
Abstract
In Saccharomyces cerevisiae, peroxisomes are the sole site of fatty acid β-oxidation. During this process, NAD+ is reduced to NADH. When cells are grown on oleate medium, peroxisomal NADH is reoxidised to NAD+ by malate dehydrogenase (Mdh3p) and reduction equivalents are transferred to the cytosol by the malate/oxaloacetate shuttle. The ultimate step in lysine biosynthesis, the NAD+-dependent dehydrogenation of saccharopine to lysine, is another NAD+-dependent reaction performed inside peroxisomes. We have found that in glucose grown cells, both the malate/oxaloacetate shuttle and a glycerol-3-phosphate dehydrogenase 1(Gpd1p)-dependent shuttle are able to maintain the intraperoxisomal redox balance. Single mutants in MDH3 or GPD1 grow on lysine-deficient medium, but an mdh3/gpd1Δ double mutant accumulates saccharopine and displays lysine bradytrophy. Lysine biosynthesis is restored when saccharopine dehydrogenase is mislocalised to the cytosol in mdh3/gpd1Δ cells. We conclude that the availability of intraperoxisomal NAD+ required for saccharopine dehydrogenase activity can be sustained by both shuttles. The extent to which each of these shuttles contributes to the intraperoxisomal redox balance may depend on the growth medium. We propose that the presence of multiple peroxisomal redox shuttles allows eukaryotic cells to maintain the peroxisomal redox status under different metabolic conditions.
Similar content being viewed by others


Introduction
Eukaryotic cells are compartmentalised into distinct, membrane-bound organelles which contain their own unique enzyme content and internal milieu. One of these is the peroxisome, a single membrane bound organelle which contains enzymes required for a wide range of reactions and pathways including oxidation reactions resultant in the production of hydrogen peroxide. Some peroxisomal functions such as β-oxidation are widely conserved across almost all eukaryotes whilst other functions may be distinct to certain eukaryotic kingdoms, species or cell types. The importance of correctly localising enzymes to peroxisomes is illustrated by the numerous devastating disorders which arise as a consequence of the mislocalisation of single or multiple peroxisomal enzymes1.
Many metabolic pathways are distributed across multiple subcellular compartments. This organisation is likely to be beneficial to cells as it allows for individual reactions in a pathway to occur within different, favourable micro-environments. Compartmentalisation of reactions allows for additional levels of spatial or temporal control and may offer cellular protection from toxic reaction intermediates or by-products. For example, peroxisomes contribute to the fine-tuning of lysine biosynthesis in Saccharomyces cerevisiae and in cells deficient in peroxisomal protein import, mRNA levels of most of the genes involved in lysine biosynthesis are upregulated2. In S. cerevisiae and many other fungi and euglenoids, L-lysine is produced via the α-aminoadipate pathway (Fig. 1), whereas bacteria, plants and some lower fungi use the diaminopimelate pathway3. Based on the localisation of GFP-tagged fusion proteins4,5 the pathway for lysine biosynthesis appears to pass through three distinct compartments (nucleus, mitochondrion, cytosol) before it concludes inside the peroxisomal matrix with the enzyme saccharopine dehydrogenase, encoded by LYS1 (Fig. 1). Metabolic pathways that are distributed across multiple compartments require transport of substrates, co-factors and products between these compartments. For peroxisomes, two types of transport have been described. The first involves transporters that mediate the movement of bulky metabolites and the second type is mediated by passive diffusion of small hydrophilic metabolites of maximally 300–400 Da through channels (porins)6.

Schematic representation of the intracellular organisation of (A) L-lysine biosynthesis in Saccharomyces cerevisiae. The subcellular localisations of the enzymes are indicated according to Saccharomyces cerevisiae Genome Database (SGD). A generic peroxisomal NADH redox shuttle for reoxidation of NADH to NAD+ is depicted in red. (B) Diagram of the malate/oxaloacetate shuttle and glycerol-3-phosphate/dihydroxyacetone phosphate shuttle, exchanging peroxisomal NADH for cytosolic NAD+. Gpd1p provides the peroxisomal glycerol-3-phosphate dehydrogenase activity. In the cytosol this may be provided by Gpd2p or by cytosolic Gpd1p.
