
Abstract
Acanthaspis cincticrus (Stål) is an assassin bug with a specialized camouflaging behavior to ambush ants in the nymphal stages. In this study, we comprehensively sequenced all the life stages of A. cincticrus, including the eggs, five nymph instars, female and male adults using Illumina HiSeq technology. We obtained 176 million clean sequence reads. The assembled 84,055 unigenes were annotated and classified functionally based on protein databases. Among the unigenes, 29.03% were annotated by one or more databases, suggesting their well-conserved functions. Comparison of the gene expression profiles in the egg, nymph and adult stages revealed certain bias. Functional enrichment analysis of significantly differentially expressed genes (SDEGs) showed positive correlation with specific physiological processes within each stage, including venom, aggression, olfactory recognition as well as growth and development. Relative expression of ten SDEGs involved in predation process was validated using quantitative real-time PCR (qRT-PCR).
Similar content being viewed by others

Introduction
Acanthaspis cincticrus (Stål) (Hemiptera: Reduviidae) is a predatory assassin bug, which feeds on ants and can be found in the vicinity of ant nests, waiting for the preys1. This species is native to Oriental Region with one generation per year. Nymphs of this species have five instars and exhibit both natural and corpse camouflaging behaviors1. They cover themselves with a range of materials found in their environment, including ants corpses and other insects, dust and soil particles, which they affix on themselves with the viscid secretions from specialized setae on the abdomen2. Camouflaging in A. cincticrus is specific to the nymphs (Fig. 1A) and is absent in the adults (Fig. 1B). This behavior has been documented in some neuropterans3. Camouflaging may benefit nymphs in two ways: by being less visible in stalking their preys and by not being obvious to potential predators. While studies on A. cincticrus thus far have focused on the morphology, biology and behaviors related to camouflaging in nymphs1,2, the molecular mechanisms underlying development and predation are unknown.

The nymph and adult of A. cincticrus. (A) A masked nymph camouflaged with ant corpses, dust and soil particles. (B) An adult male. Photographs were taken by F.K.
In predatory insects, arousing, paralyzing and sucking are important for predatory process4. These processes are influenced or regulated by several genes or pathways involved in the identification of chemical signals, and in the regulation of neuromodulators for agonistics and the digestion of the prey5,6,7. Predators locate and interact with their prey by using species-specific semiochemicals, such as the detection and discrimination of odorant-binding proteins (OBPs) and chemosensory proteins (CSPs)5. After locating the target, aggression is displayed by predaceous arthropods against their prey8. Factors that affect aggressiveness and/or fighting success in predaceous arthropods include body size9 and contestants’ age10. Previous studies reported that many genes, such as cyp6Q20, tyramine receptor, octopamine receptor and metabotropic glutamate receptor B, were likely to be involved in the regulation of complex behavioral phenotypes11,12. Tyramine receptor and octopamine receptor serve as aggression modulating neurotransmitters that affect excessive aggressiveness and impulsiveness in the biogenic amine signaling pathway11; metabotropic glutamate receptor B is a part of G protein coupled metabotropic receptors family, which is the major excitatory neurotransmitter acting through multiple independent receptors, involved in regulating aggressive behavior of mice12.
The paralyzing and digestion process of assassin bugs includes extra oral digestion (EOD)13 which is a chemical pretreatment to mobilize nutrients and is common in the predation of Heteroptera7. Venom and/or digestive enzymes have a biochemical role of typically stunning, killing prey and even aiding in prey digestion7,14. The venomous saliva of predatory reduviid bugs is known to contain a complex mixture of proteins15, peptides16 and enzymes17. The most abundant enzymes that are present in the salivary secretion include proteinases, phospholipase, trypsin like enzymes, esterase, serine proteases, etc18. Serine proteases, found in many organisms, have attracted broad interests because they have diverse physiological functions that affect processes such as digestion, immune response, cellular differentiation and prothrombin activator7,13. The presence of trypsin-like enzyme in the salivary glands of Zelus renardii indicates that it has evolved or retained trypsin-like genes for protein digestion7. Although most reduviids are common predators, studies on the factors influencing the feeding process focused on digestion7,15,16,17,18.
The transcriptomic and bioinformatic approaches presented in previous studies have been served as a foundation to explore the relationships between gene regulation and behavioral evolution in other species5,11. In this study, we performed a comprehensive transcriptome analysis during eight life stages of A. cincticrus, including egg, five instar nymphs, female and male adults. We identified 13,479 significantly differentially expressed genes (SDEGs) and also identified and characterized 115 SDEGs involved in predation, indicating differences between genders and among life stages. We performed quantitative real-time PCR (qRT-PCR) analysis to determine the expression profiles of ten SDEGs involved in predation from different life stages. The results could help elucidate the role of venom-related, aggression-related and olfactory-related genes involved in the predation of reduviids.
Results
Whole-transcriptome sequencing and annotation of the predicted proteins
cDNA libraries from all life stages of A. cincticrus yielded about 176 million clean sequence reads with an average quality value ≥ 30. GC content of the sample was averaged at 38.03% (Table S1). After assembling the clean reads, we obtained 164,745 transcripts and 84,055 unigenes with an average sequence length of 1,366 bp and 667 bp, and N50 with average lengths of 2,731 bp and 1,087 bp, respectively (Table 1). The length of unigenes ranged from 210 bp to 29,391 bp (Fig. 2A). Of which, 12, 679 (or 15.09%) unigenes had sequence length more than 1,000 bp. The result is similar to the number of unigenes reported in the moths Dendrolimus punctatus (70,664)19 and Athetis lepigone (81,356)20. A proportion of unigenes was annotated based on the available protein database for A. cincticrus. 24,402 unigenes (or 29.03%) were annotated as coding hypothetical proteins. Homology analysis of A. cincticrus unigenes showed that they best matched with the species from Hemiptera, the pea aphid Acyrthosiphon pisum and the bean bug Riptortus pedestris (Fig. 2B).

