
Abstract
Vibrio fluvialis is recognized as an emerging pathogen. However, not much is known about the mechanism of its pathogenesis, and its adaptation to a special niche such as the gall bladder. Here we describe two V. fluvialis strains that cause acute cholecystitis. It is noteworthy that both strains were susceptible to all antibiotics tested, which is in contrast to previous studies, suggesting substantial genetic diversity among V. fluvialis isolates. In agreement with their survival and growth in the gall bladder, the genomes of strains 12605 and 3663 contain a considerable number of genes that confer resistance to bile, including toxR, omp U, tolC, cmeABC, rlpB, yrbK, rpoS, damX and gltK. Furthermore, integrative and conjugative elements (ICEs), virulence factors and prophage regions were also detected in strains 12605 and 3663, reflecting their flexibility in recombination during the evolution of pathogenicity. Comparative analysis of nine available genomes of V. fluvialis revealed a core genome consisting of 3,147 genes. Our results highlight the association of V. fluvialis with a rare disease profile and shed light on the evolution of pathogenesis and niche adaptation of V. fluvialis.
Similar content being viewed by others

Introduction
Vibrio fluvialis is a halophilic Gram-negative bacterium, which is considered to be an emerging pathogen, found mostly in aquatic environments1. V. fluvialis infection is mainly associated with sporadic cases and outbreaks of gastroenteritis with cholera-like diarrhea1. Although rare, V. fluvialis can also cause extraintestinal infections, including hemorrhagic cellulitis with cerebritis2, bacteremia3, peritonitis4, acute otitis5 and endophthalmitis6. To date, only two reports have described biliary tract infection caused by this bacterium7,8. The route of V. fluvialis entry into the biliary system remains unknown, and it was assumed to be via a cutaneous lesion or due to gastrointestinal translocation upon consumption of contaminated seafood as observed in other Vibrio infections8.
Molecular characterization of this pathogen is particularly important, owing to the increasing appearance of multidrug resistant strains and their potential to cause epidemics9. Several studies have indicated that Chinese hamster ovary (CHO) cell elongation factor, enterotoxin, lipase, protease, cytotoxin, hemolysin and quorum sensing systems contribute to the pathogenicity of this organism1. Whole-genome sequences of several V. fluvialis stains have been described earlier, including two strains that cause diarrhea in human and two environmental strains10,11. These genome data help in understanding the genetic basis of the physiology, biochemical pathways and evolution of V. fluvialis. However, the mechanisms of pathogenesis and its adaptation to a special niche, such as gallbladder, are yet to be explored.
Here we describe the isolation and characterization of two V. fluvialis strains that cause acute cholecystitis. The complete genome sequence of strain 12605 and the draft genome sequence of strain 3663 were obtained by whole genome sequencing. Genes involved in bile resistance were identified in these genomes. Integrative and conjugative elements (ICEs), virulence factors and prophage regions were also found in both genomes. Comparative genomics analysis was conducted with nine V. fluvialis strains. Our findings will contribute to the understanding of the phylogenetic diversity and niche adaptation of the two strains, while also shedding light on the evolution of pathogenesis in V. fluvialis.
Results
Case record
Case 1
A 76-year-old male was admitted to our hospital with fever, vomiting and a sudden upper abdominal pain on July 2014. He reported that the recurring pain suddenly began seven days ago. Lower back pain, chills, vomiting and diarrhea were not documented during the evolution of symptoms. The medical history was remarkable, as he had a history of gallstones and had undergone surgical treatment of cholangiocarcinoma two weeks prior. Ultrasound of the biliary tract revealed a thickened gallbladder wall and pericholecystic fluid, with no visible stones, an observation consistent with the presence of acute cholecystitis. Empirical parenteral administration of 500 mg of levofloxacin every 8 h was started on the first day of hospitalization. On the third day, a curved rod shaped Gram-negative bacterium, designated as 3663, was isolated from the bile culture. The fever, abdominal pain and the clinical condition improved after antibiotic treatment. The patient was discharged two weeks later and prescribed oral cephalosporin antibiotics.