Although nicotinamide dinucleotides can be transported across the peroxisomal membrane, at least in Arabidopsis thaliana 7,8, efficient β-oxidation requires redox shuttles to locally reoxidise the NADH produced in the third step of β-oxidation. In S. cerevisiae, peroxisomal malate dehydrogenase (Mdh3p) is required to maintain the redox balance inside peroxisomes during growth on fatty acids as the sole carbon source9,10. Under these growth conditions, peroxisomes proliferate and expression of β-oxidation enzymes is induced11, including Mdh3p9. It is unclear whether Mdh3p is required for maintaining the peroxisomal redox balance under other growth conditions. Besides Mdh3p, several additional peroxisomal enzymes may influence the availability of NAD+. Glycerol 3-phosphate dehydrogenase 1 (Gpd1p) is a highly abundant enzyme that partially localises to peroxisomes in S. cerevisiae even under growth conditions where peroxisome proliferation and β-oxidation are not induced12. It has been proposed to regulate the peroxisomal redox status12,13 but evidence for this is lacking. Interestingly, glycerol-3-phosphate dehydrogenase activity has also been found to be associated with peroxisomes in mammals and Trypanosomes14,15,16,17.
We set out to investigate whether Gpd1p contributes to NAD+ availability inside peroxisomes in S. cerevisiae, under various growth conditions. In agreement with our previous studies, we found that the intraperoxisomal redox balance is maintained predominantly by Mdh3p when cells are grown on oleate medium. We provide evidence that lysine biosynthesis can be maintained by both the Mdh3p- and the Gpd1p-dependent shuttles during growth on glucose medium. We further show that when NAD+-linked saccharopine dehydrogenase is mislocalised to the cytosol, lysine biosynthesis occurs independently of the peroxisomal redox shuttles. We conclude that yeast peroxisomes contain two NAD+-linked shuttles to maintain the availability of intraperoxisomal NAD+ and that the contribution of these shuttles varies between different metabolic conditions.
Results
Gpd1p is partially localised to the peroxisomal matrix but its intraperoxisomal role has remained obscure. It has been suggested that peroxisomal Gpd1p is part of a redox shuttle required to regenerate NAD+ formed during fatty acid oxidation. However, peroxisomal malate dehydrogenase (Mdh3p) performs this role9. mdh3Δ cells are strongly, although not completely, deficient in fatty acid oxidation and show an oleate non-utilising phenotype: limited growth on media containing oleic acid as the sole carbon source (Fig. 2A). gpd1Δ cells grow on oleate medium and utilise oleate as a carbon source, this is evident from the clearance of the medium around the growth area (Fig. 2A). Growth of the mdh3/gpd1Δ double mutant appears to be slightly more affected than the mdh3Δ single mutant. As small growth differences are difficult to judge on oleate containing plates, growth curves were performed which confirmed the growth phenotypes observed on solid media (Fig. 2A). We conclude that Gpd1p is not playing a major role in intraperoxisomal NAD+ regeneration during growth on oleate which is in agreement with previous observations12.

Increased levels of peroxisomal Gpd1p can alleviate the effect of an MDH3 gene deletion. (A) Various S. cerevisiae strains were grown on oleate plates or liquid oleate medium for up to 7 days at 30 °C. (B) Fluorescence microscopy images of gpd1Δ and gpd1/pex3Δ cells expressing either Gpd1p-GFP-PTS1 or Gpd1p-GFP and gpd1Δ cells co-expressing Gpd1p-GFP and HcRed-PTS1. Bar, 5 μm. Signal for Gpd1p-GFP was enhanced compared to that of Gpd1p-GFP-PTS1. Merge is overlay of GFP and HcRed signals. (C) Western blots of subcellular fraction experiments of strains described in (B). H, Homogenate, P, 20,000 g pellet, S, 20,000 g supernatant. Cell equivalents were loaded. (D) Strains transformed with the plasmids indicated were grown at 30 °C on oleate plates or glucose/1 M NaCl plates for 7 days or 2 days, respectively. (−), control plasmid. Transformed strains were also grown on liquid oleate medium for 161 h. (E) 14C-Octanoate β-oxidation activity in intact yeast cells. All strains contained either a control plasmid (−) or expression plasmids encoding different versions of Gpd1p under control of the GPD1 promoter. Strains were grown overnight on oleate medium. WT cells were normalised to 100%. Three independent experiments were performed. Error bars represent standard error of the mean. WT activity: 4741 +/− 873 pmol/hr/OD600.
