). The growth curves revealed a higher growth rate in 5% NaCl (0.21) than in 10% (0.19), 15% (0.04), and 20% (0.0003) NaCl between 3 and 12 h, whereas no detectable growth was observed in 25% NaCl during this time (Fig. 1B). We also calculated the length of the lag phase of H. beimenensis under various NaCl conditions. The H. beimenensis lag phase in 5% and 10% NaCl was 3.8 h, whereas this phase in 15% and 20% NaCl lasted 9.5 and 23.7 h, respectively (Fig. 1C). In addition, H. beimenensis growth was inhibited at >20% NaCl (Fig. 1A). H. beimenensis has a fast growth rate in 5% NaCl; however, the maximal biomass yield was not significantly different in 5%, 10%, and 15% NaCl at 48 h (Fig. 1D). These results indicated that the optimal growth condition for H. beimenensis is 5% NaCl. Moreover, H. beimenensis exhibits halotolerance at concentrations of 10 to 20% NaCl and can thus be classified as a moderately halophilic bacterium.

Phylogenetic position and growth conditions of Halomonas beimenensis. (A) Growth curve of H. beimenensis in medium containing various concentrations of NaCl, including 0%, 5%, 10%, 15%, 20%, and 25% (w/v) NaCl. (B) The slope of growth curve in the 5%, 10%, 15%, and 20% NaCl conditions between 3 and 12 h represents the growth rate of H. beimenensis under different NaCl conditions. (C) The length of the lag phase of H. beimenensis under different NaCl conditions. (D) The concentration of H. beimenensis (OD600) under different NaCl conditions at 48 h. (E) A phylogenetic tree based on 16S rRNA sequences was constructed by the neighbor-joining method with the Juke-Cantor correction. Bootstrap values were calculated from 1,000 samplings, and values > 50% are shown. Bar, 0.02 substitutions per nucleotide position.
In a neighbor-joining-based phylogenetic tree of 16S ribosomal RNAs (rRNAs), H. beimenensis was clustered into group 121 (Fig. 1E). The mean identity for group 1 was 96.92%. In the phylogenetic tree, H. beimenensis was closest to H. koreensis (99.1% identity), the NTU-107 strain of H. beimenensis (98.8% identity), and H. organivorans (98.7% identity). NTU-111 and NTU-107 were isolated from the same location, and the phylogenetic tree results agree with the results published by Wang et al.22. In addition, identities of 92.3–96.4% were found when comparing H. beimenensis to the other Halomonas spp. in group 1 (Fig. S1, and Supplementary Table S1). In contrast, the mean 16S rRNA identity between H. beimenensis and other genera was less than 95% (Fig. 1E, Supplementary Fig. S1, and Supplementary Table S1). The phylogenetic data indicated that H. beimenensis is a new strain of Halomonas spp.
Genomic features and transcriptome profiles of H. beimenensis
The entire sequence of the H. beimenensis genome was hybrid de novo assembled with reads from Illumina MiSeq (3,719,926 reads) and PacBio SMRT (39,996 reads) by SPAdes Genome Assembler V3.5.023. One large contig of 4,053,030 bp was obtained, which contained 199 bp regions at the 5′- and 3′-ends that overlapped exactly, indicating that the contig could be completely circularized (GenBank CP021435) (Fig. 2A). A total of 3,807 identified coding sequences (CDSs) were spread across the positive strand (1,868 CDSs) and negative strand (1,939 CDSs) (Fig. 2A). The average G + C content was 68.4%, and the GC skew indicated that the replication origin and replication terminus were located at 65 kb and 2,075 kb (Fig. 2A). Gene Ontology (GO) and Clusters of Orthologous Groups (COG) categories are provided in Supplementary Figures S2 and S3.

Genome map and gene comparison of Halomonas beimenensis. (A) Genome map of H. beimenensis. Rings from the outside are as follows: 1st circle: scale marks (unit, kb); 2nd circle: predicted genes on the + strand; 3rd circle: predicted genes on the − strand; 4th circle: read counts mapping to predicted genes on the + strand in 5% NaCl; 5th circle: read counts mapping to predicted genes on the + strand in 20% NaCl; 6th circle: read counts mapping to predicted genes on the - strand in 5% NaCl; 7th circle: read counts mapping to predicted genes on the − strand in 20% NaCl; 8th circle: significantly differentially expressed genes determined based on log2 fold-changes (log2FC) in FPKM values on the + strand that were greater than 2 (up-regulated genes, red lines) or smaller than −2 (down-regulated genes, blue lines); 9th circle: significantly differentially expressed genes determined based on log2FC in FPKM values on the - strand greater than 2 (up-regulated gene, red lines) or smaller than -2 (down-regulated genes, blue lines); 10th circle: GC content, 11th circle: GC skew (above-average values in pink; below-average values in purple). (B) Venn diagram of gain and loss of genes between H. beimenensis and four Halomonas spp.: H. campaniensis, H. chromatireducens, H. elongata, and H. huangheensis.
A total of 1,530 ortholog genes were detected in Halomonas campaniensis, H. chromatireducens, H. elongata, and H. huangheensis (Fig. 2B). H. beimenensis contains 207 unique genes and lacks 1,326 genes that are present in the other 4 Halomonas spp. (Fig. 2B). The transcriptome profiles of H. beimenensis grown in 5% and 20% NaCl were used to identify differentially expressed genes (DEGs). The transcriptome reads obtained from bacteria grown in 5% (5,070,041 reads) and 20% (6,154,708 reads) NaCl were compared to the genomic sequence to determine the mapping rate and to perform fragments per kilobase of transcript per million mapped reads (FPKM) calculations. The mapped transcriptome reads are shown in the genome map (Fig. 2A). The 4th and 6th circles represent the transcriptome read counts on the + and − strands in the 5% NaCl condition, respectively, whereas the 5th and 7th circles represent the transcriptome read counts on the + and − strands in the 20% NaCl condition, respectively (Fig. 2A). Our data indicated that approximately 98.66% (5% NaCl) and 96.02% (20% NaCl) of the transcriptome reads were mapped to the genomic sequence.