Case 2
A 55-year-old man presented at an emergency department in 2013 with a four-day history of right upper quadrant pain and vomiting. He lived in the coast of Hangzhou Gulf and denied having any history of travel, but had high potential exposure to seafood. He had a history of gallstones. Abdominal computed tomography clearly showed a distended gallbladder with thickened wall and gallstones. The patient was hospitalized with a presumptive diagnosis of acute calculous cholecystitis and received cefotiam treatment. The bile cultures yielded significant growth of a curved rod shaped Gram-negative bacterium (designated as 12605). The patient was treated with intravenous fluids and broad-spectrum antibiotics for five days. His condition improved clinically, with resolution of the abdominal pain and normalization of his laboratory and ultrasonography findings. He was discharged from the hospital with a 10-day course of treatment and recovered fully.
Phenotypic and genotypic characterization of clinical isolates
MALDI-TOF analysis indicated that the strains 12605 and 3663 are highly similar to V. fluvialis from the default Bruker database (matching scores of >2.0). 16 S rRNA sequences of 12605 and 3663 were aligned by BLAST against the latest version of EzBioCloud’s database12 and the results showed that both strains share 99.86% identity with V. fluvialis NBRC 103150T. Growth curves of two V. fluvialis isolates demonstrated that bile resistance was observed in both isolates (see Supplementary Fig. S1). Antimicrobial susceptibility tests demonstrated that both strains were susceptible to all antibiotics tested (see Supplementary Table S1).
General genome features
The genomes of 12605 and 3663 were sequenced as described in material and methods. Sequencing of strain 12605 revealed the presence of two complete chromosomes (one chromosome is 3,171,566 bp in length and the other is 1,680,098 bp in length) with an average G+C content of 50.1%. The two chromosomes contain 4,395 protein coding genes, 113 tRNAs and 37 rRNAs. Sequencing of strain 3663 revealed a genome size of 4,849,960 bp with a G+C content of 49.9% (207 contigs). These contigs contain 4,441 protein coding genes, 87 tRNAs and 12 rRNAs. The genomic features of the strains 12605 and 3663, and eight other reference V. fluvialis strains are summarized in Table 1.
Phylogenetic analysis of strains 12605 and 3663
To further understand the phylogenetic relationship between the V. fluvialis strains 12605 and 3663 and other strains within this species, their genome sequences were obtained from the NCBI database and analyzed. To date, ten genomes of V. fluvialis have been deposited in the NCBI database, including strains 12605 and 3663. To obtain an estimate of the overall similarity between the ten V. fluvialis genomes, we calculated their average nucleotide identity (ANI). To our surprise, strain NCTC 11327 showed remarkably low ANI values as compared to all other strains (73.4–73.5%) (see Supplementary Fig. S2). As recommended by several early studies, ANI values of about 95–96% are considered to be the species boundary13,14,15, indicating that strain NCTC 11327 may not belong to V. fluvialis species. To further test this assumption, we compared the 16S rRNA gene of NCTC 11327 against the NCBI nr database16,17. The results showed it shared 100% sequence identity with V. vulnificus strain CAPL-B-VVF2 (KX904714.1). Thus, the genome sequence (accession no. LMTE00000000.1) deposited under the name of V. fluvialis strain NCTC 11327 is actually a member of V. vulnificus species; this genome was thus excluded from our analysis. The ANI was then recalculated among the remaining nine genomes, and the values among them were found to range between 97.1–100%, indicating a close relationship with each other (see Supplementary Fig. S3). The highest similarity with an ANI value of 100% was found between the strains ATCC 33809 and NBRC 103150. By searching against the NITE Biological Resource Center (NBRC) database, we found that NBRC 103150 is actually ATCC 33809. Therefore, the ANI value of 100% between these two genomes confirms that they actually belong to the same strain that were deposited in two different culture centers. In order to provide a high-resolution view of phylogeny, a phylogenetic tree was constructed based on the genome alignments of the nine V. fluvialis strains (Fig. 1). As expected, strains 12605 and 3663, both isolated from bile, fell into the same clade.