To test whether Gpd1p could be part of a redox shuttle and function in the regeneration of NAD+ inside peroxisomes, we decided to increase the level of Gpd1p in peroxisomes. Gpd1p-GFP is imported into peroxisomes via the PTS2 pathway but this import is rather inefficient12,13. By appending Gpd1p-GFP with a peroxisomal targeting signal type 1 (PTS1) import occurs more efficiently12. Gpd1p-deficient cells were transformed with plasmids encoding Gpd1p-GFP and Gpd1p-GFP-PTS1 under control of the GPD1 promoter. These cells were subsequently grown on oleate medium and imaged using identical camera settings. Addition of a PTS1 to Gpd1p-GFP results in a decrease in the level of cytosolic labelling concomitant with an increase in intensity of punctate fluorescence upon addition of the PTS1 (Fig. 2B). There is peroxisomal import of only a small fraction of the total Gpd1p-GFP but a punctate pattern is still visible. This is due to the relatively high concentration of Gpd1p-GFP inside peroxisomes compared to in the cytosol. Confirmation that the punctate fluorescence of Gpd1p-GFP and Gpd1p-GFP-PTS1 is due to peroxisomal localisation is evidenced by their mislocalisation to the cytosol in peroxisome-deficient pex3Δ cells. Furthermore, Gpd1p-GFP puncta co-localise with the peroxisomal marker HcRed-PTS1. In addition, homogenates of oleate-grown cells were fractionated by differential centrifugation (Fig. 2C). Gpd1p-GFP-PTS1 fractionates mainly into the 20,000 g pellet together with the peroxisomal marker 3-ketoacyl-CoA thiolase (Fox3p) in gpd1Δ cells. In contrast, Gpd1p-GFP is mainly recovered in the 20,000 g supernatant that includes cytosol and particles with low sedimentation coefficients. In peroxisome-deficient cells (gpd1/pex3Δ) both Gpd1p-GFP and Gpd1p-GFP-PTS1 end up in the 20,000 g supernatant. These experiments confirm that addition of a PTS1 to Gpd1p-GFP increases its localisation to peroxisomes in oleate grown cells.
gpd1Δ cells are sensitive to hyperosmotic growth conditions as they are impaired in the formation and accumulation of glycerol which acts as an osmolyte. As expected, mdh3/gpd1Δ cells also fail to grow on glucose medium containing 1 M NaCl (Fig. 2D). Expression of GFP tagged versions of Gpd1p rescues this growth defect (Fig. 2D) implying that the fusion proteins are enzymatically active.
We measured β-oxidation activity to further analyse the intraperoxisomal role of Gpd1p. gpd1Δ cells are able to oxidise octanoate to near WT levels. Furthermore mdh3Δ cells and mdh3/gpd1Δ cells displayed a strong β-oxidation defect with the latter mutant being slightly more affected (Fig. 2E). Expression of Gpd1p-GFP in mdh3/gpd1Δ cells fails to rescue β-oxidation or growth on oleate to the level of mdh3Δ cells. This suggests that insufficient peroxisomal Gpd1p activity is provided by the Gpd1p-GFP expression construct. However, an increase of peroxisomal Gpd1p activity by expression of Gpd1p-GFP-PTS1 partially rescues the growth defect of mdh3/gpd1Δ cells on oleate medium (Fig. 2D), and rescues fatty acid β-oxidation intermediate to that of WT and mdh3Δ cells (Fig. 2E). We conclude that, although peroxisomal Gpd1p is able to reoxidise NADH to provide intraperoxisomal NAD+, the intraperoxisomal redox balance is maintained predominantly by Mdh3p in oleate grown cells.
