
Abstract
Dynamic protein-protein interactions (PPIs) play crucial roles in cell physiological processes. The protein-fragment complementation (PFC) assay has been developed as a powerful approach for the detection of PPIs, but its potential for identifying protein interacting regions is not optimized. Recently, an ascorbate peroxidase (APEX2)-based proximity-tagging method combined with mass spectrometry was developed to identify potential protein interactions in live cells. In this study, we tested whether APEX2 could be employed for PFC. By screening split APEX2 pairs attached to FK506-binding protein 12 (FKBP) and the FKBP12-rapamycin binding (FRB) domain, which interact with each other only in the presence of rapamycin, we successfully obtained an optimized pair for visualizing the interaction between FRB and FKBP12 with high specificity and sensitivity in live cells. The robustness of this APEX2 pair was confirmed by its application toward detecting the STIM1 and Orial1 homodimers in HEK-293 cells. With a subsequent mass spectrometry analysis, we obtained five different biotinylated sites that were localized to the known interaction region on STIM1 and were only detected when the homodimer formed. These results suggest that our PFC pair of APEX2 provides a potential tool for detecting PPIs and identifying binding regions with high specificity in live cells.
Similar content being viewed by others


Introduction
Protein-protein interactions are dynamic and are precisely regulated to conduct many physiological functions within cells. Dysregulated interactions are related to dysfunction and lead to many diseases, including cancer, diabetes and neurodegenerative disorders1,2,3,4. Mapping the comprehensive protein interactome and identifying the key interaction regions between binding partners in health and disease are major tasks for life science investigations.
Proximity-dependent biotin labeling by modifying enzymes, such as tyramide5,6, ascorbate peroxidase (APEX)7,8,9, and BirA10, is a recently developed method for protein-protein interaction screening that addresses this challenge. APEX2, with quadruple mutants of soybean APEX, has an improved enzyme activity for biotinylating proteins within 20 nm and can increase electron microscope (EM) contrasts by catalyzing the polymerization of 3,3′-diaminobenzidine (DAB)7,9. When fused to the protein of interest, APEX2, in the initiation of hydrogen peroxide, can catalyze biotin-phenol to produce a biotin-phenol radical that is unstable and can covalently bind to neighboring proteins, which allows their subsequent purification and identification by mass spectrometry (MS).
Protein fragment complementation (PFC) assays have been widely used and shown to be powerful in the detection of protein-protein interactions in intact cells and in live animals11,12,13. All PFCs that are based on easy readouts, such as enzyme activity, transcription activation state or fluorescence, require stable PPIs for reconstitution11,14,15. Although they have several advantages in the detection of the interaction in the live state, none of them can directly label the interaction partners. The recently developed split HRP, which can catalyze biotinylation in live cells, has potential for direct labeling but is not suitable for interactions within cells, a consequence of spontaneous reconstitution in the endoplasmic reticulum (ER)16.
For the identification of the key regions between a protein and its partners, several approaches were developed including the X-ray crystallography, NMR spectroscopy and cross-linking of proteins combined with mass spectrometry. However, the study of the physiological binding feature still faces multiple challenges, such as the complexity of the sample purification procedures17.
In this study, taking the advantages of the APEX2 enzyme, we carried out a screen for an optimized PFC of APEX2 to enhance the specificity of the catalytic activity of the enzyme. Employing a rapamycin-induced interaction pair, FRB and FKBP12, we screened truncated APEX2 mutants based on structure and finally obtained one optimized complementary pair that lost enzymatic activity when separated and regained it only when they were brought closer together. We also validated the specificity of this strategy in two independent experiments, using homodimers of STIM1 and Orai1. Although they both can form self-associated homodimers independently of the activation state, only STIM1 can bring the PFC to reconstitute the APEX2 activity. The failure to reconstitute the activity by the Orai1 homodimer could be attributed to its C-terminal steric constraints. By use of streptavidin-based affinity purification and subsequent tandem MS, we successfully identified 18 biotinylated peptides in STIM1. More importantly, these biotinylation sites were highly enriched within the known interaction region on STIM1. Thus, this strategy is a useful tool for uncovering protein-protein interactions under specific conditions and mapping the interface of the complex in living cells.