Dendrogram of V. fluvialis strains based on genomic BLAST. The genomic BLAST file was downloaded from the NCBI database and the tree was visualized by FigTree v1.4 42.
Identification of Integrative and Conjugative Elements (ICEs)
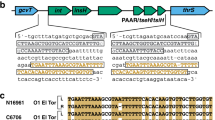
Integrative and conjugative elements (ICEs) are a diverse group of mobile elements found in bacteria18 that enable horizontal gene transfer (HGT) of virulence genes, antibiotic resistance genes, etc.19 To identify ICEs in our two bile-isolated strains (12605 and 3663), we compared their genomes against the ICEberg database19 by BLAST-2.3.0+ 20 program. Many genes belonging to various types of ICEs were shortlisted (see Supplementary Tables S2 and S3). A total of 15 genes belong to ICEVflInd1 (ICEberg ID 36) were found in both strains. To identify whether the genes of ICEVflInd1 existed in all V. fluvialis strains, we compared the complete sequences of ICEVflInd1 from GenBank database (GQ463144) with the whole genomes of the nine strains, as shown in Fig. 2. The strains carried most of genes of ICEVflInd1, with strains 539 and 560 carrying more genes than other the strains.

Comparison of integrative and conjugative elements ICEVflInd1 and V. fluvialis genomes. Genes denoted by arrows are based on the annotation of ICEVflInd1 (accession number of GQ463144).
Identification of virulence-associated genes
Further screening of the genomes for putative virulence-associated genes was conducted by aligning ORF encoded protein sequences to the virulence factor database (VFDB). Both genomes were found to contain many putative virulence factors (see Supplementary Tables S4 and S5). In addition, multiple copies of heat shock proteins including GroES, GroEL, and HspA homologs were found in both genomes and these proteins were demonstrated to be essential for bacterial survival and association for colonization in Helicobacter pylori 21. Consistent with a previous study, homologs of flagellar biosynthetic proteins (FliD, FliE, FliF, FliG, FliH, FliI, FliJ, FliK, FliL, FliM, FliN, FliO, FliP, FliQ, FliR, and FliS) were also found in these genomes22. Furthermore, we searched the genomes of 12605 and 3663 for prophage regions and found that both strains harbored several prophage regions (see Supplementary Table S6). The phage-like sequences are believed to improve cell adhesion and ability to acquire antibiotic resistance that can enable bacteria to survive in new environments and become pathogens23.
Identification of putative bile resistance-related genes
It has been demonstrated that bile salts have potent antimicrobial properties that can cause damage to bacteria24. For survival in bile and cause cholecystitis in humans, bacteria have to overcome this barrier. In response to bile salt exposure, bacteria can induce efflux systems, induce elevated resistance to bile toxicity, enhance motility, remodel outer membrane proteins, and even promote biofilm formation25. ToxR, which is a membrane-associated transcription factor, was found to play a major role in bile resistance of V. fluvialis and other Vibrio species26. Another protein GltK, which is a glutamate/aspartate transport system permease protein, was also shown to be involved in bile resistance27. Analysis of the genomes of strains 12605 and 3663 revealed the existence of genes encoding ToxR and GltK in both genomes. Furthermore, a considerable number of putative bile resistance-related proteins such as DamX (an inner membrane protein involved in bile resistance), lipopolysaccharide transporters, membrane transport systems, and polymerase sigma factor were encoded in the genomes of both 12605 and 3663. These genes may play important roles in the survival of 12605 and 3663 in the gall bladder.