Approximately 53.61% (in 5% NaCl) and 74.59% (in 20% NaCl) of the reads mapped to CDSs, whereas 40.05% (in 5% NaCl) and 21.43% (in 20% NaCl) of the reads mapped to intergenic regions (IGRs), implying that intergenic regions of the genome can also generate RNA transcripts. In 5% NaCl, the average TPM (transcripts per million reads) of CDSs and IGRs was approximately 594,952.57 and 405,047.43, respectively; in 20% NaCl, the average TPM of CDSs and IGR was approximately 811,185.16 and 188,814.83, respectively (Fig. 3A). These data suggest that CDSs exhibit higher transcriptional expression than intergenic regions and that under the high saline condition, more reads mapped to CDSs regions than to IGR regions.

Gene features of Halomonas beimenensis. (A) The average TPM (transcripts per million reads) of CDSs and the intergenic regions in the 5% and 20% NaCl conditions. (B) The differentially expressed genes (DEGs) of H. beimenensis between the 5% NaCl and 20% NaCl conditions. The pink columns represent the up-regulated DEGs, for which the log2FC of FPKM was higher than 2; the blue columns represent the down-regulated DEGs, for which log2FC in FPKM was lower than −2; the gray columns represent genes that did not significantly change (the absolute value of the log2FC of FPKM did not exceed 2). Numbers above the column represent the numbers of genes. (C) The top 5 clusters of orthologous groups (COG) categories indicated for the DEGs of H. beimenensis. (D) The top 8 clusters of Gene Ontology (GO) categories indicated for the DEGs of H. beimenensis. Up-regulated DEGs (log2FC of FPKM > 2) are represented by pink bars and numbers; the down-regulated DEGs (log2FC of FPKM < −2) are represented by blue bars and numbers; gray bars indicate genes with no significant change (−2 < log2FC of FPKM < 2). (E) The transcriptome profile of differentially expressed flagella-related genes in 5% and 20% NaCl.
We identified 614 DEGs (268 up-regulated genes and 346 down-regulated genes) showing a log2 fold-change (log2FC) in FPKM >2 between the 5% and 20% NaCl conditions (Fig. 3B and Table S2). The top COG and GO categories, with the corresponding number of DEGs, are highlighted in Fig. 3C and D, respectively. The COG data indicated that 34 DEGs related to “cell motility”, 30 DEGs related to “energy production and conversion”, and 22 DEGs related to “inorganic ion transport and metabolism” were up-regulated in 20% NaCl, whereas 30 DEGs related to “energy production and conversion” were down-regulated under such conditions (Fig. 3C and Supplementary Table S2). Moreover, the GO category analysis revealed that 16 DEGs related to “cell motility” were up-regulated (Fig. 3D). Therefore, cell motility plays an important role in halotolerance. In fact, many flagellum-related H. beimenensis genes were up-regulated in 20% NaCl (Fig. 3E). Detailed information on the genes corresponding to various COG and GO categories is provided in the Supplementary material. The transcriptome profiles of H. beimenensis in 5% and 20% NaCl are available at the ContigViews database (www.contigviews.bioagri.ntu.edu.tw).
Halotolerance-related genes
Of the 1,256 Tn5 transposition clones, 22 showed a loss of or decreased tolerance to 15% NaCl. In these 22 clones, 16 unique genes were interrupted by the Tn5 transposon (Fig. 4A and Fig. S4). The transcriptome profile showed that 11 out of these 16 genes were up-regulated in 20% NaCl, including the potassium transporter gene trkA2, the sodium-translocating NADH:quinone oxidoreductase gene nqrA, the ATP synthase gene atpC, the NAD-specific glutamate dehydrogenase gene gdhB, the quinolinate synthetase gene nadA, the dTDP-4-dehydrorhamnose 3,5-epimerase gene rfbC, the undecaprenyl-phosphate galactosephosphotransferase gene rfbP, the (p)ppGpp synthetase/guanosine-3′,5′-bis(diphosphate) 3′-diphosphatase gene spoT, the ATP-dependent protease gene lon, the 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase gene mtnN2, and the acetyltransferase gene lacA, whereas the anti-anti-sigma factor gene rsbV, the PrkA family serine protein kinase gene prkA, the twin-arginine translocation protein gene tatB, and the TetR family transcriptional regulator gene acrR1 were down-regulated. Notably, lon, mtnN2, and nqrA showed only a slight up-regulation in the transcriptome profiles (Fig. 4B, upper panel).

Growth and gene expression levels of 16 halotolerance-related genes in the wild-type and Tn5 mutants of Halomonas beimenensis under different salt conditions. (A) The growth of wild-type (WT) H. beimenensis and 16 Tn5 mutant lines under 5% and 15% NaCl conditions. (B) The expression of 16 halotolerance-related genes in WT H. beimenensis was determined by the FPKM values from the transcriptome data and qRT-PCR analysis of bacteria grown in 5% and 20% NaCl conditions. (C) The highlighted gene expression of the 16 genes in the WT bacteria and the 16 Tn5 mutants of H. beimenensis under 5% and 15% NaCl conditions analyzed by qRT-PCR. Each data point was compared with the gene expression in WT bacteria grown in 5% NaCl.
We used quantitative reverse transcription polymerase chain reaction (qRT-PCR) to verify the expression of the 16 genes, and the obtained data agreed with the NGS transcriptome profile (Fig. 4B and C). Notably, the tmRNA-binding protein gene smpB did not exhibit significantly different expression between the 5% and 20% NaCl conditions (Fig. 4B). In the COG classifications, among the 16 genes, the atpC, and nqrA were in the “energy production and conversion” category, whereas trkA2 was in the “inorganic ion transport and metabolism” category. rfbP and rfbC were in the “cell wall/membrane/envelope biogenesis” category, whereas gdhB was in the “amino acid transport and mechanisms”. By contrast, the nadA, and spoT were categorized as the “small molecule metabolic process” category according to GO, whereas prkA, rsbV, and spoT were categorized under the “signal transduction mechanisms” category according to COG. acrR1 was in the “transcription” category according to COG and the “DNA binding” category according to GO. lon and smpB were in the “posttranslational modification, protein turnover, chaperones” category according to COG, implying these genes play roles in protein translation. The COG and GO categories of the remaining three genes did not indicate clear biological functions. Interestingly, previous studies have demonstrated that AtpC, NqrA, and TrkA2 are involved in energy production and potassium uptake, whereas NadA is involved in compatible solute biosynthesis3,10,24,25,26. Mutation of the trkA and nqr genes in H. elongata and V. cholerae, respectively, results in loss of halotolerance3,27. Therefore, these results were confirmed by examining the halotolerance of the Tn5 mutants, and it was indicated that energy production and ion transportation play important roles in halotolerance. The COG and GO categories for the 16 genes are listed in Supplementary Table S3.