The statistics of assembly and homology analyses. (A) Size distribution of unigenes. (B) Species distribution of the BLASTX against NCBI-NR database, proportions of more than 1% were shown.
Functional classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis
The functions of unigenes were predicted by Gene Ontology (GO) analysis at the macro level. In total, 11,682 unigenes were classified into 58 sub-categories belonging to three GO functional categories: cellular component (CC), molecular function (MF) and biological process (BP) (Fig. S1). The GO classification of unigenes indicated that ‘cell part’ (20.79%) and ‘cell’ (20.68%) were the dominant sub-categories in the CC category, ‘catalytic activity’ (42.40%) and ‘binding’ (38.83%) were the most dominant sub-categories in the MF category, and ‘metabolic process’ (24.23%) and ‘cellular process’ (21.81%) were the most dominant sub-categories in the BP category.
We also annotated the unigenes by searching the Cluster of Orthologous Groups (COG) database to classify the functions of the predicted proteins. A total of 6,822 unigenes had a COG classification. Among the 25 COG categories (Fig. S2), the cluster for ‘general function prediction only’ was the most dominant group (18.25%), followed by ‘translation, ribosomal structure and biogenesis’ (8.66%) and ‘post-translational modification, protein turnover, chaperones’ (8.58%).
In order to identify the biological pathways represented in the transcriptome of A. cincticrus, 7,193 unigene sequences were mapped to 193 KEGG pathways (Table S2). The most prominent metabolic pathways were protein processing in endoplasmic reticulum (4.25%), oxidative phosphorylation (3.99%) and RNA transport (3.70%).
The SDEGs in different life stages
We evaluated the gene expression levels to measure the genetic landscape in different life stages of A. cincticrus. We performed nine comparisons of the transcriptomes from pairs of life stages (e.g. egg vs 1st instar, 1st vs 2nd instar, 2nd vs 3rd instar, 3rd vs 4th instar, 4th vs 5th instar, 1st vs 5th instar, 5th instar vs adult male, 5th instar vs adult female, male vs female adults) (Table S3). The results yielded 13,479 SDEGs (Table S4), which were then hierarchically clustered in a heat map (Fig. S4). In this heat map, the gene expression profiles differed through all life stages with each comprising a main cluster of high expression genes. In the egg stage, many crucial embryogenesis related genes also comprised maternal genes such as kruppel, hunchback, nanos-like protein and serine protease snake-like, protein takeout-like, maelstrom, pumilio homolog 1-like and staufen (Table S5). The nymph and adult stages also had their own high expression unigenes, including cuticular proteins, histone H2A, cytochrome P450s, ecdysone-induced protein and juvenile hormone binding proteins in nymph stages, while vitellogenin and sperm flagellar protein 1-like in the adults. A previous study reported that these maternal genes, e.g. snake and nanos, might be involved in the embryonic development of Oncopeltus fasciatus 21.
The distribution of upregulated, downregulated and uniqueness of SDEGs in the different comparisons were shown in the Table S6. We also analyzed the changes in gene expression between the nymph stages and displayed the distribution of SDEGs using a Venn diagram that illustrated the intersection between the expressed genes in various developmental nymph stages (Fig. S4). In total, 38 SDEGs were shared by all nymph stages. 203, 937, 333 and 614 SDEGs were uniquely expressed in 1st vs 2nd instar, 2nd vs 3rd instar, 3rd vs 4th instar, and 4th vs 5th instar indicating that different life stages had different highly expressed unigenes that participated in specific life activities.
In the comparison of egg vs 1st instar, upregulated SDEGs with the high variation (10+ fold) were serine protease, trypsin-like protease, OBPs, cuticular protein, etc. The downregulated SDEGs (3+ fold) were annotated to early cuticle protein 6, hunchback, nanos-like protein, serine protease snake-7, vitellogenin receptor, etc.
In the comparisons of 1st vs 2nd instar and 2nd vs 3rd instar, the most enriched GO terms were oxidation-reduction process (BP) and structural constituent of ribosome (MF). Lipid particle (CC) and cytoplasm (CC) were respectively enriched in 1st vs 2nd instar and 2nd vs 3rd instar (Table S7). Oxidative phosphorylation, ribosome and lysosome were the top three enrichment pathways in the KEGG pathway analysis (Table S8). When comparing 3rd vs 4th instars, most upregulated genes were enriched for GO categories including protein metabolic process (BP), intracellular (CC) and catalytic activity (MF) (Table S7). According to KEGG enrichment analysis, the upregulated genes were enriched for pathways such as arginine and proline metabolism, wnt signaling pathway and glutathione metabolism (Table S8). When comparing 4th vs 5th instar, most of the SDEGs were categorized into nucleosome assembly (BP), intracellular (CC) and catalytic activity (MF) (Table S7). The KEGG analysis indicated that arginine and proline metabolism, glutathione metabolism and progesterone-mediated oocyte maturation were the top enriched pathways (Table S8).
We also performed a comparison of 1st vs 5th instar. The upregulated genes with the high expression variation (5+ fold) were pupal cuticle protein precursor, defensin, histone, etc. The downregulated genes with high expression variation were ecdysone-induced protein, juvenile hormone binding protein-like precursor, etc.
Between the 5th instar nymph vs adult male transcriptomes, the upregulated genes with the high expression variation (5+ fold) included venom serine protease, odorant-binding protein, methylmalonyl-CoA decarboxylase, etc. The downregulated genes included digestive and metabolic genes such as histone H2A, prostaglandin reductase, cytochrome P450 monooxygenase CYP6 X 1v1, etc.