Results
Screening the PFC of APEX2 through the rapamycin-induced protein interaction
The rapamycin-binding domain of the mammalian target of rapamycin (FRB) can bind to FK506-binding protein 12 (FKBP) in the presence of rapamycin with high affinity18. Employing this property, we constructed sets of truncations for optimizing the APEX2 complementary pairs which fused to FRB and FKBP12. Basically, the different truncation pairs that fused to the FRB or FKBP12 were spaced by a linker12. Because the APEX2 enzyme can catalyze the H2O2-dependent polymerization of DAB, leading to localized precipitation19,20, it is easy to visualize the complementation by common light microscopy (Fig. 1A). The criterion is that DAB staining is exclusively produced when the PFC of APEX2 bind together in the presence of rapamycin, while there is no detectable signal when expressed alone or when both are expressed but without rapamycin.

Optimization of the N-terminal APEX2(NA)/C-terminal APEX2(CA) complementation pair in cells. (A) Schematic of protein-fragment complementation (PFC) of APEX2. The interaction between proteins 1 (P1) and 2 (P2) brings inactive NA and CA into close proximity to reconstitute the peroxidase activity. This enzyme can biotin-label neighboring proteins within 20 nm for MS and catalyze the polymer of DAB for EM imaging. (B) The initial PFCs fused with FRB-flag and myc-FKBP12 to generate incremental truncation libraries. (C) Structure of wild-type soybean ascorbate peroxidase (1OAG). The NA and CA fragments are highlighted in green and yellow, respectively. (D) DAB staining for the rapamycin-induced FRB/FKBP12 association in live cells. HEK-293 cells were transfected with FRB-flag-NA and CA-myc-FKBP12, followed by treatment with rapamycin or DMSO as indicated. DAB staining was performed as described. Scale bars, 10 μm. (E) Biotin labeling was tested using the confocal image. HEK-293 cells were transfected with different plasmids as indicated and treated with rapamycin or DMSO. Biotin labeling was initiated with H2O2 following biotin-phenol incubation. Scale bars, 10 μm. (F) Western blot to test biotin labeling in the live cells. Cells were treated as in (E). The whole lysates were resolved by 10% SDS-PAGE and tested by streptavidin-HRP for biotinylated proteins, anti-flag antibody for FRB-NA and anti-myc antibody for CA-FKBP12. Equal amount of protein in each lane was loaded as quantified by β-actin. The original blots were presented in the Supplementary Fig. S3.
The crystal structures of APEX from the pea and soybean demonstrate the heme-binding sites and ascorbate-binding sites form the key catalytic region21,22,23. The Cys32 and Arg172 residues are ascorbate-binding sites, and there are 14 helices and 4 beta sheets in the whole APEX2 (1OAG) protein secondary structure23. Based on this information, we began with four N-terminal truncations (NA151, NA181, NA201 and NA240), all of which avoided disruptions to the helices, except NA240, together with two C-terminal truncations (CA102 and CA202) that had some overlap with the N-terminal mutants (Fig. 1B). We were lucky to observe that the NA201 and CA202 pairs worked well, while the others were not satisfactory (Supplementary Fig. S1). To determine if there were any other sites suitable for the PFC, we carried out a second screening around NA201 and Arg 172, which played a major role in the stabilization of substrate binding (Supplementary Fig. S1). In the end, we obtained 3 NAs (NA201, NA215, NA230) that could reconstitute the enzyme with CA202 without spontaneous activity. Among these, we selected the NA201/CA202 pair, hereafter referred to as NA/CA, which had the highest enzyme activity with the lowest background.
The optimized NA and CA are indicated in green and yellow in the map of the APEX2 structure (Fig. 1C). There were significant DAB signals in the HEK-293 cells co-expressing the FRB-flag-NA/CA-myc-FKBP12 in the presence of rapamycin, while few detectable signals over the background were evident with the DMSO treatment (Fig. 1D). We then tested the biotinylation ability of this pair. The biotinylated proteins in the HEK-293 cells expressing both FRB-flag-NA and CA-myc-FKBP12 were predominantly detectable with streptavidin-cy5 only in the presence of rapamycin compared to the cells treated with DMSO (Fig. 1E). These results were confirmed by a western blot experiment with a streptavidin-HRP probe (Fig. 1F). The expression levels of FRB-flag-NA/CA-myc-FKBP12 were tested using anti-flag and anti-myc antibodies, respectively (Fig. 1F). These results confirmed that the NA/CA pair had specific enzymatic activity in both DAB staining and biotin labeling. We then applied the pair to two other independent cases of protein-protein interaction to examine the specificity of our complementary fragments of APEX2 for the detection of dimerization.