S. cerevisiae peroxisomes have been functionally linked to lysine biosynthesis2 and recently, the enzyme required for the ultimate step in the lysine biosynthesis pathway, saccharopine dehydrogenase, was shown to localise to peroxisomes5. Based on a variety of global localisation studies and more detailed analyses, our current model of the spatial organisation of the lysine biosynthetic pathway is summarised in Fig. 1. Saccharopine dehydrogenase, encoded by the LYS1 gene, requires NAD+ for the production of lysine. We tested whether lysine biosynthesis requires a peroxisomal pool of Gpd1p or Mdh3p. Both mdh3Δ cells and gpd1Δ cells grow well on minimal glucose medium in either the presence or the absence of lysine. Interestingly, an mdh3/gpd1Δ double mutant is a lysine bradytroph (Fig. 3A). Both the addition of lysine to this medium or the reintroduction of Gpd1p expression restored growth (Fig. 3B,C). Gpd1p lacking its peroxisomal targeting signal did not restore growth of mdh3/gpd1Δ cells in lysine-deficient glucose medium (Fig. 3B). Furthermore, we have previously shown that replacing the active site residue lysine 245 with alanine, has a deleterious effect on activity. Although the mutant is stably expressed18 and targeted to peroxisomes to a similar extent as wild type Gpd1p-GFP (Fig. 3D), expression of Gpd1p K245A-GFP was unable to restore lysine biosynthesis in mdh3/gpd1Δ cells (Fig. 3E). We conclude that both Mdh3p and peroxisomal Gpd1p activity are required for efficient lysine biosynthesis in glucose grown cells.

Mdh3p and Gpd1p are involved in lysine biosynthesis. (A,B,E) Growth of the strains indicated on solid complete synthetic glucose medium (CSM) either in the presence or absence of L-lysine for 2 days at 30 °C. Gpd1 versions encoded by plasmids were tagged with GFP. (CP), control plasmid. (C) Doubling time of indicated strains during the exponential growth phase in batch cultures on 2% glucose medium lacking lysine. n = 3, error bars indicate standard error of the mean. (D) Fluorescence microscopy images of gpd1Δ cells expressing either Gpd1p-GFP or Gpd1p K245A-GFP grown on glucose medium. Bar, 5 μm.
Besides saccharopine dehydrogenase, an additional NAD+-dependent dehydrogenase (homoisocitrate dehydrogenase encoded by the LYS12 gene) in the pathway contains the C-terminal tripeptide SRL that resembles the prototypic PTS119 and is identical to the PTS1 of Lys1p. This putative PTS1 is conserved in related yeast species, but Lys12p also contains a typical N-terminal mitochondrial targeting signal (MTS) and Lys12p-GFP localises to mitochondria4. Since a PTS1 only acts at the extreme C-terminus of a protein, the use of a C-terminal GFP tag would interfere with peroxisomal targeting. Indeed, tagging proteins at the N-terminus instead of the C-terminus has uncovered many new peroxisomal proteins5. We tagged Lys12p at the N-terminus in a yeast expression plasmid and transformed it into WT cells. GFP-Lys12p did not colocalise with the peroxisomal marker HcRed-PTS1, in contrast to GFP-Lys1p in glucose grown cells. The N-terminal tag does interfere with the mitochondrial targeting signal of Lys12p, hence GFP-Lys12p mislocalised to the cytosol (Fig. 4A). Based on these observations we postulate that the lysine biosynthesis defect in mdh3/gpd1Δ cells is a consequence of a decrease in saccharopine dehydrogenase activity.

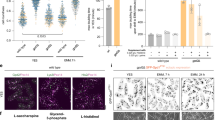
Mdh3p and Gpd1p are required for lysine biosynthesis only when saccharopine dehydrogenase is localised to peroxisomes. (A) Fluorescence microscopy of cells expressing GFP-tagged Lys12p or Lys1p with the peroxisomal matrix marker HcRed-PTS1. Bar, 5 μm. (B) The ratio of saccharopine/lysine in indicated strains grown overnight on 2% glucose medium lacking lysine. (C,D) Growth of the strains indicated on solid complete synthetic glucose medium (CSM) either in presence or absence of L-lysine for 2 days at 30 °C. (E) Doubling time of indicated yeast strains during the exponential growth phase in batch cultures on 2% glucose medium lacking lysine. n = 3, error bars indicate standard error of the mean. (F) Fluorescence microscopy of mdh3/gpd1Δ cells with Lys1p tagged in the genome at its C-terminus with mCherry.