When comparing female vs male adults, the upregulated genes (5+ fold) included venom serine protease, vitellogenin receptor, etc. The downregulated genes included sperm flagellar protein 1-like, ejaculatory bulb-specific protein 3, heat shock protein 60, etc.
The three comparisons (egg vs 1st instar, 1st vs 5th instar, 5th instar vs adult male), including two developmental transitions (eggs to nymphs, nymphs to adults), shared two pathways for the upregulated SDEGs: wnt signaling pathway and neuroactive ligand-receptor interaction (Fig. 3A), which are important pathways in processing information22. Three pathways, including arginine and proline metabolism, purine metabolism and oxidative phosphorylation (Fig. 3A), were enriched in two comparisons (egg vs 1st instar, 5th instar vs adult male). The oxidative phosphorylation pathway plays a critical role in the supply of energy23. The enrichment analysis indicated that energy metabolism increased during development in A. cincticrus. In addition, other genes in this pathway encoding transport oxidoreductase and heat shock proteins (hsps) were upregulated as well. Glutathione metabolism was downregulated in two comparisons (5th instar vs adult male, adult male vs adult female), and arginine and proline metabolism were downregulated in two comparisons (1st vs 5th instar, adult male vs adult female). Inositol phosphate metabolism and phosphatidylinositol signaling system were downregulated in egg vs 1st instar nymph (Fig. 3B). The changes in phosphatase in Triatoma saliva could interfere with anti-haemostatic response and facilitate their feeding24.

Enrichment KEGG pathways for the upregulated (A) and downregulated (B) SDEGs in comparisons of different life stages.
In the GO enrichment of BP, the most enrichment of upregulated SDEGs, including metabolic process, cellular process, single-organism cellular process, signal transduction, proteolysis, and oxidative phosphorylation, were shared by two comparisons (egg vs 1st instar and 5th instar vs adult male) (Fig. S5A). The most enrichment of upregulated SDEGs in the CC (Fig. S5B) included integral component of membrane, cytoplasm, membrane, lipid particle, plasma membrane and microtubule associated complex, were shared in two comparisons (egg vs 1st instar and 5th instar vs adult male). As for the MF (Fig. S5C), two comparisons (egg vs 1st instar and 5th instar vs adult male) revealed five common GO enrichment categories of upregulated SDEGs, including catalytic activity, binding, transferase activity, nucleic acid binding and ATP binding. Four comparisons (egg vs 1st instar, 1st vs 5th instar, 5th instar vs adult male, adult male vs adult female) shared one common GO term (catalytic activity). Serine protease, involved in general digestion and protein metabolism7,25, was identified in catalytic activity indicating that the organism was undergoing continuous growth.
Together, the KEGG pathway and GO enrichment analyses identified a diverse group of development-related genes that undergo differential transcriptional responses during the developmental stages and predation process. These genes include maternal genes, cuticular proteins, metabolite synthases, hormone-related genes (Table S5), venom-related genes, aggression-related genes and olfactory-related genes (Table S8).
Genes putatively involved in predation
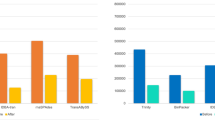
By comparing the transcriptomes from different life stages, we identified 115 genes involved in predation, including 37 venom-related, 23 aggression-related and 55 chemosensory-related genes (Table S9). The venom-related genes included esterase, trypsin-like protease, trypsin precursor, lipase, venom serine protease 34-like, serine carboxypeptidase 1, lysosomal acid phosphatase precursor, etc. It appeared that these genes exhibited low expression level in egg stage and relatively high expression level in nymph and adult stages. The transcript levels of trypsin-like protease, trypsin precursor and esterase showed the similar changing trend to that of the most venom-related genes, except esterase being low in 5th instar (Fig. 4A–C). The majority of these genes were enriched in hydrolase activity, catalytic activity and peptidase activity.

Temporal distribution of ten SDEGs involved in predation. (A–C) Three venom-related genes. (D–F) Three aggression-related genes. (G–J) Four olfactory-related genes. The transcript levels of SDEGs were calculated as the log10 FPKM of each comparison in transcriptome analysis and were shown on the y-axis.
The aggression-related genes included tyramine receptor, octopamine receptor, cyp6Q20, deaf1, metabotropic glutamate receptor B, progesterone, androgens, serotonin, etc. Apparently egg had the relatively low expression of these genes compared to other stages. As in the nymph stages, no obvious trends were observed regarding the expression of most aggression-related genes. 5th instar nymph had the relatively low expression of tyramine receptor (Fig. 4D) and highest expression level of octopamine receptor (Fig. 4E). Androgens and serotonin with a significant downregulated expression in the 3rd instar. Adult male had the higher expression of most aggression-related genes, e.g. tyramine receptor, metabotropic glutamate receptor B (Fig. 4F), deaf1, trp, progesterone, androgens, and serotonin (Table S9), than adult female. The enrichment GO categories were signal transducer activity, cell communication and G-protein coupled receptor signaling pathway.
We identified 22 OBPs, 17 CSPs, 11 olfactory receptors (ORs), four sensory neuron membrane proteins (SNMPs) and one pheromone-binding protein (PBP)-like transcripts. OBPs and CSPs exhibited relatively low expression in egg stage and high expression in nymph and adult stages (Fig. 4G,H). PBP exhibited relatively low expression in egg, 2nd instar and 5th instar, and showed high expression in the other life stages (Fig. 4I). SNMPs seemed to have a constant expression during all life stages (Fig. 4J). Adult male had the higher expression levels of most ORs than other stages. These genes were mainly enriched for odorant binding, response to stimulus, phosphorylation as well as in the olfactory receptor pathway.