Detection of homodimer interaction
To lend support to the specificity of the APEX2 pair, we checked its performance in two cases: homodimers of stromal interacting molecule 1 (STIM1) and Orai1, respectively.
STIM1 Homodimerization
STIM1 is a key modulator of the store operated calcium channel entry (SOCE) at ER. It acts as an ER-resident Ca2+ sensor that can be activated by the depletion of ER Ca2+ stores24. STIM1 was reported as a dimer in the resting and activated states25. Because the C-terminus is composed of flexible fragments, we tested whether this could help the reassembly of the enzyme. We fused NA or CA to the cytoplasmic C-terminus of STIM1 to construct STIM1-NA and STIM1-CA (Fig. 2A,B). When we co-transfected the constructs into HEK-293 cells, there was a significant DAB signal both in the resting condition and after depletion of Ca2+ stores with thapsigargin (TG), while no signal was detectable over the background with single mutant expression (Fig. 2C upper). For comparison, we transfected the cells with STIM1-APEX2 and detected a strong signal condensed below the plasma membrane and some mild signals in the cytoplasm (Fig. 2C bottom, left). However, the signal produced by the STIM1-NA/STIM1-CA was more specific.

Examination of homo-dimerization of STIM1 using protein-fragment complementation (PFC) of APEX2. (A) Schematic of STIM1-NA, STIM1-CA, STIM1-APEX2, ORAI1-NA, ORAI1-CA and ORAI1-APEX2. APEX2 and NA are tagged with flag, while CA is tagged with myc. (B) Schematic of the topology of STIM1-PFC and ORAI1-PFC of APEX2. (C) HEK-293 cells were transfected with various plasmids as indicated. NA indicates STIM1 or Orai1 fused with flag-NA, CA indicates STIM1 or Orai1 fused with CA-myc. Cells were treated with TG or 2Ca2+ for 15 minutes. DAB staining was performed as in Fig. 1(D). Scale bar: 10 μm. (D) HEK-293 cells plated on glass were transfected with different plasmids as indicated. Live cells were stimulated with TG or DMSO as above. Biotin labeling was initiated as in Fig. 1(E). Scale bar: 10 μm.
To evaluate the biotin labeling efficiency, we incubated the cells expressing the PFC of APEX2 with biotin-phenol (BP) and initiated with H2O2. A similar distribution of the biotinylated proteins was produced to that of DAB in cells co-expressing both STIM1-NA and STIM1-CA with or without TG; there was no detectable signal in the cells expressing either mutant alone. The localized staining pattern was apparent compared to the widely dispersed signal produced by cells expressing STIM1-APEX2 (Fig. 2D). An independent western blot experiment further confirmed the biotinylation capability reconstituted by STIM1-NA and STIM1-CA (Fig. 3A). These results further demonstrate that the complementary fragments can reconstitute the APEX2 enzyme when they are brought together in close proximity.

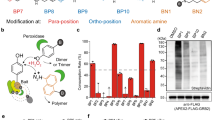
Biotinylation analysis of reconstituted enzyme by STIM1-NA/CA. (A) The cells were transfected with the different plasmids as indicated. Biotinylation was initiated as in Fig. 1(E). Whole cell lysates were resolved by 10% SDS-PAGE, and detected by western blot using streptavidin-HRP, anti-flag, anti-myc and anti-actin (left). The original blots were presented in the Supplementary Fig. S4. Enrichment was performed with streptavidin-beads, and proteins were separated by SDS-PAGE gel (right). These experiments were separately repeated twice. (B) Example of an MS/MS spectrum for a peptide with biotin-phenol on tyrosine. Fragment ions assigned to both y-ions and b-ions are labeled. (C) Schematic of the topology of different domains in STIM1. Red letters a-e indicate the approximate positions of identified biotin-labeled sites. (D) The biotin-labeled sites and their counts of the MS2 spectra. Each existing biotinylated amino acid is shown in red. Single*: cells expressed only STIM1-NA or STIM1-CA; Both*: cells expressed both STIM1-NA/STIM1-CA, which were in 2 mM calcium.