Gene expression profiles of the Tn5 mutant lines
To study the adaptation of the 16 Tn5 mutants that exhibited gene expression differences in 5% and 20% NaCl, we evaluated gene expression in wild-type (WT) H. beimenensis and the 16 Tn5 mutants grown in 15% NaCl by qRT-PCR (Fig. 4C). To highlight the importance of these 16 genes in halotolerance regulation, each gene was assigned a specific cut-off threshold based on the significant p-value (Table S4) for up- or down-regulation compared with the gene expression in the WT at 5% NaCl, and these expression patterns are shown in Fig. 4C.
With the exception of gdhB gene, the remaining 15 genes could be classified as “positive”, “negative”, or “bifunctional” regulatory genes in the halotolerant response. In some cases, the gene mutation did not affect the expression of other genes, but any gene that was affected by mutation of another gene was classified as a “regulated gene”. For instance, mutation of prkA resulted in the down-regulation of many other genes in 5% and 15% NaCl, suggesting that prkA is a positive regulatory gene (Fig. 4C). By contrast, gdhB and tatB mutations led to many genes being up-regulated in 5% and 15% NaCl, suggesting that these two genes are negative regulatory genes (Fig. 4C). The spoT was classified as a bifunctional regulatory gene because the mutant caused the up- or down-regulation of various genes in 5% and 15% NaCl (Fig. 4C).
In 5% NaCl, lon was classified as a positive regulatory gene, whereas nadA, rsbV, acrR1, and lacA were classified as negative regulatory genes (Fig. 4C). Moreover, rfbP, nqrA, mtnN2, atpC, rfbC, smpB, and trkA2 were classified as bifunctional regulatory genes (Fig. 4C). However, the genes originally observed to be positive or negative regulatory genes in 5% NaCl might play different roles in 15% NaCl to trigger halotolerance. For instance, rfbP, nqrA, lacA, smpB, and trkA2 were classified as positive regulatory genes, whereas rfbC and atpC were classified as negative regulatory genes in 15% NaCl (Fig. 4C). Moreover, nadA, rsbV, and lon were classified as bifunctional genes, whereas mtnN2 and acrR1 were classified as regulated genes (Fig. 4C).
According to the gene expression observed in the WT and mutants, we constructed gene-for-gene networks to summarize the relationships between these critical genes (Fig. 5). The regulatory network of these 16 genes was more complex in 5% NaCl (Fig. 5A) than in 15% NaCl (Fig. 5B). Regarding the promotion of gene expression, MtnN2 promoted rfbP, trkA2, smpB, and lacA expression, whereas Lon promoted smpB and rfbC expression in 5% NaCl (Fig. 5A). Moreover, RfbP promoted trkA2 expression (Fig. 5A). In contrast, most of the examined genes exhibited complex negative regulatory relationships in 5% NaCl (Fig. 5A). nqrA, atpC, and tatB were the most negatively regulated genes in the 5% NaCl condition (Fig. 5A). Interestingly, prkA exhibited feedback control with smpB, lacA, and rfbC (Fig. 5A). trkA2 also exhibited feedback with atpC and nqrA (Fig. 5A).

The network of 16 halotolerance-related genes of Halomonas beimenensis. (A) The putative network in the 5% NaCl condition. (B) The putative network in the 15% NaCl condition. The symbols for each gene suggest involved functions, e.g., (K) potassium; (B) betaine; (E) ectoine; (P) proline; (T) trehalose; (M) cell motility; and (MK) potassium-dependent cell motility.
Surprisingly, most of the negative relationships observed in the 5% NaCl condition were not active in 15% NaCl (Fig. 5B). There are several important changes that must be highlighted. First, in 15% NaCl, MtnN2 ceased promoting expression of the other examined genes, and its expression was promoted by RfbP, NqrA, LacA, SmpB, and TrkA2 (Fig. 5B). Second, TatB acted as an indirect repressor to negatively affect nqrA, lon, lacA, spoT, and acrR1 expression, whereas tatB was negatively regulated in 5% NaCl (Fig. 5B). TatB is involved in the translocation of folded proteins across the cytoplasmic membrane28; therefore, loss of its function might have consequences for many aspects of cell metabolisms. Third, several genes involved in transcription/translation and signaling affected the expression of genes related to energy conversion and inorganic ion transport, compatible solute or translation in 15% NaCl. For instance, SpoT and RfbC specifically affected atpC, and SmpB promoted the expression of nadA and lon (Fig. 5B). Finally, trkA2 exerted a bilateral promotion effect together with rfbP (Fig. 5B). These dynamic variations suggest adaptation to high-salinity conditions.
Chemical growth complementation of the Tn5 mutants
Accumulations of KCl and organic compatible solutes is a critical approach used by halophilic bacteria to overcome lower external water activity under high-salinity conditions29. We hypothesized that some of the Tn5 mutants may have lost the ability to maintain ionic strength or to regulate the biosynthesis of organic compatible solutes. Therefore, we examined whether the application of additional KCl and compatible solutes could help Tn5 mutants to adapt to high-salinity stress. In WT H. beimenensis, treatment with 20 mM ectoine, 20 mM glutamate, and 200 mM KCl slightly enhanced bacterial growth, whereas the other compatible solutes did not affect H. beimenensis growth (Fig. 6).

Chemical complementation of wild-type and Tn5-mutated Halomonas beimenensis in 15% NaCl. (A) The complementation analysis of the wild-type bacteria with a variety of compatible solutes. (B) The growth of Tn5 mutant lines in which the mutant phenotype was slightly or fully rescued by a variety of compatible solutes. (C) The growth of Tn5 mutant lines that were inhibited by a variety of compatible solutes. (D) The growth of Tn5 mutant lines in which the mutant phenotype was slightly or fully rescued by 200 mM KCl.