Trend analysis
To understand the expression patterns of the 13,479 SDEGs, gene data from all life stages were clustered into 50 model profiles (Fig. S6). 4,183 genes represented 13 significant gene expression patterns, and 33 of 115 SDEGs related to predation could be clustered into five profiles with significance (p < 0.05). The genes assigned to most profiles showed variation in nymph stages and demonstrated a bias between genders, e.g. trypsin precursor, trypsin-like protease and esterase showing a bias towards females; metabotropic glutamate receptor B and ORs was observed as a male-biased expression.
qRT-PCR validation
To further confirm the quality of the transcriptome, we compared the expression patterns of the seven libraries, including those from 1st to 5th instar nymphs, male and female adults, of ten selected SDEGs (three venom-related, three aggression-related, four olfactory-related genes) involved in predation using qRT-PCR (Fig. 5). The expression trend of seven SDEGs in qRT-PCR analysis was consistent with that detected in the transcriptome analysis earlier (Figs 4 and 5). Inconsistencies were found in the three SDEGs, tyramine receptor, OBP and SNMP, which were different in some stages between qRT-PCR and transcriptome analysis. This difference in gene expression might be caused by the difference in the accuracy of these two assay methods. The SDEGs analyzed in this study were unigenes obtained from transcriptome that were assembled and mapped, while qRT-PCR showed a lower sensitivity than transcriptome sequencing. Nevertheless, qRT-PCR analysis confirmed the direction of change detected by transcriptome analysis, indicating that our results are reliable.

The dynamic expression patterns of ten SDEGs by using qRT-PCR. (A–C) Three venom-related genes. (D–F) Three aggression-related genes. (G–J) Four olfactory-related genes. The relative transcript level of each SDEGs was shown as the mean ± SE. Significant differences among different life stages were indicated by different letters (p < 0.05, t-test).
Discussion
In this study, we sequenced and characterized the transcriptome from the different life stages of A. cincticrus, with a particular focus on SDEGs involved in the development and the predation process. The transcripts displayed a variety of interesting differential expression patterns observed across all life stages. The characteristics of gene expression patterns between life stages and predation related unigenes were discussed below.
Genes involved in development
During its life cycle, A. cincticrus undergoes incomplete metamorphosis. Many crucial genes and transcriptional factors, such as nanos-like protein, hunchback and serine protease snake-like, participate in embryonic development of model organisms. For instance, the nanos and snake gene are core components of the germplasm and are involved in the formation of anterior-posterior body axis during early embryogenesis21. These genes were categorized in the GO terms of ‘catalytic activity’ and ‘hydrolase activity’, which are the essential first steps in the eggs activation cascade26. In this study, the high expression of genes related to embryonic development revealed the ongoing drastic cellular differentiation process in the eggs.
The nymphs should continually prey on ants to sustain the energy for molting through five nymph stages. They begin movement right after hatching from the eggs and feed on ants to store energy for development and various activities. Genes participating in muscle growth27 including histone H2A and genes pertaining to metabolic detoxification28 including glycosidases and cytochrome P450s were identified in the transcriptomes of nymphs. Glycosidases have been previously proposed to aid in the digestion of endosymbiont cell walls in Rhodnius prolixus 29. Cytochrome P450s are highly expressed in the “oxidoreductase activity”, and are known to be involved in steroid and lipid metabolism30 and can participate in insecticide resistance31. Insect molting is rigorously choreographed and is coordinated by fluctuations in the juvenile hormone (JH) and ecdysone. JH plays a key role in regulating development and metamorphosis32, while ecdysone-regulated genes play key roles during molting and metamorphosis of Oncopeltus fasciatus 21. We discovered that the expression of ecdysone-induced protein and juvenile hormone binding protein was high in the early nymphal stages. In general, the wings in reduviids become externally visible during the 2nd instar nymph, and they grow larger at each successive instar, then eventually they are fully developed when reduviids molt into adulthood33. We herein found notch, which is critical for wing disc development by secreting signal molecules34,35, were activated and highly expressed in the 2nd instar nymph of A. cincticrus.
In the final stage of development, adults reach sexual maturity followed by the behaviors like courtship, mating and oviposition. The hsps in Drosophila 36 potentially mediate the cellular response to thermal stress and indirectly influence the downregulation of sex steroid receptors, resulting in repressing the proliferation of growing follicles in proestrus, and therefore impact the organism’s fecundity and longevity37. Expression of hsps was lower in adult female than adult male. The KEGG enrichment term “oocyte meiosis” was enriched among the upregulated genes in the comparison of transcriptomes from male vs female adults, with vitellogenin highly expressed in the females. Vitellogenin encodes the vitellin in oocytes, and is expressed as an egg yolk precursor in female insects and sterile worker bees, as well as in other insects37.
Genes involved in predation
The predatory process of reduviids involves the following steps: arousing, locating, approaching, paralyzing, sucking, releasing and cleaning4. Neuroactive ligand-receptor interaction, wnt signaling pathway, glutathione metabolism, arginine and proline metabolism are important metabolic pathways in managing information and protein digestion. These processes are coordinated by a series of genes. SDEGs involved in predation were enriched in these pathways in nymphs and adults.