Orai1 homodimer
Orai1, located on the plasma membrane, is another crucial pore-forming component of the SOCE channel. Orai1 has been reported as a dimer in the resting state and a tetramer or hexamer when activated26,27,28. The structure-function study shows that each Orai1 has 4 transmembrane helices (TM1-4) which are linked with the N and C extensions that face the cytosol. TM1 lines the pore, while TM4 and the downstream C-terminus are located at the periphery of the gate with an anti-parallel orientation between two isoforms28. We fused the NA and CA to the C-terminus of Orai1 to check whether or not the far distance of the Orai1 C- terminus would prevent reconstitution of the enzyme activity (Fig. 2A,B). Both the DAB staining and biotin labeling experiment showed that no signal was detectable when Orai1-NA and Orai1-CA were co-expressed with or without TG stimulation, while cells expressing Orai1-APEX2 exhibited a strong signal mainly at the plasma membrane (Fig. 2C,D). These results demonstrate that the complementary pair can not reconstitute the enzyme when fused to the Orai1 C-terminus. The failure to reconstitute the enzyme may result from the large distance between the APEX2 pairs fused to Orai1; it is suggested from the crystal structure of Orai1 that the C-termini of the neighboring Orai1s form an alternating pair28,29. These results provide further evidence that the reconstitution by the PFC of APEX2 requires a close distance and orientation match in three-dimensional space.
Biotin enrichment and identification of the binding sites in the STIM1 homodimer
To further exploit the advantages of APEX2-based proximity-labeling, we aimed to identify the nearby proteins of the STIM1 dimer through biotin labeling, affinity purification using streptavidin beads and subsequent MS analysis. The biotinylation efficiency was verified by western blot with HRP-streptavidin and further confirmed by affinity enrichment using streptavidin beads (Fig. 3A). The gels were subjected to in-gel digestion with trypsin, and the resulting peptides were analyzed by MS. All results for the biotinylated peptides were confirmed by manually checking the MS/MS spectra. In total, 36 biotinylated peptides were specifically identified in STIM1-NA/STIM1-CA co-expressing cells, rather than in either the STIM1-NA or STIM1-CA expressing cells, of which there were 23 peptides belonged to STIM1 itself, representing five different biotinylated sites (Supplementary Table S1). This indicated that the high specificity of biotinylation catalyzed by the reassembled peroxidase. Moreover, further mapping of these five biotinylated sites revealed that all of them were localized to the cytoplasmic domains of STIM1 (Fig. 3C). While one site was located in the C-terminal flexible region (518hSDSESSLHmSDR), the other four sites, located in the 316-to-394 amino acid region, were sterically adjacent to each other according to the reported STIM1 fragment structure of CC1-CC2 and SOAR30. These results strongly demonstrate that the complementary STIM1-NA and STIM1-CA can fully reconstitute the peroxidase activity and biotinylate proteins that are nearby.
Discussion
Protein-protein interactions play vital roles in the life of the cell. The PFC assay is a useful strategy that can provide high specificity and sensitivity because of its absolute requirement for the correct three-dimensional structure of a resultant fragment hybrid11. In this study, we developed a PFC of APEX2 based on a readily observable biological readout, DAB staining. To achieve this, we fused N-terminal APEX2 (NA) to FRB and C-terminal APEX2 (CA) to FKBP12. The NA and CA do not spontaneously reassemble. The interaction between FRB and FKBP12 induced by rapamycin can force the reconstitution of the two halves of APEX2, leading to full enzyme activity that can biotinylate neighboring proteins and catalyze the DAB polymer(Fig. 1, Supplementary Fig. S2). We identified 3 NAs (NA201, NA215, NA230) that could complement to CA202.
The 201th amino acid lies in the 14th turn between the 11th and 12th alpha helix, the 215th amino acid lies in the 15th turn, and the 230th amino acid lies at the beginning of the 14th alpha helix. Other N-terminal mutants that were larger than these three could not show the enzyme activity (Supplementary Fig. S1) when coupled with CA202. These results demonstrate that the correct secondary structure of the N-terminus is essential for its function. It is thus essential to choose the proper truncation site that will not disrupt the 3D structure of the PFC pair.