Treatment with 20 mM betaine and 20 mM ectoine compensated for the loss of halotolerance in the spoT, tatB, and trkA2 mutants grown in 15% NaCl, resulting in growth similar to that of WT bacteria (Fig. 6A and B, and Table 1). Moreover, 20 mM ectoine can compensate for an smpB mutation (Fig. 6A and B, and Table 1). For the nadA and gdhB mutants, betaine only slightly rescued the mutant phenotype in 15% NaCl (Fig. 6B and Table 1). Treatment with 20 mM trehalose fully rescued the spoT and rsbV mutants but only slightly compensated for the rfbC, smpB, mtnN2 and nqrA mutations (Fig. 6A and B, and Table 1). However, 20 mM glutamate and 20 mM proline inhibited rather than rescued growth in the mutants (Fig. 6C), and only the lacA mutants exhibited slightly recovered growth when treated with proline (Fig. 6B). Furthermore, 20 mM ectoine inhibited growth of the lon mutant (Fig. 6B). These data suggested that various compatible solutes have different effects that crosstalk with these genes in compensating for the loss of halotolerance.
In the KCl compensation experiment, 200 mM KCl enhanced the halotolerance of WT bacteria in 15% NaCl (Fig. 6D, and Table 1). Similar to the compensation effects of the compatible solutes, 200 mM KCl fully rescued the mutant phenotype in the prkA, smpB, atpC, trkA2, and rsbV mutants but had only a slight effect on the mtnN2, tatB, and nqrA mutants (Fig. 6D, and Table 1). By contrast, the growth of the lon mutant was repressed in medium containing 15% NaCl with 200 mM KCl, suggesting that KCl has different effects on the various Tn5 mutants (Fig. 6D, and Table 1). In summary, we found that betaine and ectoine exert similar compensatory effects on mutants, whereas KCl and trehalose exert similar compensatory effects on the mtnN2, rsbV, and nqrA mutants (Table 1), implying the existence of type 2 halotolerance in H. beimenensis. Nevertheless, these genes belong to the transcription/translation, signaling, and energy production categories.
Cell motility
Bacterial motility is an important behavior used to seek out a suitable environment30. The motility of the rfbP, rfbC, lon, gdhB, and nqrA mutants was completely abolished in 5% and 15% NaCl, suggesting these genes might be positively correlated with motility (Fig. 7A and Table 1). In 15% NaCl, the spoT and prkA mutants showed only slightly reduced motility, whereas the motility of the other mutants and even the WT was clearly inhibited, suggesting that spoT and prkA, which are categorized as signaling genes, might be negative regulators of motility under saline stress (Fig. 7B). Notably, the spoT mutant showed slightly reduced motility in 5% NaCl but slightly enhanced motility in 15% NaCl (Fig. 7A). Interestingly, the regulatory networks of spoT were different between 5% and 15% NaCl, implying that SpoT plays different roles in 5% and 15% NaCl (Fig. 5).

Motility and auto-aggregation phenotype of wild-type and 16 Tn5 mutants of Halomonas beimenensis. (A) Motility phenotype assay of the wild-type (WT) and 16 Tn5 mutants. (i) to (vi) were assayed on 3% agar plates containing 5% NaCl for 24 h. (v) to (viii) were assayed on 3% agar plates containing 15% NaCl for 48 h. (B) Motility phenotype assay of WT bacteria and 16 Tn5 mutants with 200 mM KCl. (i) to (vi) were assayed on 3% agar plates containing 5% NaCl for 24 h. (v)-(viii) were assayed on 3% agar plates containing 15% NaCl for 48 h. (C) Auto-aggregation phenotype of the rfbP and rfbC mutants under different salt conditions.
Potassium-dependent enhancement of cell motility
We wondered whether KCl could also compensate for the loss of motility in these mutants. Compared with treatment with 15% NaCl alone, treatment with 200 mM KCl enhanced and slightly enhanced the motility of WT H. beimenensis in 5% NaCl and in 15% NaCl, respectively (Fig. 7A and B), suggesting potassium-dependent enhancement of cell motility (Fig. 7A and B, and Table 1). However, the motility of the rfbP, rfbC, lon, gdhB, and nqrA mutants was not recovered by treatment with 200 mM KCl, suggesting that these genes might be involved in the potassium-dependent enhancement of cell motility (Fig. 7B panels i to iv, and Table 1). To summarize observations regarding KCl compensation and cell motility, KCl enhanced or recovered both the growth and cell motility of the smpB, trkA2, rsbV, and atpC mutants in 15% NaCl, whereas KCl recovered cell motility but only slightly recovered the growth of the mtnN2 and tatB mutants in 15% NaCl (Table 1).
Cell surface properties
Furthermore, mutation of the rfbP has been demonstrated to alter cell surface properties, resulting in aggregation of bacteria in liquid culture31. Aggregation of the rfbP and rfbC mutants was observed after the liquid cultures were incubated on the laboratory bench for a few minutes in 5% or 15% NaCl (Fig. 7C). These data agreed with our transcriptome data, indicating that rfbP and rfbC might play a role in cell wall, cell membrane, and envelope biogenesis (Supplementary Table S3).
Discussion
NGS has been widely used in many genomics, functional genetics, and epigenetics studies32. However, AT-rich repeated sequences within genomic sequences can make it difficult to fill the gaps between contigs33,34. The long reads (3,000–37,335 bp/read) obtained by PacBio SMRT sequencing in this study can act as a scaffold to link two contigs35. Indeed, our results indicated that the completed genomic sequence of H. beimenensis was de novo assembled from a single large contig. Therefore, it is possible to obtain complete genomes of prokaryotic bacteria through one-time de novo assembly using both short and long reads. Furthermore, we predicted 3,807 genes in the H. beimenensis genome, which provided information for whole-transcriptome and Tn5 transposon mutagenesis analyses to identify candidate halotolerance-related genes and study halotolerance mechanisms.
Halophilic bacteria maintain the ionic strength of the cytoplasm at a level equal to that of the external environment by increasing potassium concentrations to overcome saline stress36. Nevertheless, most eubacteria exclude ionic solutes and reduce the water activity of the cytoplasm by accumulating organic compatible solutes, which do not disrupt normal cell functions37. In H. beimenensis, KCl and ectoine slightly enhanced WT bacterial growth but compensated for the loss of halotolerance in particular mutants, suggesting that various compatible solutes exert different effects that exhibit crosstalk with these genes in the compensation of halotolerance. Indeed, NqrA is a subunit of Na+-NQR, whereas TrkA2 is involved in the symport of hydrogen ions by the Trk system for potassium uptake3,9,10. In E. coli, atpC encodes subunit ε of ATP synthase, which facilitates the production of a proton gradient across the cell membrane while producing ATP from ADP24. Orthologous genes of nqrA, trkA2, and atpC have been found to be associated with energy conversion and inorganic ion transport, and it is therefore not surprising to find that mutation of the three genes results in loss of halotolerance. Based on transcriptome and proteome20 profiles, the expression of Na+-NQR is up-regulated or increased in high salinity, suggesting sodium ion efflux. Notably, H. elongata does not encode an ortholog of trkA2 of H. beimenensis.