Changes in the redox state in a particular cellular compartment can be determined by measuring changes in the ratio of the concentrations of the oxidised and reduced metabolites of a compartment-specific NAD+-linked dehydrogenase20. A disruption of the peroxisomal NAD+/NADH ratio as a consequence of a block in the redox shuttles should lead to an increase in the Lys1p substrate/product ratio. Indeed, the saccharopine/lysine ratio increases from 0.633 ± 0.03 × 10−3 in WT cells to 51.4 ± 1.98 × 10−3 in mdh3/gpd1Δ cells, an increase of more than 80 fold. In mdh3Δ and gpd1Δ cells the ratios were only slightly increased compared to WT (1.07 ± 0.05 × 10−3 and 1.51 ± 0.02 × 10−3, respectively) (Fig. 4B).
Localisation of Lys1p to peroxisomes is not essential for lysine biosynthesis as peroxisome-deficient cells are not lysine bradytrophs2. We hypothesised that if we mislocalised Lys1p to the cytosol in mdh3/gpd1Δ cells, the cytosolic pool of NAD+ would support Lys1p activity and lysine biosynthesis would be restored. In agreement with our hypothesis, growth on lysine-deficient medium was restored in cells that lack peroxisomes (mdh3/gpd1/pex3Δ cells) and in cells that are deficient in import of PTS1-containing proteins (mdh3/gpd1/pex5Δ cells). In mdh3/gpd1/pex7Δ cells, where PTS2 import is selectively blocked, no growth restoration occurs (Fig. 4C) as Lys1p is imported via the PTS1 pathway. Subsequently, Lys1p was tagged with mCherry at the C-terminus in the genome, which inactivates its PTS1. This results in its cytosolic localisation (Fig. 4F) and as we hypothesised, restores growth on lysine-deficient medium (Fig. 4D,E). This indicates that a decrease of saccharopine dehydrogenase activity (Lys1p-activity) causes lysine bradytrophy in mdh3/gpd1Δ cells. We conclude that two independent shuttles operate to maintain the availability of peroxisomal NAD+. Each shuttle can be sufficiently active in the absence of the other shuttle to support lysine biosynthesis to a level that does not increase cell doubling time in glucose medium.
Discussion
We set out to discern the role of glycerol-3-phosphate dehydrogenase inside peroxisomes. Our experimental data implicates this enzyme in the reoxidation of intraperoxisomal NADH to NAD+. Peroxisomal NAD+ is required to support NAD+-dependent dehydrogenation reactions. Gpd1p is not the only enzyme inside peroxisomes that can fulfil this role. Malate dehydrogenase encoded by the MDH3 gene was previously shown to perform this role when cells are growing on fatty acids as the sole carbon source. Under these growth conditions, cells depend on fatty acid β-oxidation for growth. In mdh3Δ cells, β-oxidation is blocked at the level of 3-hydroxyacyl-CoA dehydrogenase9. By increasing the peroxisomal pool of Gpd1p, we were able to partially overcome this block which illustrates that Gpd1p can substitute for Mdh3p, but normally does not.
Our further studies showed that Gpd1p together with Mdh3p are required for lysine biosynthesis, in particular to maintain saccharopine dehydrogenase activity inside peroxisomes. Two NAD+-dependent dehydrogenases in the lysine biosynthesis pathway (Lys1p and Lys12p) contain a C-terminal tripeptide that fits the PTS1 consensus. Indeed GFP-Lys1p localises to peroxisomes but GFP-Lys12p does not. The presence of a putative PTS1 in Lys12p is intriguing and could point to it being imported into peroxisomes under conditions different from the ones used in this study. Alternatively, the context of the C-terminus of Lys12p could be preventing its recognition by the PTS1 receptor. Two additional observations support our conclusion that during growth on glucose medium, saccharopine dehydrogenase (Lys1p) is the only lysine biosynthetic enzyme that is dependent on the availability of intraperoxisomal NAD+. Firstly, by mislocalising Lys1p to the cytosol in mdh3/gpd1Δ cells, lysine biosynthesis is restored and secondly, in mdh3/gpd1Δ cells the substrate/product ratio for Lys1p is 80 fold increased.