SOCE mediated by STIM1 and ORAI1 plays a critical role in calcium homeostasis in most mammalian cells. Both STIM1 and ORAI1 can form homodimers in the resting and active states. We fused the complementary APEX2 pair to each of their C-termini and obtained a space-limited signal in the STIM1 homodimer but not in the ORAI1 homodimer, suggesting the specificity of the PFC of APEX2. Many studies using the STIM1 cytosolic domain have made clear that the CCI and SOAR regions are responsible for the intermolecular interaction and that the C-terminal region remains flexible31,32. These features are consistent with the successful reconstitution of enzyme activity by attaching the PFC pair of APEX2 to the C-terminus of STIM1 and with biotinylation near the interaction region. Although the Orai1 homodimer may bring the complementary fragments close in the resting and activate states, the C-terminus of Orai1 that forms alternating pairs may enlarge the distance of the complementary pairs, thereby leading to the failed reassembly of the enzyme activity.
The limitation of PFC of APEX2 is that the reconstituted enzyme activity may be relatively lower than APEX2. We did not identify STIMATE, a protein reported to physically bind to the STIM1 juxtamembrane CC1 instead of the SOAR domain. This is probably because the distance between STIMATE and STIM1 is beyond the active region of the reconstituted enzyme33. In addition, the localization of the complemented pair, expression level and the expression ratio should be considered when assessing the results.
In conclusion, the PFC of APEX2 that we identified can reconstitute full enzymatic activity when the domains are directed in proximity to each other. This strategy enhances the specificity of the proximity-tagging method where biotinylation only occurs at the right time and the right site when the protein complex is formed. By taking advantage of the unstable nature of the phenoxyl radical generated during oxidation of the reconstituted enzyme, we can obtain the limited interaction regions between proteins under a specific condition in live cells.
Methods
Chemicals and reagents
PCDNA3-flag-APEX2 was purchased from Addgene (Cambridge). The following reagents were purchased from Sigma: rapamycin, DAB, 30% (w/w) hydrogen peroxide (H2O2), the anti-flag antibody, and standard reagents for gel electrophoresis and western blotting including Ponceau S HCl, urea, biotin, the 100x protease inhibitor cocktail, sodium deoxycholate, sodium dodecyl sulfate, sodium ascorbate, Trolox, and sodium azide. The anti-myc antibody was purchased from Cell Signaling Technology. Streptavidin-HRP was purchased from CWBIO. Biotin-phenol was purchased from Iris-Biotech (CAS-NO: 41994-02-9). Lipofectamine 2000 was purchased from Life Technologies. Coomassie and sequencing grade modified trypsin were purchased from Promega. Streptavidin magnetic beads were purchased from Pierce (Catalog No. 88817).
Construction of incremental truncations and interaction protein pairs
The initial N- and C-terminal fragments of APEX2 were amplified from PCDNA3-flag-APEX2. The rapamycin-binding domain (FRB) of human mTOR and human FK506-binding protein 12 (FKBP) were generated by PCR amplification from plasmids in our lab. A flexible linker was added as described12. Briefly, to separate the mutant and targets, we introduced linker 1 (QISYASRGGGSSGGG) for NA and linker 2: (GGGSSGGGQISYASRG) for CA. All mutants and the wild-type flag-APEX2 were amplified by PCR to tag targets as indicated.
Cell culture, transfection and treatment
HEK-293 cells were cultured in DMEM supplemented with 10% FBS at 37 °C with 5% CO2. Transfections were performed as described in the protocol. Briefly, cells were seeded to different plates, and the next day, the DNA and Lipofectamine were mixed and added to the cells. Forty hours later, expression was detected by western blot or immunofluorescence. For imaging experiments, cells were grown on small glass coverslips that were pretreated with 10 μg/ml polylysine for 20 minutes in 24-well plates. For the STIM1 activation, cells transfected with indicated plasmids were treated with either 2-Ca2+ Ringer’s (155 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM D-glucose, and 5 mM Hepes, pH 7.3 with NaOH) or 0-Ca2+ Ringer’s (155 mM NaCl, 4.5 mM KCl, 3 mM MgCl2, 10 mM D-glucose, 5 mM Hepes, and 1 mM EGTA, pH 7.3.) plus 1 μM TG for 15 minutes24. To stimulate the interaction between FRB and FKBP12, 200 nM rapamycin were added 6 hours before experiments.