Comparative genomic analysis of V. fluvialis strains
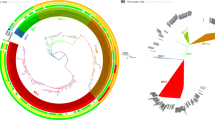
The circular maps and BLAST visualization of the nine V. fluvialis genomes are illustrated in Fig. 3. The results were in agreement with our phylogenetic tree. Pan-genome analysis of the nine genomes revealed that the core genome consists of 3,147 genes from all 40,602 total genes, which constituted the pan-genome. The pan-genome size of these nine strains was 7,625 and their increased tendency showed the pan-genome could be taken as an open pan-genome. (Fig. 4). The ratio of core-genome in each species ranged from 22% to 44% and was correlated with the total coding sequence (CDS) numbers (see Supplementary Table S7). The ratio of core/pan-genome with nine strains was 41.3%. We also analyzed the functional classifications of ortholog clusters using the COG database. The results obtained are summarized in Table 2. With the exception of poorly characterized or uncharacterized COGs, the most abundant category in the pan genome was [K] transcription. The next abundant COGs were [E] Amino acid transport and metabolism, followed by [T] Signal transduction mechanisms. In the core genome, [E] Amino acid transport and metabolism was the largest gene family, followed by [K] transcription and [T] Signal transduction mechanisms. Subsequently, the proportion of the conserved groups (core, dispensable, and specific genomes) in each category was investigated further (see Supplementary Fig. S4).

BLAST atlas of the genomes of various V. fluvialis strains. The circles from inside to outside: GC content, and GC skew of V. fluvialis 12605; BLASTN pairwise comparison of the V. fluvialis genomes: 12605, 3663, ATCC 33809, PG41, I21563, 560 539, S1110 and NBRC 103150. The white and colored regions of the outer rings indicate regions absent and present, respectively.

Comparative genomic analysis of V.fluvialis strains. (A)Pan-genome and core genome profiles, the numbers of new genes in the V. fluvialis pan-genome and core genome are plotted against the number of genomes added; (B) Venn diagram showing the number of species-specific gene families in the genome of each strain. The number of core genomes is represented in the center.
Discussion
Though the emerging human pathogen V. fluvialis was known to cause diarrheal illness and extraintestinal infections for quite some time, in depth studies on the pathogenesis of V. fluvialis have not been performed so far22. To our knowledge, there are only two known cases of V. fluvialis-associated biliary tract infections and no reports of acute cholecystitis since its first description in human infection. Very little is known about the clinical importance of V. fluvialis in biliary tract infection and its niche adaptation to bile salts. In this study, we report two cases of acute cholecystitis. Since only V. fluvialis was detected from the bile samples of the affected individuals, we conclude that the inflammatory responses were caused by V. fluvialis infection. However, the source of infection is difficult to trace. We speculate that the patients were brought in contact with pathogens in an aquatic environment, since V. fluvialis occurs widely in the aquatic realm and our patients may have potentially been exposed to seafood and aquatic environment1.
Identification of the specific species is essential for appropriate antimicrobial treatment and improved clinical care. However, this remains a major challenge with the API20E and Vitek 2 systems due to similarities in the phenotypic characteristics of V. fluvialis and other Vibrio species1. In the current study, we employed MALDI-TOF and 16 S rRNA sequencing methods for rapid identification of V. fluvialis, which confirmed the combined strategy as a reliable method for accurate identification.
In this study, the MICs of all antibiotics tested were found to be within the susceptible range. This finding may explain the improved recovery of two patients following treatment with antibiotics. In contrast, Liang et al. reported that most of the V. fluvialis strains from China were resistant to β-lactams, azithromycin, and sulfamethoxazole, and that the clinical isolates showed higher resistance in general than the environmental isolates9.
To further ascertain the adaptive features and essential genes of V. fluvialis 12605 and 3663 for survival in the gallbladder, we performed WGS and comparative genomic analysis. Bile salts can damage membrane lipids and cause misfolding and denaturation of intracellular proteins28. Therefore, bacteria must survive in high concentrations of bile salts before invading the gall bladder epithelial cells. In agreement with their survival and growth in the gall bladder, the genomes of 12605 and 3663 were found to encode several bile resistance-related genes. These include toxR, ompU, ompT, tolC, which are essential for bile resistance in V. cholera 26,29; cmeABC, which mediates enhanced resistance to bile in Campylobacter 30; gltK, which has been confirmed to be involved in bile resistance in Enterococcus faecium 27; rlpB, yrbK and rpoS, which have been reported to be involved in bile resistance in Salmonella 31; and damX, which encodes an inner membrane protein involved in bile resistance in S. enterica 32.