Because reduviids can only ingest liquid food, the salivary secretions play a critical role in paralyzing and feeding of their prey. The toxic protein content of venomous saliva in reduviids varies with respect to males and females38. Records of adult female reduviids paralyzing their prey more rapidly than adult males reinstate the above fact39. The esterase was abundant in the saliva of A. cincticrus may likely aid in paralysis of prey18. In the salivary glands, trypsin-like mRNA is dominant which is involved in cleaving the proteins at lysine and arginine residues40. The transcript levels of most SDEGs were low in eggs, relatively high in nymph and adult stages and exhibited a bias towards adult female. This result was consistent with the eggs being in a static state, while the nymphs feed on mass of ants for development and the females reserve energy to oviposition. The females have high expression of trypsin precursor, trypsin-like protease and esterase, leading to the stronger toxicity of the saliva than males and obtaining more ants due to their ability in paralyzing the prey more rapidly than the males. The activation and upregulation of these genes are critical for successful paralyzing and digestion of the prey.
Success rate of aggression can be influenced by several factors, such as physical disparities (e.g. size, strength and weapon)41,42, as well as physical exertion and experience in previous fights43,44. For example, different size of the parasitoid wasps has a different fighting propensity and weak-fighting ones usually stay hidden to avoid the stress of fighting45. Males usually are more likely to respond aggressively than females10 due to the limited opportunities to mate46. Monoamines, neuropeptides, and pheromones have been implicated as important neuromodulators for agonistic displaying in insects47. The expression of cyp6Q20 in olfactory sensory organs has also been reported to regulate hereditary-related and environmental-related aggression in Drosophila melanogaster48, suggesting a function in response to aggression-related stimuli. These results suggest that changes in octopamine receptor of nymph during an aggressive confrontation may be related to sufficient amounts of octopamine that induce the initiation, level and duration of aggressive behaviors49. We found that the metabotropic glutamate receptor B was highly abundant in A. cincticrus adult male, which is implicated in the regulation of aggression in honey bee11 and in the competition of reproduction in Nasonia vitripennis 9. The progesterone synthesized by the adrenocortical tissue was the highest in males, and may act indirectly on the brain after local conversion to other steroids that affect aggressive behavior. Allopregnanolone may be synthesized largely in males and is a neurosteroid that has been shown to play a role in regulating mammalian aggressive behavior50,51. These results of gene expression are consistent with the behavior that occurs in nature, i.e., A. cincticrus males exhibiting stronger aggressive behaviors against ants and other males to obtain food and win the mating chance. Similarly, A. cincticrus adults are generally larger in body size and inhabit in larger range of space suggesting they may exhibit stronger aggression than nymphs. Nymphs usually live underneath the stone, most often gregariously, likely consistent with their aggressive ability to avoid stress of fighting. Thus, it is not difficult to understand the camouflaging behavior in nymphs to protect themselves when facing danger or their enemies.
Insect predation is known to activate a series of mechanisms in order to locate prey, accompanied by sensitive identification of odor. Signal transmission from receptors to higher central nervous system rely on certain proteins, such as olfactory receptors (ORs) and OBPs52, that could discern stimulants of insects. For instance, green lacewing Chrysopa pallens use CpalOBP2 to detect and recognize the alarm pheromone in aphid species5. Hull et al.53 speculated that the plant bug Lygus lineolaris presented more OBP-like transcripts because of its broad host range. However, in pea aphid, there were relatively small number of OBPs owing to its parasitic lifestyle and specialized ecology54. Hekmat-Scafe et al.55 suggested that a moderate number of OBPs could act in a combinatorial manner with a moderate number of ORs to greatly increase the recognition power of an insect’s olfactory system. ORs were found highly expressed in male antennae of Asian corn borer due to its specific olfactory behavior and finding the emerging female56. Antennae-enriched CSPs may be involved in C. pallens identifying and binding volatile from pests, such as aphids, or pest-damaged plants57. Interestingly, the transcript abundance of these genes varied in different life stages of A. cincticrus. The transcripts were abundant in nymph and adult stages, suggesting a potential role in the detection and discrimination of odors. Surprisingly, greater sex bias was seen for some transcripts between genders, and OBPs were differentially expressed in nymph stages. The majority of these olfactory genes are highly expressed in the olfactory organ of female antennae than that of adult male, in contrast to their usual function for adult male to detect mates at long range, may be useful for females to detect pheromones released from ants in order to locate them accurately. Male-biased expression was observed in ORs in antennae, suggesting that they may mediate olfactory behavior specific to males, for example finding emerging adult female. The high expression of OBPs in A. cincticrus nymphs might suggest the unique functions in chemoreception. Compared with the egg stage, the nymphs and adults are active and had the need to detect odorants for their survival and reproduction in the environment.
Methods
Collection and colony maintenance
The colony of A. cincticrus used in this study was originally obtained from Yu County (39.9465°N, 114.9379°E), Hebei, China, then was maintained in a light incubator at 25 ± 2 °C, 60 ± 5% relative humidity (RH) under a 16 h: 8 h light/dark cycle. Specimens of eight life stages were collected as eggs, 1st to 5th instar nymphs, male and female adults. These samples were immediately frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
RNA isolation, cDNA library construction and Illumina sequencing
Total RNA was isolated using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocols. RNA integrity was assessed using the RNA Nano 6000 Assay Kit in the Agilent Bioanalyzer 2100 system (Agilent Technologies, USA). The extracted RNA was used to establish a cDNA library for transcriptome sequencing using an Illumina Hiseq. 2500 sequencer platform at Biomarker Technologies Corporation (Beijing, China).
Raw reads cleaning and quality control
In order to guarantee high-quality and clean reads, raw reads containing more than 5% unknown nucleotides and low-quality reads containing more than 50% of bases with a Q-value ≤ 20% were excluded from the sequence assembly. The clean reads were subjected to subsequent analyses and deposited in the NCBI SRA database under accession numbers: SRR5099968 (egg), SRR5099969-73 (1st to 5th instar nymphs), SRR5106081 (adult female) and SRR5106242 (adult male).