Why S. cerevisiae contains two distinct parallel pathways to reoxidise intraperoxisomal NADH is still unclear. The presence of multiple peroxisomal redox shuttles appears to be widespread throughout the eukaryotic kingdom. For instance, in addition to glycerol 3-phosphate dehydrogenase, mammalian liver peroxisomes contain malate dehydrogenase and lactate dehydrogenase that have been suggested to act as redox shuttles. However, evidence for a role in vivo is lacking21,22,23,24. T. brucei, in its mammalian host, relies exclusively on glycolysis for energy production. Under this condition, the glycerol 3-phosphate dehydrogenase/DHAP shuttle regenerates NAD+ 15. In the insect or procyclic form of T. brucei, intra-glycosomal NAD+ is regenerated via three different pathways, of which the glycerol 3-phosphate dehydrogenase/DHAP shuttle appears to be a back-up pathway25,26.
The requirement for multiple redox shuttles is also seen when considering the distinct shuttles which are operational between the mitochondrial matrix and the cytosol. These shuttles allow exchange of reduction equivalents under varying growth conditions27. Likewise, the presence of multiple peroxisomal shuttles may allow for maintenance of the NAD+/NADH ratio under a wide range of metabolic conditions with the contribution of each shuttle differing as conditions vary. For instance, the malate/oxaloacetate shuttle is important during growth of S. cerevisiae on oleate, a condition where Mdh3p and the glyoxylate cycle enzymes (including Mdh2p) are induced and oxaloacetate will be available. Under these gluconeogenic conditions, either the DHAP availability or the level of Gpd1p inside peroxisomes may be too low for an effective operation of the glycerol 3-phosphate/DHAP shuttle. In contrast, both shuttles operate when cells are grown in glucose medium. The dehydrogenase of one of the shuttles uses the PTS1 import route whilst the dehydrogenase of the other shuttle is imported via the PTS2 pathway. This may further allow for flexibility in the regulation of the two shuttles.
Expression of Gpd1p-GFP in glucose grown mdh3/gpd1Δ cells restored growth on lysine deficient medium, normalised the saccharopine/lysine ratio and supported growth under hyperosmotic conditions. This suggests that the Gpd1p-GFP expression construct is fully functional under these growth conditions. However, on oleate medium it did not seem to be fully active. The reason for this is unclear but may reflect a decrease in peroxisomal import or limited activity of the fusion protein that only becomes apparent under certain growth conditions.
Our study emphasises a central role of peroxisomes in the regulation of NAD+ metabolism. Gpd1p co-imports the nicotinamidase Pnc1p13,18,28. Its role inside peroxisomes is unclear as was the role of Gpd1p. Further experiments are under way to investigate whether the co-import of Pnc1p with Gpd1p is linked to the regulation of the peroxisomal NAD+ pool.
Experimental procedures
Yeast strains, media and growth conditions
Cells were grown at 30 °C in any of the following media: YPD (1% yeast extract, 2% peptone, 2% glucose), minimal media (YM2) for the selection of the uracil (URA3) prototrophic marker (2% glucose, 0.17% yeast nitrogen base without amino acids, 0.5% ammonium sulphate, 1% casamino acids) or minimal media (YM1) for the selection of all prototrophic markers except uracil (2% glucose, 0.17% yeast nitrogen base without amino acids, 0.5% ammonium sulphate). For analysis of growth characteristics, cells were grown on 2% glucose in complete synthetic medium lacking lysine (Formedium, UK). In some cases, as indicated in figure legends, L-lysine was added at 6 mg/ml. Liquid oleate media constituted of 0.5% potassium phosphate buffer pH 6.0, 0.1% yeast extract, leu− drop out (Formedium), 0.17% yeast nitrogen base without amino acids, 0.1% oleate, 0.2% Tween 40. For growth curves, cell density (OD600) was measured after washing samples twice in water.