In Halomonas spp., betaine, ectoine, and proline are important for halotolerance, although certain species also exhibit trehalose uptake, implying that various compatible solutes might facilitate halotolerance in different Halomonas species38,39,40. Ectoine synthesis enzymes of H. elongata (EctA, EctB, and EctC) are increased in optimal salinity compared with low salinity20. However, the levels of these enzymes did not change significantly in H. beimenensis in 20% NaCl, suggesting that the amounts of these enzymes were sufficient for ectoine synthesis under high salinity. 2-Oxoglutarate is a precursor of proline that is produced from glutamate by NAD-GDH25. However, the application of additional proline did not rescue the growth of the gdhB mutant in this study. In addition, NAD is a cofactor in the catalysis of the interconversion of 2-oxoglutarate and glutamate by NAD-GDH, and NadA is involved in the biosynthesis of quinolinic acid (QA), which is a substrate for NAD+ biogenesis in E. coli and Salmonella typhimurium 25,26. Therefore, mutations in nadA and gdhB affect the biosynthesis of compatible solutes.
The identification of new genes involved in other pathways and crosstalk with halotolerance was the main purpose of this study. In transcription, rsbV encodes an anti-anti-sigma factor (an anti-sigma factor antagonist) in various species that controls the activation of sigma factor B (SigB) under osmotic stress41,42. SigB binds an anti-sigma factor under normal conditions; however, under osmotic stress, the unphosphorylated form of RsbV competes with SigB to bind the anti-sigma factor, resulting in the release of SigB, which then associates with the core RNA polymerase to activate stress response genes41,42.
In translation surveillance, E. coli lon encodes an ATP-dependent protease that acts as a heat-shock regulon and is involved in selective intracellular proteolysis to control protein quality and maintain cellular homeostasis43,44. SmpB forms a ribonucleoprotein complex (tmRNP) with transfer-messenger RNA (tmRNA), elongation factor Tu (EF-Tu), and ribosomal protein S1 to monitor trans-translation45. Therefore, SmpB and Lon play roles in translation surveillance and homeostasis, respectively, to control halotolerance.
TatB of E. coli is involved in the twin-arginine translocation (TAT) protein export system, and synthesis of H7 flagellin is abolished in a the triple mutant of TAT (ΔtatABC)28. According to our NGS data, 27 of 37 genes associated with flagellar biosynthesis were up-regulated under high-salinity conditions. The question of whether a tatB mutation in H. beimenensis affects flagellum synthesis will be an interesting topic for future research. In Pectobacterium atrosepticum and Salmonella enterica, rfbP encodes an undecaprenyl-phosphate galactose phosphotransferase, whereas rfbC encodes a biosynthetic precursor of O-antigen, which is involved in the synthesis of dTDP-L-rhamnose and influences bacterial motility31,46. RfbP and RfbC are related to cell envelope biogenesis; mutation of the corresponding genes could cause deficits in cell envelop develop and, consequently, various dysfunctions, resulting in the misregulation of genes and loss of osmoregulation. The mutation of rfbP and rfbC in H. beimenensis, S. enterica and P. atrosepticum results in reduced swimming or swarming ability31,46.
With respect to quorum sensing, in V. cholera, mtnN is a 5′-methylthioadenosine nucleosidase that plays a role in S-adenosylmethionine-related quorum sensing pathways, which might facilitate bacterial communication, motility, gene transfer, and secondary metabolites47,48. The mutation of mtnN2 in H. beimenensis influences bacterial growth and motility. Notably, no findings have demonstrated a correlation between quorum sensing and halotolerance, and many genes promote mtnN2 expression in 15% NaCl (Fig. 5B).
spoT of E. coli encodes a guanosine-3′,5′-(bis)pyrophosphate (ppGpp) synthetase and hydrolase that plays roles in the control of starvation and DNA repair49,50. Our data indicated that SpoT plays significant roles in the regulation of compatible solutes and cell motility. E. coli lacA encodes a galactoside O-acetyltransferase that is associated with the lactose operon lacZYA, which is involved in carbohydrate metabolism51,52. This study is the first to report that LacA influences halotolerance in H. beimenensis.
Finally, the functions of PrkA and AcrR1 are unknown; however, our bioinformatics data suggest that PrkA is involved in signaling and that AcrR1 is involved in translation. prkA is down-regulated under high-salinity conditions in H. beimenensis and M. alhagi 53. However, the prkA mutant of H. beimenensis shows lower tolerance to saline stress, whereas the M. alhagi prkA mutant exhibits tolerance53, suggesting that PrkA is associated with the regulation of salt stress but exhibits different functions in different bacterial species. Moreover, AcrA and AcrB of E. coli are positive regulators of osmotic stress, whereas AcrR is a repressor of the acrA and acrB genes54. However, the acrR1 mutant of H. beimenensis lost its saline adaptation ability, implying the existence of unknown mechanism for AcrR1-regulated halotolerance.
Figure 5B summarizes gene functions and interactions in halotolerance and cell motility. Based on the network associated with treatment with 15% NaCl, a halotolerance signal could be communicated via quorum sensing by mtnN2 expression triggered by other functions, such as cell motility, energy and ion transport. Na+-NQR and ATPase, which are involved in sodium pumps and energy production, respectively, are negatively regulated by the flagellum. Moreover, NadA and GdhB could be involved in compatible solute production for saline adaptation. Motility assays indicated that PrkA and SpoT negatively regulate cell motility but that genes in the cell motility and compatible solute categories, among others, promote cell motility. Interestingly, these cell motility-related genes exhibit high overlap with the potassium-dependent enhancement of motility. Finally, compatible solute production and response genes were identified through bioinformatics and experimental approaches in this study. The complete genome of H. beimenensis and the 16 mutants can be employed as a model system to further investigate mechanisms of halotolerance in the future. From an industry perspective, H. beimenensis became an attractive strain in fermentation because of underlying mechanisms of halotolerance. Moreover, this study can be useful in informing strain selection for fermentation application.