Phage-mediated horizontal gene transfer is known to drive virulence and genomic diversification of bacteria. The sequences of the prophage regions from strains 12605 and 3663 were found to be homologous to those of Vibrio prophages. Integrative and conjugative elements (ICEs) are a diverse group of mobile elements found in bacteria18 that enable horizontal gene transfer (HGT) of virulence genes, antibiotic resistance, etc19. According to the ICEberg database, there are two well-characterized ICEs found in V. fluvialis, ICEVflH-08942 in V. fluvialis H-08942 (ICEberg ID 149) and ICEVflInd1 in V. fluvialis Ind1(ICEberg ID 36). ICEVflInd1was first discovered in V. fluvialis Ind1with resistance profile (dfr18, floR, strBA and sul2) and notable variable genes (toxin-antitoxin system33). A number of genes belonging to ICEVflInd1 (ICEberg ID 36) were found in the nine V. fluvialis strains. Interestingly, strains 539 and 560 carry more ICE genes when compared to other strains. V. fluvialis strains 539 and 560, which inhabit Crassostrea rhizophorae and Anomalocardia brasiliana respectively, were also reported to possess a variant of the ICE SXT elements for the first time in Brazil10. SXT is a member of the ICE SXT/R391 family and was first described in clinical isolates of V. cholerae O13934.
Comparative analysis of the nine available genomes of V. fluvialis allowed us to determine the global gene repertoire of the species. The genome comparisons also revealed a ‘pan-genome’ that includes a core genome consisting of 3,147 genes common to all strains. This core-genome represents 71.6% of the genome of strain 12605 and 70.9% of the genome of strain 3663. Phylogenetic analysis divided the dataset into distinct populations, with the bile-isolated strains 12605 and 3663 being grouped into the same clade. Two animal-associated strains (539 and 560) were clustered together, showing a distinct separation from strains ATCC 33809 and NBRC 103150 isolated from human fecal samples. Strains PG41, I21563 and S1110 formed separate lineages.
Our study describes two cases of acute cholecystitis caused by V. fluvialis and provides new insights into the genomic architecture of the pathogen. The genomic information obtained in this study not only increases our understanding of the genetic basis of bile resistance, virulence, and adaptation mechanisms in V. fluvialis, but also helps in the identification of V. fluvialis core genes that can facilitate the detection of V. fluvialis in clinical samples.
Methods
Ethics approval statement
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Clinical Ethics Committee of the first Affiliated Hospital of Zhejiang University. Written informed consent of this case report was obtained from the patient for the publication
Identification of isolates
Both isolates were isolated from Mueller-Hinton broth agar with 5% sheep blood. The initial identification of strains 3663 and 12605 was done by MALDI-TOF MS analysis as previously described35. Overnight grown cultures were subjected to analysis using a Microflex MALDI-TOF mass spectrometer (BrukerDaltonics, Germany). The raw spectra were then analyzed using the MALDI Biotyper 2.0 database (BrukerDaltonics, Germany). The 16S rRNA nucleotide sequences of the two strains were obtained by PCR and sequencing. The sequences were then compared against NCBI database and EzTaxon-e database.
Growth conditions and bile treatment
V. fluvialis strains were grown anaerobically at 37 °C in FEM medium (10 g of Bacto-Peptone (Difco) and 40 g of NaCl adjusted to pH 8.5, per liter) with or without 0.3% ox bile solution treatment; three biological replicates were performed.
Antimicrobial susceptibility testing
Testing for susceptibility to amikacin, aztreonam, ciprofloxacin, meropenem, piperacillin, gentamicin, levofloxacin, cefepime, amoxicillin/clavulanic acid, imipenem, cefotaxime, ceftazidime, chloramphenicol, cefoperazone and minocycline was done by the disc diffusion method and interpreted with reference to a previous study9. Escherichia coli ATCC 25922 was used as control.