Assembly and functional gene annotation
The clean reads were assembled using Trinity (http://trinityrnaseq.sourceforge.net/) with default assembly parameters. Clean reads with a certain overlap length were initially combined to form long fragments without N (named contigs). Related contigs were clustered using the TGICL software58 to yield unigenes (without N) that cannot be extended on either ends, and redundancies were removed to acquire non-redundant unigenes.
Subsequently, non-redundant unigenes were analyzed and annotated. Unigenes were mapped against the NCBI-NR, SwissProt and COG databases using the BLASTX algorithm with an e-value cut-off of 10−5 to retrieve functional annotations based on sequence similarity. High-priority databases (followed by NBCI-NR, SwissProt, and KEGG) were selected to determine the direction of the unigene sequences. According to the best alignment results, the coding sequences were generated. The ESTScan software59 was used to determine the sequence direction of the unigenes that could not be aligned to any of the above databases. Functional gene annotations were collected for all unigene sequences ≥ 150 bp using Blast2GO60. To determine the distribution of gene functions at the macro level, Blast2GO was used to retrieve the GO terms for each sequence by searching the NCBI-NR database. The WEGO software61 was used to plot the distribution of the GO functional classification of the unigenes. Functional pathway analysis was performed using the KAAS webserver62 from KEGG.
Differential gene expression and trend analysis
Gene expression levels were calculated by RSEM63. The read counts were adjusted by the eDEGR program package through one scaling normalized factor. The DEGSeq package was used to analyze the gene expression difference between two groups. DEGSeq provided statistical routines for determining differential expression in unigenes using a model based on the negative binomial distribution. P-values were adjusted using the Benjamini-Hochberg’s approach for calculating the false discovery rate (FDR). Fragments per kilobase of exon per million fragments mapped (FPKM) was used to quantify the expression level of unigenes. Genes with an adjusted p < 0.05, FDR ≤ 0.01 and the absolute value of log2 Fold change (FC) ≥ 1 were set as the thresholds to identify the SDEGs. FC is the ratio of FPKM between the two samples. GO enrichment analysis of SDEGs was carried out based on an algorithm provided by GOstat64, with the complete annotation results set as the background. The p-value was approximated using the Chi-square test. Fisher’s exact test was used when any expected value was below 5. This program was implemented as a pipeline65.
STEM (Short Time-Series Expression Miner) software was used to profile gene expression time series and identify significant expression tendencies66 in these life stages based on FPKM values. Based on different signal density of genes under different situations, we identified a set of unique model expression tendencies. The raw expression values were converted into log2 ratios. Some unique profiles were further defined using a strategy for clustering short time-series gene expression data. Each model profile includes the actual or the expected number of genes. Fisher’s exact test and multiple-comparison tests were used to determine whether the significant profiles had higher probability than expected67.
qRT-PCR validation
To validate the quality of the transcriptome data and estimate the relative expression, qRT-PCR was performed using the SYBR Premix Ex Taq (Takara, Japan) according to the manufacture’s protocols and a CFX96 real-time PCR detection system (Bio-Rad, USA). Primer sequences of selected genes were designed by DNAMAN and were listed in Table S10. Total RNA was extracted as described in the RNA isolation and cDNA was synthesized by Promega Goscript Reverse Transcription Systems (Fisher Scientific, USA). All qRT-PCR reactions had three biological and three technical replicates. The average threshold cycle (Ct) was calculated from values in all six replicates per sample. The actin gene was chosen as an endogenous control to normalize expression between different samples, as has been used in other insects68. The 2−△△CT method69 was used to evaluate the relative gene expression. All data were statistically analyzed by independent sample students t-test using SPSS 17.0. The relative expression level of each unigene was presented as mean ± standard error (SE).
References
Cao, L. M., Rédei, D., Li, H. & Cai, W. Z. Revision of the genus Acanthaspis Amyot & Serville (Hemiptera: Heteroptera: Reduviidae: Reduviinae) from China, with new records of species to adjacent countries. Zootaxa 3892, 1–66 (2014).
Weirauch, C. Anatomy of disguise: camouflaging structures in nymphs of some Reduviidae (Heteroptera). Amer. Mus. Novi. 3542, 1–18 (2006).
Eisner, T., Hicks, K., Eisner, M. & Robson, D. S. “Wolf-in-sheep’s-clothing” strategy of a predaceous insect larva. Science 199, 790–794 (1978).
Li, H., Zhao, G. Y., Cao, L. M., Xu, K. & Cai, W. Z. Taxonomic and bionomic notes on the white spot assassin bug Platymeris biguttatus (Linnaeus) (Hemiptera: Reduviidae: Reduviinae). Zootaxa 2644, 47–56 (2010).
Li, Z. Q. et al. Odorant-binding proteins display high affinities for behavioral attractants and repellents in the natural predator Chrysopa pallens. Comp. Biochem. Phys. A. 185, 51–57 (2015).
Roeder, T., Seifert, M., Kähler, C. & Gewecke, M. Tyramine and octopamine: antagonistic modulators of behavior and metabolism. Arch. insect Biochem. 54, 1–13 (2003).
Cohen, A. C. Organization of digestion and preliminary characterization of salivary trypsin-like enzymes in a predaceous heteropteran Zelus renardii. J. Insect Physiol. 39, 823–829 (1993).
Lorenz, K. Z. On Aggression. Harcourt, Brace & World, New York (1966).
Tsai, Y. J. J., Barrows, E. M. & Weiss, M. Pure self-assessment of size during male-male contests in the parasitoid wasp Nasonia vitripennis. Ethology 120, 816–824 (2014).
Tsai, Y. J. J., Barrows, E. M. & Weiss, M. Why do larger and older males win contests in the parasitod wasp Nasonia vitripennis? Anim. Behav. 91, 151–159 (2014).