Methods
Bacterial strain and growth conditions
The H. beimenensis NTU-111 strain used in this study was originally isolated from brine samples from the Beimen saltern in southern Taiwan22. H. beimenensis was grown in basal medium containing 5 g/L yeast extract (Bacto, BD), 5 g/L Casamino acids (Bacto, BD) and 5 g/L MgSO4·7H2O (Wako), pH 7.5. To determine the optimal growth conditions for H. beimenensis, the bacteria were grown in basal medium with various concentrations of NaCl, including 0%, 5%, 10%, 15%, 20%, and 25% NaCl (w/v), and were incubated at 37 °C with shaking at 220 rpm. The concentration of H. beimenensis (OD600) was monitored by a spectrophotometer (Libra S4, Biochrom) every 6 h until 72 h with three replicates.
Genomic DNA and total RNA extraction
H. beimenensis was grown in basal medium with 5% NaCl at 37 °C for 16 h (OD600 = 2.0), and the genomic DNA was extracted using a Qiagen Gentra Puregene Kit (Qiagen) according to the manufacturer’s instructions. For total RNAs purification, H. beimenensis cells (OD600 = 2.0) were grown in medium containing 5%, 15%, and 20% NaCl, and RNA was extracted using a Total RNA Purification Kit (Geneaid) according to the manufacturer’s instructions.
Phylogenetic tree analysis
For phylogenetic tree analysis, MEGA v7 was used to compare 16 S rRNA sequences of the Halomonadaceae family (Supplementary material) were compared via the neighbor-joining method with 1,000 bootstrap replicates55. Here, Escherichia coli (U00006) and Pseudomonas aeruginosa (X06684) were selected as out groups.
Genomic DNA and whole-transcriptome sequencing
The whole genome sequencing of H. beimenensis was performed on the MiSeq (2 × 300) paired-end sequencing (Illumina) and SMRT (20k) (PacBio) platforms (Genomics, BioSci & Tech Co.). The whole-transcriptome sequencing was performed using MiSeq (2 × 300) paired-end strand-specific sequencing (Illumina) (Genomics, BioSci & Tech Co.). The raw reads reported in this paper are available in the NCBI Short Read Archive under accession numbers SRR5572247 (genomic DNA for PacBio), SRR5572216 (genomic DNA for Illumina), SRR5572264 (transcriptome for 5% NaCl), and SRR5572268 (transcriptome for 20% NaCl).
Genomic DNA assembly, gene prediction, annotation, and gene comparison
The whole-genome sequence of H. beimenensis was de novo assembled by the SPAdes Genome Assembler v3.5.0 using hybrid assembly of the Illumina short reads and the PacBio long reads with default parameters23. The web-based software Rapid Annotation using Subsystem Technology (RAST; version 2.0) on the PARTIC platform (www.patricbrc.org/portal/portal/patric/Home) was used for gene prediction and annotation56,57. GC content was also calculated with the PARTIC platform. Transfer RNAs and rRNAs were predicted with tRNAscan58 and RNAmmer59, respectively. The whole-genome graphical representation was constructed using CIRCOS (version 0.67–7)60. For GO, gene sequences were analyzed using BLAST2GO (version 3.3.5) with default settings61. For COG analysis, gene sequences were analyzed using NCBI’s COG database (version 2014) (www.ncbi.nlm.nih.gov/COG/)62. For analysis of the gain and loss of genes in H. beimenensis, the coding genes of four Halomonas spp., H. campaniensis, H. chromatireducens, H. elongata, and H. huangheensis were used for ortholog group clustering by OrthoMCL (orthomcl.org/orthomcl/) (Li et al., 2003).
Transcriptome data processing and differential gene expression analysis
For gene expression analysis, clean transcriptomic reads from the 5% and 20% NaCl samples were aligned to the whole genome sequence of H. beimenensis using Bowtie2 software (version 2.2.5)63. The transcript expression levels were represented using FPKM values evaluated with eXpress (version 1.5.1)64. Genes were considered to be differentially expressed if the absolute value of the log2FC in FPKM exceeded 2.
Tn5 transposon mutagenesis
Mutagenesis was performed using the EZ-Tn5TM < R6Kγori/KAN-2 > Tnp TransposomeTM Kit (Epicentre) to generate mutants according to the manufacturer’s protocol. Electrocompetent H. beimenensis cells were prepared through Choi’s approach, with some modifications65. The Tn5 transposon was introduced into the H. beimenensis cells in a 2-mm cuvette with 2.5 kV for 5 ms. After electroporation, the cells were suspended in 900 mL of basal medium containing 5% (w/v) NaCl and recovered for 1 h at 37 °C with shaking at 220 rpm and then plated the electroporated cells on basal medium agar containing 5% NaCl and 50 µg/mL kanamycin. The Tn5-transformed colonies were subcultured in basal medium containing 5% or 15% NaCl for halotolerance evaluation.
Mutants that exhibited decreased or lost halotolerance in 15% NaCl were selected. To identify Tn5 insertion positions, the genomic DNA from the mutants was digested with Hyp99I, EagI, BanI, BglII, BsaHI, SfoI, Hyp99I, or BsaI, and then self-ligated and transformed into TransforMaxTM EC100DTM pir + Electrocompetent E. coli cells (Epicentre) that were plated on LB agar containing 50 µg/mL kanamycin for recovery of the Tn5 insertion fragment. Rescued plasmid DNA was extracted and sequenced using the EZ-Tn5 <R6Kγori/KAN-2> transposon-specific primers.
Validation of gene expression
qRT-PCR was used to validate gene expression in mutants and WT H. beimenensis. Primers were designed using Primer3 and the BLAST tools available from NCBI (www.ncbi.nlm.nih.gov/tools/primer-blast/), and the primer sequences are listed in Supplementary Table S5.
Complementation of the Tn5 mutants and evaluation of cell motility
The chemical complementation of the Tn5 mutants was performed by adding 200 mM KCl, 20 mM betaine, 20 mM ectoine, 20 mM glutamate, 20 mM proline, or 20 mM trehalose to basal medium containing 5% or 15% NaCl. To analyze cell motility, 20 µL of bacterial cells (WT or mutants) (OD600 = 2.0) were incubated at 37 °C on semi-solid basal medium agar plates (3% agar) with 5% or 15% NaCl. For the 5% NaCl condition, the bacteria were incubated up to 24 h, whereas the bacteria were incubated up to 48 h for the 15% condition. The diameter of each colony was determined.
References
Yin, J., Chen, J. C., Wu, Q. & Chen, G. Q. Halophiles, coming stars for industrial biotechnology. Biotechnol. Adv. 33, 1433–1442 (2015).