Sequencing, assembly and annotation
Whole genome sequencing of strain 12605 was carried out using PacBio RS II Sequencing System and genome assembly was done by SMRT Analysis 2.2.1 36. Whole genome sequencing of strain 3663 was carried out using the Illumina Hiseq. 2000 sequencer (Illumina, USA) with a high-throughput 2 × 100 bp pair end sequencing strategy. Prior to analysis, read sets were filtered, which involved deleting reads with low-quality base calls or similarity to Illumina adaptors. Subsequently, the raw reads were trimmed and assembled using Velvet. PAGIT flow was used to assemble the contigs and correct sequencing errors as described previously37. The two genomes were finally annotated by RAST server38.
Identification of Integrative and Conjugative Elements (ICEs) and virulence-associated genes
Integrative and conjugative elements (ICEs) were identified using BLAST-2.3. 0+ program20 against the ICEberg database19 with an e-value cutoff of 1e-10 and an identity threshold of 60%. Virulence factors were annotated using BLAST-2.3. 0+ program20 against the virulence factor database (VFDB)39 with an e-value cutoff of 1e-10 and an identity threshold of 80%. Putative phage sequences were identified by PHAST40.
Comparative genomic analysis
ANI values among V. fluvilas strains were calculated using OrthoANI41. Genomic BLAST file of V. fluvilas strains was downloaded from NCBI (https://www.ncbi.nlm.nih.gov/genome/2299) and the dendrogram was visualized using FigTree v.1.4 42. Multiple genome alignment was performed using Mauve43 and BLAST Ring Image Generator (BRIG)44. For the pan-genome computation, PGAP v1.12 was used as described earlier45. The function of ortholog clusters was classified according to clusters of orthologous groups (COGs)46.
Accession numbers
The 16S rRNA sequences of V. fluvialis 12605 and 3663 have been deposited in GenBank with the accession numbers KP780091 and KP780090. The complete genome sequence of V. fluvialis 12605 has been deposited in DDBJ/EMBL/GenBank with the accession numbers CP019118 and CP019119. Whole Genome Shotgun data of V. fluvialis 3663 has been deposited in DDBJ/EMBL/GenBank with the accession number JXXQ00000000.
References
Ramamurthy, T., Chowdhury, G., Pazhani, G. P. & Shinoda, S. Vibrio fluvialis: an emerging human pathogen. Front Microbiol 5, 91, https://doi.org/10.3389/fmicb.2014.00091 (2014).
Huang, K. C. & Hsu, R. W. Vibrio fluvialis hemorrhagic cellulitis and cerebritis. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 40, e75–77, https://doi.org/10.1086/429328 (2005).
Albert, M. J. et al. A fatal case associated with shigellosis and Vibrio fluvialis bacteremia. Diagnostic microbiology and infectious disease 14, 509–510 (1991).
Lee, J. Y. et al. Acute infectious peritonitis caused by Vibrio fluvialis. Diagnostic microbiology and infectious disease 62, 216–218, https://doi.org/10.1016/j.diagmicrobio.2008.05.012 (2008).
Chen, P. J., Tseng, C. C., Chan, H. T. & Chao, C. M. Acute Otitis due to Vibrio fluvialis after Swimming. Case reports in emergency medicine 2012, 838904, https://doi.org/10.1155/2012/838904 (2012).
Hassan, I. J., MacGowan, A. P. & Cook, S. D. Endophthalmitis at the Bristol Eye Hospital: an 11-year review of 47 patients. The Journal of hospital infection 22, 271–278 (1992).
Yoshii, Y., Nishino, H., Satake, K. & Umeyama, K. Isolation of Vibrio fluvialis, and unusual pathogen in acute suppurative cholangitis. The American journal of gastroenterology 82, 903–905 (1987).