DAB staining
DAB staining was performed on transiently transfected HEK-293 cells as described34. Briefly, 40 hours after the transfection, cells were washed with PBS and fixed with 2% glutaraldehyde (Electron Microscopy Sciences) in PBS buffer, pH 7.4 for 30 minutes. Then, cells were rinsed with PBS twice and quenched with 20 mM glycine in PBS for 5 minutes, followed by 2 washes with PBS. DAB staining was initiated with the addition of freshly diluted 0.5 mg/ml DAB with 10 mM H2O2 in PBS. After 15–30 minutes, reactions were stopped by removal of DAB with two washes. Cells were imaged by bright-field microscopy. Acquisition times ranged from 50–100 ms.
Image analyses
All acquired images were analyzed using the ImageJ (NIH) software.
Biotin-phenol labeling in live cells
Biotin-phenol (BP) labeling was performed as described9. Briefly, genes were transfected into HEK-293 cells using Lipofectamine 2000. After 40 hours, biotin-phenol labeling was initiated by the addition of 0.5 mM BP, which was incubated at 37 °C in 5% CO2 for 30 minutes, accompanied by the addition of 1 mM H2O2 for 1 minute. The reaction was stopped with quenching buffer containing 10 mM sodium azide, 10 mM sodium ascorbate, and 5 mM Trolox. Cells were washed 6 times with PBS containing the 3 inhibitors followed by 3 washes with PBS. All washes were performed on ice. Cells were then detected by immunofluorescence or western blot.
Immunofluorescence microscopy
After initiations with BP and H2O2, cells were washed with ice-cold quenching buffer and fixed with 4% PFA for 20 min at room temperature (RT) followed by permeabilization with PBS with 0.1% Triton(PBST) for 5 minutes. They were then blocked with 3% BSA in PBS. The NA was detected with the flag antibody, CA was detected with anti-myc, and the biotin signal was detected with cy5-streptavidin after one hour incubations at RT or overnight incubations at 4 °C. After three washes with PBST, cells were incubated with secondary Alexa Fluor 488-goat anti-mouse IgG or Alexa Fluor cy3-goat anti-rabbit IgG for 1 h at RT. Cells were washed six times with PBST. Confocal imaging was performed using an Olympus FV1200. The confocal head contained a quad-band notch dichroic mirror (405/488/561/647 nm).
Western blot analysis
Biotinylated cells were washed, and whole-cell lysates were prepared in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 1 × protease cocktail, 1 mM PMSF, 10 mM sodium azide, 10 mM sodium ascorbate, and 5 mM Trolox). After centrifugation to remove insoluble materials, proteins were separated by SDS-PAGE and analyzed using streptavidin-HRP, anti-flag antibody, and anti-myc antibody. A small fraction of the cells that were washed and lysed without the quenchers was used for protein concentration measurements by BCA.
After washing, cells were washed with the quencher solution for six times, lysed with 800 μL of freshly prepared RIPA lysis buffer on ice, and ultra-sonicated. Lysates were clarified by centrifugation at 13,000 rpm for 10 minutes at 4 °C. Then, we put the supernatants into 3k columns (balanced with RIPA first), centrifuged at 13,000 rpm for 30 minutes at 4 °C and washed the columns with RIPA 6 times. The sample was thoroughly recovered to a new EP tube. The suspensions were incubated with 150 μL streptavidin-coated magnetic beads at room temperature for 1 hour. Streptavidin beads were then washed with 2 × 1 mL RIPA lysis buffer, once with 1 M KCl, 0.1 M Na2CO3 and 2 M Urea, and again with 2 × 1 mL RIPA lysis buffer. Biotinylated proteins were eluted by incubating the beads with 40 μL 1x NuPAGE LDS Sample Buffer supplemented with 20 mM DTT and 2 mM biotin and heating to 95 °C for 5 minutes.