DasSarma, S. & DasSarma, P. In eLS Vol. https://doi.org/10.1002/9780470015902.a0000394.pub3 (John Wiley & Sons, Ltd, 2012).
Kraegeloh, A., Amendt, B. & Kunte, H. J. Potassium transport in a halophilic member of the bacteria domain: identification and characterization of the K+ uptake systems TrkH and TrkI from Halomonas elongata DSM 2581T. J. Bacteriol. 187, 1036–1043 (2005).
Shivanand, P. & Mugeraya, G. Halophilic bacteria and their compatible solutes - osmoregulation and potential applications. Curr. Sci. 100, 1516–1521 (2011).
Kraegeloh, A. & Kunte, H. J. Novel insights into the role of potassium for osmoregulation in Halomonas elongata. Extremophiles 6, 453–462 (2002).
Schlosser, A., Hamann, A., Bossemeyer, D., Schneider, E. & Bakker, E. P. NAD+ binding to the Escherichia coli K(+)-uptake protein TrkA and sequence similarity between TrkA and domains of a family of dehydrogenases suggest a role for NAD+ in bacterial transport. Mol. Microbiol. 9, 533–543 (1993).
Kuhlbrandt, W. Biology, structure and mechanism of P-type ATPases. Nature reviews. Molecular cell biology 5, 282–295, https://doi.org/10.1038/nrm1354 (2004).
Fagerbakke, K. M., Norland, S. & Heldal, M. The inorganic ion content of native aquatic bacteria. Can. J. Microbiol. 45, 304–311 (1999).
Steuber, J. et al. Central role of the Na(+)-translocating NADH:quinone oxidoreductase (Na(+)-NQR) in sodium bioenergetics of Vibrio cholerae. Biol. Chem. 395, 1389–1399 (2014).
Muras, V., Dogaru-Kinn, P., Minato, Y., Hase, C. C. & Steuber, J. The Na+ -translocating NADH:Quinone oxidoreductase enhances oxidative stress in the cytoplasm of Vibrio cholerae. J. Bacteriol. 198, 2307–2317 (2016).
Yin, L., Xue, Y. & Ma, Y. Global microarray analysis of alkaliphilic halotolerant bacterium Bacillus sp. N16-5 salt stress adaptation. PloS one 10, e0128649 (2015).
Zhou, A. et al. Characterization of NaCl tolerance in Desulfovibrio vulgaris Hildenborough through experimental evolution. ISME J. 7, 1790–1802 (2013).
Cheng, B. et al. Alkaline Response of a Halotolerant Alkaliphilic Halomonas Strain and Functional Diversity of Its Na+ (K+)/H+ Antiporters. The Journal of biological chemistry 291, 26056–26065, https://doi.org/10.1074/jbc.M116.751016 (2016).
Liu, X., Luo, Y., Mohamed, O. A., Liu, D. & Wei, G. Global transcriptome analysis of Mesorhizobium alhagi CCNWXJ12-2 under salt stress. BMC Microbiology 14, 319 (2014).
Yaakop, A. S. et al. Characterization of the mechanism of prolonged adaptation to osmotic stress of Jeotgalibacillus malaysiensis via genome and transcriptome sequencing analyses. Sci. Rep. 6, 33660 (2016).
Jiang, J. Q. et al. Salt-tolerance genes involved in cation efflux and osmoregulation of Sinorhizobium fredii RT19 detected by isolation and characterization of Tn5 mutants. FEMS Microbiol. Lett. 239, 139–146 (2004).
Nagarajan, T., Vanderleyden, J. & Tripathi, A. K. Identification of salt stress inducible genes that control cell envelope related functions in Azospirillum brasilense Sp7. Mol. Genet. Genomics 278, 43–51 (2007).
Wei, W., Jiang, J., Li, X., Wang, L. & Yang, S. S. Isolation of salt-sensitive mutants from Sinorhizobium meliloti and characterization of genes involved in salt tolerance. Lett. Appl. Microbiol. 39, 278–283 (2004).
Zuleta, L. F., Italiani, V. C. & Marques, M. V. Isolation and characterization of NaCl-sensitive mutants of Caulobacter crescentus. Appl. Environ. Microbiol. 69, 3029–3035 (2003).
Kindzierski, V. et al. Osmoregulation in the Halophilic Bacterium Halomonas elongata: A Case Study for Integrative Systems Biology. PloS one 12, https://doi.org/10.1371/journal.pone.0168818 (2017).
Arahal, D. R., Ludwig, W., Schleifer, K. H. & Ventosa, A. Phylogeny of the family Halomonadaceae based on 23S and 165 rDNA sequence analyses. Int. J. Syst. Evol. Microbiol. 52, 241–249, https://doi.org/10.1099/00207713-52-1-241 (2002).
Wang, C. Y., Wu, S. J., Ng, C. C., Tzeng, W. S. & Shyu, Y. T. Halomonas beimenensis sp. nov., isolated from an abandoned saltern. Int. J. Syst. Evol. Microbiol. 62, 3013–3017 (2012).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Ruhle, T. & Leister, D. Assembly of F1F0-ATP synthases. Biochim. Biophys. Acta. 1847, 849–860 (2015).
Bonete, M. J., Perez-Pomares, F., Diaz, S., Ferrer, J. & Oren, A. Occurrence of two different glutamate dehydrogenase activities in the halophilic bacterium Salinibacter ruber. FEMS Microbiol. Lett. 226, 181–186 (2003).
Ollagnier-de Choudens, S., Loiseau, L., Sanakis, Y., Barras, F. & Fontecave, M. Quinolinate synthetase, an iron-sulfur enzyme in NAD biosynthesis. FEBS Lett. 579, 3737–3743 (2005).
Hase, C. C. & Barquera, B. Role of sodium bioenergetics in Vibrio cholerae. Biochim. Biophys. Acta. 1505, 169–178 (2001).
Pradel, N. et al. Contribution of the twin arginine translocation system to the virulence of enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 71, 4908–4916 (2003).
Roberts, M. F. Organic compatible solutes of halotolerant and halophilic microorganisms. Saline Systems 1, 5 (2005).
Mitchell, J. G. & Kogure, K. Bacterial motility: links to the environment and a driving force for microbial physics. FEMS Microbiol. Ecol. 55, 3–16 (2006).