Liu, W. L., Chiu, Y. H., Chao, C. M., Hou, C. C. & Lai, C. C. Biliary tract infection caused by Vibrio fluvialis in an immunocompromised patient. Infection 39, 495–496, https://doi.org/10.1007/s15010-011-0146-0 (2011).
Liang, P., Cui, X., Du, X., Kan, B. & Liang, W. The virulence phenotypes and molecular epidemiological characteristics of Vibrio fluvialis in China. Gut pathogens 5, 6, https://doi.org/10.1186/1757-4749-5-6 (2013).
de Oliveira Veras, A. A. et al. Draft Genome Sequences of Vibrio fluvialis Strains 560 and 539, Isolated from Environmental Samples. Genome announcements 3, doi:https://doi.org/10.1128/genomeA.01344-14 (2015).
Khatri, I., Mahajan, S., Dureja, C., Subramanian, S. & Raychaudhuri, S. Evidence of a new metabolic capacity in an emerging diarrheal pathogen: lessons from the draft genomes of Vibrio fluvialis strains PG41 and I21563. Gut pathogens 5, 20 (2013).
Yoon, S.-H., et al. Introducing EzBioCloud: A taxonomically united database of 16S rRNA and whole genome assemblies. International journal of systematic and evolutionary microbiology (2016).
Goris, J. et al. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. International journal of systematic and evolutionary microbiology 57, 81–91 (2007).
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proceedings of the National Academy of Sciences 106, 19126–19131 (2009).
Chun, J. & Rainey, F. A. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. International journal of systematic and evolutionary microbiology 64, 316–324 (2014).
Zhang, Z., Schwartz, S., Wagner, L. & Miller, W. A greedy algorithm for aligning DNA sequences. Journal of Computational biology 7, 203–214 (2000).
Morgulis, A. et al. Database indexing for production MegaBLAST searches. Bioinformatics 24, 1757–1764 (2008).
Burrus, V., Pavlovic, G., Decaris, B. & Guédon, G. Conjugative transposons: the tip of the iceberg. Molecular microbiology 46, 601–610 (2002).
Bi, D. et al. ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic acids research 40, D621–D626 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC bioinformatics 10, 421 (2009).
Schauer, K. et al. The Helicobacter pylori GroES cochaperonin HspA functions as a specialized nickel chaperone and sequestration protein through its unique C-terminal extension. Journal of bacteriology 192, 1231–1237 (2010).
Lu, X. et al. Identification of genetic bases of vibrio fluvialis species-specific biochemical pathways and potential virulence factors by comparative genomic analysis. Appl Environ Microbiol 80, 2029–2037, https://doi.org/10.1128/AEM.03588-13 (2014).
Casjens, S. Prophages and bacterial genomics: what have we learned so far? Molecular microbiology 49, 277–300 (2003).
Merritt, M. E. & Donaldson, J. R. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. Journal of medical microbiology 58, 1533–1541 (2009).
Hay, A. J. & Zhu, J. Host intestinal signal-promoted biofilm dispersal induces Vibrio cholerae colonization. Infection and immunity 83, 317–323 (2015).
Provenzano, D., Schuhmacher, D. A., Barker, J. L. & Klose, K. E. The Virulence Regulatory Protein ToxR Mediates Enhanced Bile Resistance in Vibrio cholerae and Other PathogenicVibrio Species. Infection and immunity 68, 1491–1497 (2000).
Zhang, X. et al. Functional genomic analysis of bile salt resistance in Enterococcus faecium. BMC genomics 14, 299 (2013).
Begley, M., Gahan, C. G. & Hill, C. The interaction between bacteria and bile. FEMS microbiology reviews 29, 625–651 (2005).
Bina, J. E. & Mekalanos, J. J. Vibrio cholerae tolC is required for bile resistance and colonization. Infection and immunity 69, 4681–4685 (2001).
Lin, J., Sahin, O., Michel, L. O. & Zhang, Q. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infection and immunity 71, 4250–4259 (2003).