Alaux, C. et al. Honey bee aggression supports a link between gene regulation and behavioral evolution. P. Natl. Acad. Sci. USA 106, 15400–15405 (2009).
Castro, V. D., Martínlópez, M. & Navarro, J. F. Effects of Ly35740, a selective agonist for glutamate metabotropic receptors of group II, on aggressive behavior in mice. Univ. Psychol. 9, 617–625 (2010).
Cohen, A. C. Review: extra-oral digestion in predaceous terrestrial arthropod. Annu. Rev. Entomol. 40, 85–103 (1995).
Castilla, A. M., Huey, R. B., Calvete, J. J., Richer, R. & Al-Hemaidi, A. H. M. Arid environments: opportunities for studying co-evolutionary patterns of scorpion venoms in predator-prey systems. J. Arid Environ. 112, 165–169 (2015).
Ambrose, D. P. & Maran, S. P. M. Polymorphic diversity in salivary and haemolymph proteins and digestive physiology of assassin bug Rhynocoris marginatus (Fab.) (Het., Reduviidae). J. Appl. Entomol. 124, 315–317 (2000).
George, P. J. E. & Ambrose, D. P. Polymorphic adaptive insecticidal resistance in Rhynocoris marginatus (Fabr.) (Het., Reduviidae) a non-target biocontrol agent. J. Appl. Entomol. 125, 207–209 (2001).
Edwards, J. S. The action and composition of the saliva of an assassin bug Platymeris rhadamanthus Gaerst. (Hemiptera, Reduviidae). J. Exp. Biol. 38, 61–77 (1961).
Evangelin, G., Bertrand, H., Muthupandi, M. & John, W. S. Venomous saliva of non-haematophagous reduviid bugs (Heteroptera: Reduviidae): a review. Biolife 2, 615–626 (2014).
Yang, C. H., Yang, P. C., Li, J., Yang, F. & Zhang, A. B. Transcriptome characterization of Dendrolimus punctatus and expression profiles at different developmental stages. PLoS ONE 11, e0161667 (2016).
Li, L. T., Zhu, Y. B., Ma, J. F., Li, Z. Y. & Dong, Z. P. An analysis of the Athetis lepigone transcriptome from four developmental stages. PLoS ONE 8, e73911 (2013).
Ewen-Campen, B. et al. The maternal and early embryonic transcriptome of the milkweed bug Oncopeltus fasciatus. BMC Genomics 12, 61 (2011).
Su, S. Y. et al. Transcriptomic analysis of EGb 761-regulated neuroactive receptor pathway in vivo. J. Ethnopharmacol. 123, 68–73 (2009).
Gibson, J. D., Niehuis, O., Verrelli, B. C. & Gadau, J. Contrasting patterns of selective constraints in nuclear- encoded genes of the oxidative phosphorylation pathway in holometabolous insects and their possible role in hybrid breakdown in Nasonia. Heredity 104, 310–317 (2010).
Erneux, C., Govaerts, C., Communi, D. & Pesesse, X. The diversity and possible functions of the inositol polyphosphate 5-phosphatases. Biochim. Biophys. Acta 1436, 185–199 (1998).
Neurath, H. Evolution of proteolytic enzymes. Science 224, 350–357 (1984).
Horner, V. & Wolfner, M. Transitioning from egg to embryo: triggers and mechanisms of egg activation. Dev. Dynam. 237, 527–544 (2008).
Li, S. W. et al. Transcriptome and gene expression analysis of the rice leaf folder, Cnaphalocrosis medinalis. PLoS ONE 7, e47401 (2012).
Sparks, M. E., Shelby, K. S., Kuhar, D. & Gundersen-Rindal, D. E. Transcriptome of the invasive brown marmorated stink bug, Halyomorpha halys (Stål) (Heteroptera: Pentatomidae). PLoS ONE 9, e111646 (2014).
Ribeiro, J. M. C. & Pereira, M. E. A. Midgut glycosidases of Rhodnius prolixus. Insect Biochem. 14, 103–108 (1984).
Feyereisen, R. Insect P450 enzymes. Annu. Rev. Entomol. 44, 507–533 (1999).
Enayati, A. A., Ranson, H. & Hemingway, J. Insect glutathione transferases and insecticide resistance. Insect Mol. Biol. 14, 3–8 (2005).
Staal, G. B. Insect growth regulators with juvenile hormone activity. Entomology 20, 417–460 (1975).
Truman, J. W. & Riddiford, L. M. The origins of insect metamorphosis. Nature 401, 447–452 (1999).
Tabata, T. & Takei, Y. Morphogens, their identification and regulation. Development 131, 703–712 (2004).
de Celis, J. F. & Garcia-Bellido, A. Roles of the Notch gene in Drosophila wing morphogenesis. Mech. Dev. 46, 109–122 (1994).
Hu, J. T., Chen, B. & Li, Z. H. Thermal plasticity is related to the hardening response of heat shock protein expression in two Bactrocera fruit flies. J. Insect Physiol. 67, 105–113 (2014).
Zhu, J., Indrasith, L. S. & Yamashita, O. Characterization of vitellin, egg-specific protein and 30 kDa protein from Bombyx eggs, and their fates during oogenesis and embryogenesis. Biochim. Biophys. Acta (BBA) Gen. Sujects 882, 427–436 (1986).
Sahayaraj, K. & Vinothkanna, A. Insecticidal activity of venomous saliva from Rhynocoris fuscipes (Reduviidae) against Spodoptera litura and Helicoverpa armigera by microinjection and oral administration. J. Venom. Anim. Toxins. 17, 486–490 (2011).