In-gel protein digestion and extraction
Enriched biotinylated proteins by magnetic streptavidin beads were separated on 10% SDS-PAGE gels and stained with Coomassie G-250 (Invitrogen). Lanes for each replicate were manually cut into 3–4 gel bands. Each band was destained with 40% acetonitrile/50 mM NH4HCO3. The gels were dehydrated with 100% acetonitrile and dried for 5 min using a SpeedVac. Disulfide bonds were reduced with DTT (10 mM, 56 °C, 45 min), and the free sulfhydryl groups were alkylated with iodoacetamide (55 mM, 25 °C, 60 min in the dark). Gel pieces were sequentially washed with 50 mM NH4HCO3, 50% acetonitrile/50 mM NH4HCO3, and dehydrated with 100% acetonitrile. After drying with a SpeedVac, the gel was rehydrated using 100 ng/μL trypsin (Promega, V5113) on ice for 30 min, and the digestion was carried out at 37 °C overnight and quenched with 1.0% TFA. The tryptic peptides were extracted twice with 60% acetonitrile containing 0.1% TFA, and the combined digest solution was dried using a SpeedVac.
Mass spectrometry
The nano-LC-MS/MS experiments were performed on a Q Exactive mass spectrometer (Thermo Scientific) coupled to an Easy-nLC 1000 HPLC system (Thermo Scientific). The dried peptides were resuspended in 0.1% formic acid/2% acetonitrile and loaded onto a 100 μm id × 2 cm-fused silica trap column packed in-house with reversed-phase silica (Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch GmbH) and then separated on a 75 μm id × 20 cm C18 column packed with reversed-phase silica (Reprosil-Pur C18 AQ, 3 μm, Dr. Maisch GmbH). The loaded peptides were eluted with a 78-min gradient. Solvent A consisted of 0.1% FA in a water solution, and solvent B consisted of 0.1% FA in an acetonitrile solution. The segmented gradient was 4–12% B, 5 min; 12–22% B, 50 min; 22–32% B, 12 min; 32–95% B, 1 min; 95% B, 7 min at a flow rate of 280 nl/min.
The mass spectrometer was operated in the data-dependent acquisition mode, and full-scan MS data were acquired in the Orbitrap with a resolution of 70,000 (m/z 200) across the mass range of 300–1600 m/z. The target value was 3.00E + 06 with a maximum injection time of 60 ms. The raw MS data were processed with Proteome Discovery (version 1.4, Thermo Scientific). Briefly, peptide identification was performed with the Sequest HT search engine against a UniProt database supplemented with all the frequently observed contaminants in MS. The following parameters were used for database searching: 10 ppm precursor mass tolerance, 0.02 Da fragment ion tolerance, up to two missed cleavages, carbamidomethyl cysteine, biotin-phenol on tyrosine, histone, cysteine and tryptophan, and oxidized methionine as the variable modification. The peptide confidence was set at a high level (q-value < 0.01) for the peptide filter. A manual check was performed for the biotinylated peptides to avoid suspicious assignments with unclear MS/MS spectra.
Change history
11 June 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
20 April 2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
St George-Hyslop, P. et al. Alzheimer’s disease and possible gene interaction. Science. 263, 537 (1994).
Taylor, I. W. et al. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat Biotechnol. 27, 199–204 (2009).
Shangary, S. & Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 49, 223–241 (2009).
Virkamaki, A., Ueki, K. & Kahn, C. R. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 103, 931–943 (1999).
Li, X. W. et al. New insights into the DT40 B cell receptor cluster using a proteomic proximity labeling assay. J Biol Chem. 289, 14434–14447 (2014).
Rees, J. S., Li, X. W., Perrett, S., Lilley, K. S. & Jackson, A. P. Selective Proteomic Proximity Labeling Assay Using Tyramide (SPPLAT): A Quantitative Method for the Proteomic Analysis of Localized Membrane-Bound Protein Clusters. Curr Protoc Protein Sci. 80(19), 27 11–18 (2015).
Lam, S. S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 12, 51–54 (2015).
Hung, V. et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell. 55, 332–341 (2014).
Rhee, H. W. et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 339, 1328–1331 (2013).
Chapman-Smith, A. & Cronan, J. E. Jr. Molecular biology of biotin attachment to proteins. J Nutr. 129, 477S–484S (1999).
Luker, G. D., Sharma, V. & Piwnica-Worms, D. Visualizing protein-protein interactions in living animals. Methods. 29, 110–122 (2003).
Luker, K. E. et al. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA 101, 12288–12293 (2004).
Michnick, S. W., Ear, P. H., Manderson, E. N., Remy, I. & Stefan, E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 6, 569–582 (2007).
Toby, G. G. & Golemis, E. A. Using the yeast interaction trap and other two-hybrid-based approaches to study protein-protein interactions. Methods. 24, 201–217 (2001).