Hernández, S. B., Cota, I., Ducret, A., Aussel, L. & Casadesús, J. Adaptation and preadaptation of Salmonella enterica to bile. PLoS Genet 8, e1002459 (2012).
López-Garrido, J., Cheng, N., García-Quintanilla, F., García-del Portillo, F. & Casadesús, J. Identification of the Salmonella enterica damX gene product, an inner membrane protein involved in bile resistance. Journal of bacteriology 192, 893–895 (2010).
Wozniak, R. A. et al. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS genetics 5, e1000786 (2009).
Daccord, A., Ceccarelli, D. & Burrus, V. Integrating conjugative elements of the SXT/R391 family trigger the excision and drive the mobilization of a new class of Vibrio genomic islands. Molecular microbiology 78, 576–588 (2010).
Espinal, P., Seifert, H., Dijkshoorn, L., Vila, J. & Roca, I. Rapid and accurate identification of genomic species from the Acinetobacter baumannii (Ab) group by MALDI-TOF MS. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases 18, 1097–1103, https://doi.org/10.1111/j.1469-0691.2011.03696.x (2012).
Liao, Y.-C., Lin, S.-H. & Lin, H.-H. Completing bacterial genome assemblies: strategy and performance comparisons. Scientific reports 5, 8747 (2015).
Zhang, F. et al. Permanent draft genome sequence of Bacillus flexus strain T6186-2, a multidrug-resistant bacterium isolated from a deep-subsurface oil reservoir. Marine genomics 18 Pt B, 135–137, https://doi.org/10.1016/j.margen.2014.09.007 (2014).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics 9, 75, https://doi.org/10.1186/1471-2164-9-75 (2008).
Chen, L., Xiong, Z., Sun, L., Yang, J. & Jin, Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic acids research 40, D641–645, https://doi.org/10.1093/nar/gkr989 (2012).
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J. & Wishart, D. S. PHAST: a fast phage search tool. Nucleic acids research 39, W347–W352 (2011).
Lee, I., Kim, Y. O., Park, S.-C. & Chun, J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. International journal of systematic and evolutionary microbiology 66, 1100–1103 (2016).
Rambaut, A. 2006-2012. Fig. Tree. Tree Figure Drawing Tool, version 1.4. 0. University of Edinburgh: Institute of Evolutionary Biology [on-line]. Available from tree. bio. ed. ac. uk/ (accessed 30 January 2015).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS one 5, e11147 (2010).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402, https://doi.org/10.1186/1471-2164-12-402 (2011).
Kim, J.-N. et al. Comparative genomics reveals the core and accessory genomes of Streptomyces species. J. Microbiol. Biotechnol 25, 1599–1605 (2015).
Tatusov, R. L. et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29, 22–28 (2001).
Acknowledgements
This study was partially funded by grants from the National Basic Research Program of China (No. 2015CB554201); the National Natural Science Foundation of China (Nos 81361138021, 81711530049, 81301461 and 41406140); the National Key Research and Development Program of China (Nos 2016YFD0501105); the Zhejiang Provincial Key Research and Development Program (No. 2015C03032); the Zhejiang Provincial Natural Science Foundation of China (No. LY17H190003); and the opening foundation of the State Key Laboratory for Diagnosis and Treatment of Infectious Diseases (2010KF04).
Author information
Authors and Affiliations
Contributions
B.Z., X.J., Y.X. and L.L. designed the study. B.Z., X.J., H.C., J. Z., H.X., X.Y., and C.H. performed experiments. B.Z., X.J., H.C., and L.G. analyzed data. Y.X. and L.L. contributed reagents, materials and analysis tools. B.Z., X.J., and Y.X. wrote the manuscript that was revised by all co-authors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, B., Jiang, X., Cheng, H. et al. Genome characterization of two bile-isolated Vibrio fluvialis strains: an insight into pathogenicity and bile salt adaption. Sci Rep 7, 11827 (2017). https://doi.org/10.1038/s41598-017-12304-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12304-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