S. cerevisiae strains used in this study are listed in supplementary Table 1. BY4741 or BY4742 wild type strains and deletion mutants marked with KanMX cassette sequence were obtained from Euroscarf. Any strains created in this study were modified from the BY4741 or BY4742 using the HphMX4 cassette29 and selected on YPD containing 300 µg/ml Hygromycin B (Melford, UK).
Plasmids
Most of the plasmids used in this study, were constructed by gap repair30 in Ycplac derivatives31. LYS1 and LYS12 were amplified by PCR containing 18 nucleotide flanking regions identical to each side of the intended insertion site and inserted into a Tpi1 promoter driven N-terminal expression vector derived from Ycplac33. All GPD1 expression constructs contained 523 bp upstream from the ATG as their promoter regions and have been described previously18. In brief, Gpd1p-mCherry-PTS1 was constructed by fusion of mCherry-SKL to the C-terminus of Gpd1p. ∆PTS2-Gpd1p was constructed by deletion of the first twenty-one amino acids from the N-terminus and reintroduction of a start codon. Peroxisomal marker plasmid HcRed-PTS1 was described previously32.
Image acquisition
Image acquisition and processing was performed as described previously33. Cells were analysed with a microscope (Axiovert 200 M; Carl Zeiss MicroImaging, Inc.) equipped with an Exfo X-cite 120 excitation light source, band pass filters (Carl Zeiss MicroImaging, Inc. and Chroma Technology Corp.), an α-Plan-Fluar 100x/1.45 NA, Plan-Apochromat 63x/1.4 NA or A-plan (Carl Zeiss MicroImaging, Inc.) and a digital camera (Orca ER; Hamamatsu). Image acquisition was performed using Volocity software (Improvision). Fluorescence images were collected as 0.5 μm Z-stacks using exposures of up to 200 ms, merged into one plane in Openlab and processed further in Photoshop (Adobe). Bright field images were collected in one plane.
Subcellular fractionation
Cells grown overnight on oleate medium were converted to spheroplasts with Zymolyase 20T (5 mg/g cells). The spheroplasts were washed twice in ice cold 1.2 M sorbitol/5 mM 2(N-morpholino)ethane sulfonic acid (MES) (pH 6)/1 mM EDTA/1 mM KCl before resuspension in 0.65 M sorbitol/5 mM MES (pH 6)/1 mM EDTA/1 mM KCl (fractionation buffer) containing 1 mM PMSF and Protease inhibitor cocktail. Cell breakage was achieved by careful resuspension of spheroplasts and incubation on ice for 10 min. Intact cells and nuclei were removed by centrifugation (800 g for 10 min). From this homogenate (Hom), 1 ml was transferred to a new Eppendorf tube and centrifuged at 20,000 g for 20 min. The supernatant (S) was collected and the pellet (P) was resuspended in 1 ml fractionation buffer. Equivalent volumes of these fractions were analysed by SDS–PAGE and immunoblotting. Monoclonal anti-GFP antibody was obtained from Roche (11814460001), and anti-Pgk1 22C5 from Invitrogen. Anti-yeast Thiolase was a kind gift from Ben Distel. Secondary antibody was HRP-linked Goat anti-mouse polyclonal (BioRad 1706516). Blots were blocked in 2% (w/v) fat-free Marvel™ milk in TBS-Tween 20 (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% (v/v) Tween 20). Immunoreactive proteins were detected by enhanced chemiluminescence (Biological Industries) and blots were imaged with a G box (Syngene).