Bowden, S. D. et al. Surface swarming motility by Pectobacterium atrosepticum is a latent phenotype that requires O antigen and is regulated by quorum sensing. Microbiology 159, 2375–2385, https://doi.org/10.1099/mic.0.070748-0 (2013).
Loman, N. J. & Pallen, M. J. Twenty years of bacterial genome sequencing. Nat. Rev. Microbiol. 13, 787–794 (2015).
Bodi, K. et al. Comparison of commercially available target enrichment methods for next-generation sequencing. J. Biomol. Tech. 24, 73–86, https://doi.org/10.7171/jbt.13-2402-002 (2013).
Nakamura, K. et al. Sequence-specific error profile of Illumina sequencers. Nucleic. Acids. Res. 39, e90 (2011).
Williamson, A., De Santi, C., Altermark, B., Karlsen, C. & Hjerde, E. Complete genome sequence of Halomonas sp. R5–57. Stand. Genomic Sci. 11, 62 (2016).
Martin, D. D., Ciulla, R. A. & Roberts, M. F. Osmoadaptation in Archaea. Appl. Environ. Microbiol. 65, 1815–1825 (1999).
Sleator, R. D. & Hill, C. Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol. Rev. 26, 49–71 (2002).
Cummings, S. P. & Gilmour, D. J. The effect of NaCl on the growth of a Halomonas species: accumulation and utilization of compatible solutes. Microbiology 141, 1413–1418 (1995).
Kempf, B. & Bremer, E. Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Archives of microbiology 170, 319–330 (1998).
Yun, S. H., In, S. B. & Park, D. H. Influence of NaCl on the growth and metabolism of Halomonas salina. J. Microbiol. Biotechnol. 15, 118–124 (2005).
Pane-Farre, J. et al. Role of RsbU in controlling SigB activity in Staphylococcus aureus following alkaline stress. J. Bacteriol. 191, 2561–2573 (2009).
Lee, E. J., Cho, Y. H., Kim, H. S., Ahn, B. E. & Roe, J. H. Regulation of sigmaB by an anti- and an anti-anti-sigma factor in Streptomyces coelicolor in response to osmotic stress. J. Bacteriol. 186, 8490–8498 (2004).
Rotanova, T. V. et al. Classification of ATP-dependent proteases Lon and comparison of the active sites of their proteolytic domains. Eur. J. Biochem. 271, 4865–4871 (2004).
Tsilibaris, V., Maenhaut-Michel, G. & Van Melderen, L. Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 157, 701–713 (2006).
Wower, J., Wower, I. K., Kraal, B. & Zwieb, C. W. Quality control of the elongation step of protein synthesis by tmRNP. J. Nutr. 131, 2978s–2982s (2001).
Bogomolnaya, L. M. et al. Identification of novel factors involved in modulating motility of Salmonella enterica serotype typhimurium. PloS one 9, e111513 (2014).
Gutierrez, J. A. et al. Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 5, 251–257 (2009).
Montgomery, K., Charlesworth, J. C., LeBard, R., Visscher, P. T. & Burns, B. P. Quorum sensing in extreme environments. Life 3, 131–148 (2013).
Kamarthapu, V. et al. ppGpp couples transcription to DNA repair in E. coli. Science 352, 993–996 (2016).
Magnusson, L. U., Farewell, A. & Nystrom, T. ppGpp: a global regulator in Escherichia coli. Trends. Microbiol. 13, 236–242 (2005).
Lo Leggio, L., Dal Degan, F., Poulsen, P., Andersen, S. M. & Larsen, S. The structure and specificity of Escherichia coli maltose acetyltransferase give new insight into the LacA family of acyltransferases. Biochemistry 42, 5225–5235 (2003).
Libby, E. A., Roggiani, M. & Goulian, M. Membrane protein expression triggers chromosomal locus repositioning in bacteria. Proc. Natl. Acad. Sci. USA 109, 7445–7450 (2012).
Liu, X., Luo, Y., Li, Z. & Wei, G. Functional analysis of PrkA - a putative serine protein kinase from Mesorhizobium alhagi CCNWXJ12-2 - in stress resistance. BMC Microbiol. 16, 227 (2016).
Ma, D., Alberti, M., Lynch, C., Nikaido, H. & Hearst, J. E. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol. Microbiol. 19, 101–112 (1996).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008).
Snyder, E. E. et al. PATRIC: the VBI PathoSystems Resource Integration Center. Nucleic. Acids. Res. 35, D401–406 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic. Acids. Res. 35, 3100–3108 (2007).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome. Res. 19, 1639–1645 (2009).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic. Acids. Res. 43, D261–269 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Roberts, A. & Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 10, 71–73 (2013).
Choi, K. H., Kumar, A. & Schweizer, H. P. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397 (2006).
Acknowledgements
We thank Dr. Ming-Che Shih for supporting Tn5 mutagenesis. The National Center for High-Performance Computing, National Applied Research Laboratories provided computing power for this project. This study was supported by a grant from the Ministry of Science and Technology of Taiwan under contract number (MOST 106-2321-B-002-008-).
Author information
Authors and Affiliations
Contributions
Y.H.C. performed the experiments and experimental data analysis and wrote the manuscript. C.W.L. performed the bioinformatics analysis for the genome assembly and gene identification. S.S.L. and Y.T.S. are the corresponding authors in charge of the project design, experimental data analysis, and manuscript preparation.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, YH., Lu, CW., Shyu, YT. et al. Revealing the Saline Adaptation Strategies of the Halophilic Bacterium Halomonas beimenensis through High-throughput Omics and Transposon Mutagenesis Approaches. Sci Rep 7, 13037 (2017). https://doi.org/10.1038/s41598-017-13450-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13450-9
This article is cited by
-
Establishment of an optimized electroporation method for Halomonas sp. YK44 and its application in the coproduction of PHB and isobutanol
Biotechnology and Bioprocess Engineering (2024)
-
Chemoautotrophic production of gaseous hydrocarbons, bioplastics and osmolytes by a novel Halomonas species
Biotechnology for Biofuels and Bioproducts (2023)
-
Genome analysis of haloalkaline isolates from the soda saline crater lake of Isabel Island; comparative genomics and potential metabolic analysis within the genus Halomonas
BMC Genomics (2023)
-
Metabolic engineering of Halomonas campaniensis strain XH26 to remove competing pathways to enhance ectoine production
Scientific Reports (2023)
-
Halomonas flagellata sp. nov., a halophilic bacterium isolated from saline soil in Xinjiang
Archives of Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