Sahayaraj, K., Kumara, S. S. & Balasubramaniam, R. Prey influence on the salivary gland and gut enzymes qualitative profile of Rhynocoris marginatus (Fab.) and Catamirus brevipennis (Serville) (Heteropetera: Reduviidae). J. Entomol. 4, 331–336 (2007).
Law, J. H., Dunn, P. E. & Kramer, K. J. Insect proteases and peptidases. Adv. Enzymol. 45, 389–425 (1977).
Arnott, G. & Elwood, R. W. Assessment of fighting ability in animal contests. Anim. Behav. 77, 991–1004 (2009).
Briffa, M. Decisions during fights in the house cricket, Acheta domesticus: mutual or self assessment of energy, weapons and size? Anim. Behav. 75, 1053–1062 (2008).
Brown, W. D., Smith, A. T., Moskalik, B. & Gabriel, J. Aggressive contests in house crickets: size, motivation and the information content of aggressive songs. Anim. Behav. 72, 225–233 (2006).
Stevenson, P. A. & Schildberger, K. Mechanisms of experience dependent control of aggression in crickets. Curr. Opin. Neurobiol. 23, 318–323 (2013).
Hartley, C. S. & Matthews, R. T. The effect of body size on male-male combat in the parasitoid wasp Melittobia digitata Dahms (Hymenoptera: Eulophidae). J. Hymenopt. Res. 12, 272–277 (2003).
Barry, K. L. & Kokko, H. Male mated choice: why sequential choice can make its evolution difficult. Anim. Behav. 80, 163–169 (2010).
Bubak, A. N., Grace, J. L., Watt, M. J. & Renner, K. J. & Swallow, J. G. Neurochemistry as a bridge between morphology and behavior: perspectives on aggression in insects. Curr. Zool. 60, 778–790 (2014).
Wang, L., Dankert, H., Perona, P. & Anderson, D. J. A common genetic target for environmental and heritable influences on aggressiveness in Drosophila. P. Natl. Acad. Sci. USA 105, 5657–5663 (2008).
Grohmann, L. et al. Molecular and functional characterization of an octopamine receptor from honeybee (Apis mellifera) brain. J. Neurochem. 86, 725–735 (2003).
Stevenson, P. A., Hofmann, H. A., Schoch, K. & Schildberger, K. The fight and flight responses of crickets depleted of biogenic amines. J. Neurobiol. 43, 107–120 (2000).
Fish, E. W., DeBold, J. F. & Miczek, K. A. Aggressive behavior as a reinforcer in mice: activation by allopregnanolone. Psychopharmacology 163, 459–466 (2002).
Fan, J., Francis, F., Liu, Y., Chen, J. L. & Cheng, D. F. An overview of odorant-binding protein functions in insect peripheral olfactory reception. Genet. Mol. Res. 10, 3056–3069 (2011).
Hull, J. J., Perera, O. P. & Snodgrass, G. L. Cloning and expression profiling of odorant-binding proteins in the tarnished plant bug, Lygus lineolaris. Insect Mol. Biol. 23, 78–97 (2014).
Zhou, J. J. et al. Genome annotation and comparative analyses of the odorant-binding proteins and chemosensory proteins in the pea aphid Acyrthosiphon pisum. Insect Mol. Biol. 19, 113–122 (2010).
Hekmat-Scafe, D. S., Scafe, C. R., McKinney, A. J. & Tanouye, M. A. Genome-wide analysis of the odorant-binding protein gene family in Drosophila melanogaster. Genome Res. 12, 1357–1369 (2002).
Zhang, T. T. et al. Male- and female- biased gene expression of olfactory-related genes in the antennae of Asian corn borer, Ostrinia furnacalis (Guenée) (Lepidoptera: Crambidae). PLoS ONE 10, e0128550 (2015).
Li, Z. Q. et al. First transcriptome and digital gene expression analysis in Neuroptera with an emphasis on chemoreception genes in Chrysopa pallens (Rambur). PLoS ONE 8, e67151 (2013).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Iseli, C., Jongeneel, C. V. & Bucher, P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 99, 138–148 (1999).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, 293–297 (2006).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, 182–185 (2007).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Beißbarth, T. & Speed, T. P. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 20, 1464–1465 (2004).
Chen, S. et al. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS ONE 5, e15633 (2010).
Ernst, J. & Bar-Joseph, Z. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics 7, 191 (2006).
Ramoni, M. F., Sebastiani, P. & KohaneI, S. Cluster analysis of gene expression dynamics. P. Natl. Acad. Sci. USA 99, 9121–9126 (2002).
Zou, D. Y. et al. Nutrigenomics in Arma chinensis: transcriptome analysis of Arma chinensis fed on artificial diet and Chinese oak silk moth Antheraea pernyi pupae. PLoS ONE 8, e60881 (2013).
Livak, K. J. & Schmitten, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△CT method. Methods 25, 402–408 (2001).
Acknowledgements
This work was funded by the National Basic Research Program of China (No. 2013CB127600), the National Key Technology R & D Program of the Ministry of Science and Technology (No. 2012BAD19B00) and the Special Fund for Scientific Research (No. 2012FY111100).
Author information
Authors and Affiliations
Contributions
F.K., H.L. and W.C. designed and performed the research. F.K. and Y.Z. collected samples. F.K. and Y.Z. performed the molecular work. F.K. and H.L. analyzed the data. All authors discussed results and implications. F.K., H.L., S.L., Y.Z., X.Z. and W.C. wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kou, F., Li, H., Li, S. et al. Temporal transcriptomic profiling of the ant-feeding assassin bug Acanthaspis cincticrus reveals a biased expression of genes associated with predation in nymphs. Sci Rep 7, 12691 (2017). https://doi.org/10.1038/s41598-017-12978-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12978-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