Feinberg, E. H. et al. GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron. 57, 353–363 (2008).
Martell, J. D. et al. A split horseradish peroxidase for the detection of intercellular protein-protein interactions and sensitive visualization of synapses. Nat Biotechnol. 34, 774–780 (2016).
Sinz, A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 25, 663–682 (2006).
Choi, J., Chen, J., Schreiber, S. L. & Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 273, 239–242 (1996).
Connolly, C. N., Futter, C. E., Gibson, A., Hopkins, C. R. & Cutler, D. F. Transport into and out of the Golgi complex studied by transfecting cells with cDNAs encoding horseradish peroxidase. J Cell Biol. 127, 641–652 (1994).
Li, J., Wang, Y., Chiu, S. L. & Cline, H. T. Membrane targeted horseradish peroxidase as a marker for correlative fluorescence and electron microscopy studies. Front Neural Circuits. 4, 6 (2010).
Jones, D. K., Dalton, D. A., Rosell, F. I. & Raven, E. L. Class I heme peroxidases: characterization of soybean ascorbate peroxidase. Arch Biochem Biophys. 360, 173–178 (1998).
Patterson, W. R. & Poulos, T. L. Crystal structure of recombinant pea cytosolic ascorbate peroxidase. Biochemistry. 34, 4331–4341 (1995).
Sharp, K. H., Mewies, M., Moody, P. C. & Raven, E. L. Crystal structure of the ascorbate peroxidase-ascorbate complex. Nat Struct Biol. 10, 303–307 (2003).
Wu, M. M., Buchanan, J., Luik, R. M. & Lewis, R. S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 174, 803–813 (2006).
Yang, X., Jin, H., Cai, X., Li, S. & Shen, Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc Natl Acad Sci USA 109, 5657–5662 (2012).
Penna, A. et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 456, 116–120 (2008).
Balasuriya, D., Srivats, S., Murrell-Lagnado, R. D. & Edwardson, J. M. Atomic force microscopy (AFM) imaging suggests that stromal interaction molecule 1 (STIM1) binds to Orai1 with sixfold symmetry. FEBS Lett. 588, 2874–2880 (2014).
Hou, X., Pedi, L., Diver, M. M. & Long, S. B. Crystal structure of the calcium release-activated calcium channel Orai. Science. 338, 1308–1313 (2012).
Palty, R., Stanley, C. & Isacoff, E. Y. Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 25, 963–980 (2015).
Stathopulos, P. B. et al. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat Commun. 4, 2963 (2013).
Korzeniowski, M. K., Manjarres, I. M., Varnai, P. & Balla, T. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci Signal. 3, ra82 (2010).
Ma, G. et al. Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat Commun. 6, 7826 (2015).
Jing, J. et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca(2)(+) influx. Nat Cell Biol. 17, 1339–1347 (2015).
Martell, J. D. et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 30, 1143–1148 (2012).
Acknowledgements
We thank the staff at the Institute of Biophysics Core Facilities. We specifically thank Yan Teng for her technical support with confocal imaging, Dr. Jifeng Wang and Mrs. Mengmeng Zhang for MS operation, Junfeng Hao for light microscopy imaging, Dr. Sher Ali for his great technical support and Xiaolan Xu for suggestions about the manuscript. This work was supported by grants from the National Natural Science Foundation of China (31570765, 31400703), the National Key Basic Research Project of China (2015CB910303).
Author information
Authors and Affiliations
Contributions
T.X. and L.Z. conceived the project. M.M.X. carried out the experiments including plasmid construction, transfection, DAB staining, biotin labeling and immunofluorescence. J.J.H. performed the proteomic experiments and MS data analysis. L.L.W. and D.W.C. performed plasmid construction. J.Z. L prepared all the materials required for the experiment. L.Z. and T.X. wrote the manuscript with help from all authors. All authors read the manuscript and discussed the interpretation of results.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, M., Hou, J., Wang, L. et al. Optimizing the fragment complementation of APEX2 for detection of specific protein-protein interactions in live cells. Sci Rep 7, 12039 (2017). https://doi.org/10.1038/s41598-017-12365-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12365-9
This article is cited by
-
The development of proximity labeling technology and its applications in mammals, plants, and microorganisms
Cell Communication and Signaling (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