β-Oxidation activity measurements
β-oxidation assays in intact yeast cells were performed as described previously9 and optimized for the pH and the amount of protein. Oleate-grown cells were washed in water and resuspended in 0.9% NaCl (OD600 = 1.0). Aliquots of 20 μl of cell suspension were used for β-oxidation measurements in 200 μl of 50 mM MES (pH 6.2) and 0.9% (w/v) NaCl supplemented with 10 μM [1-14C]-octanoate. After 1 hr at 28 °C, the incubation was stopped with 50 μl 2.6 M perchloric acid (PCA). Subsequently, [1-14C]-CO2 was trapped with 500 μl 2 M NaOH. The 14C labelled β-oxidation products were then collected after extracting the acidified material with cloroform/methanol/heptane and quantified in a liquid scintillation counter. The quantification of the rate of fatty acid oxidation was measured by the summation of the amount of [1-14C]-CO2 and 14C-labelled β-oxidation products. Results are presented as percentage relative activity to the rate of oxidation of wild type cells.
Saccharopine/lysine ratio measurement
Yeast cells were grown overnight on 2% glucose and intact cells were harvested at 4 °C. 0.5 OD600 units of cells were resuspended in 50 μl PBS and transferred to a 1.5 ml Eppendorf tube. 0.5 ml of 100% ACN plus 20 µl of internal standard mixture (containing 32 nmol d4-lysine) were added and homogenized for 15 sec by vortexing. Samples were centrifuged for 10 min at 4 °C at a speed of 12,000 g. The supernatant was transferred to a 4 ml glass vial and evaporated under a stream of nitrogen at 40 °C. After evaporation, 110 µl of 0.01% heptafluorobutyric acid was added to dissolve the residue and mixed by vortexing. This suspension was transferred to a Gilson vial for HPLC-MS/MS analysis as described previously34. For the calculation of AA concentrations, 50 µl standards containing 0, 30, 60, 125 or 250 μM lysine or saccharopine in PBS were added to the internal standard (20 µl, same composition as mentioned above) and analyzed as described above.
References
Waterham, H. R., Ferdinandusse, S. & Wanders, R. J. Human disorders of peroxisome metabolism and biogenesis. Biochim Biophys Acta 1863, 922–933 (2016).
Breitling, R., Sharif, O., Hartman, M. L. & Krisans, S. K. Loss of compartmentalization causes misregulation of lysine biosynthesis in peroxisome-deficient yeast cells. Eukaryot Cell 1, 978–986 (2002).
Xu, H., Andi, B., Qian, J., West, A. H. & Cook, P. F. The alpha-aminoadipate pathway for lysine biosynthesis in fungi. Cell Biochem Biophys 46, 43–64 (2006).
Huh, W. K. et al. Global analysis of protein localization in budding yeast. Nature 425, 686–691 (2003).
Yofe, I. et al. One library to make them all: streamlining the creation of yeast libraries via a SWAp-Tag strategy. Nat Methods 13, 371–378 (2016).
Antonenkov, V. D. & Hiltunen, J. K. Transfer of metabolites across the peroxisomal membrane. Biochim Biophys Acta 1822, 1374–1386 (2012).
Bernhardt, K., Wilkinson, S., Weber, A. P. & Linka, N. A peroxisomal carrier delivers NAD(+) and contributes to optimal fatty acid degradation during storage oil mobilization. Plant J 69, 1–13 (2012).
van Roermund, C. W. et al. The Peroxisomal NAD Carrier from Arabidopsis Imports NAD in Exchange with AMP. Plant Physiol 171, 2127–2139 (2016).
van Roermund, C. W., Elgersma, Y., Singh, N., Wanders, R. J. & Tabak, H. F. The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions. EMBO J 14, 3480–3486 (1995).
Visser, W. F., van Roermund, C. W., Ijlst, L., Waterham, H. R. & Wanders, R. J. Metabolite transport across the peroxisomal membrane. Biochem J 401, 365–375 (2007).
Veenhuis, M., Mateblowski, M., Kunau, W. H. & Harder, W. Proliferation of microbodies in Saccharomyces cerevisiae. Yeast 3, 77–84 (1987).
Jung, S., Marelli, M., Rachubinski, R. A., Goodlett, D. R. & Aitchison, J. D. Dynamic changes in the subcellular distribution of Gpd1p in response to cell stress. J Biol Chem 285, 6739–6749 (2010).
Kumar, S., Singh, R., Williams, C. P. & van der Klei, I. J. Stress exposure results in increased peroxisomal levels of yeast Pnc1 and Gpd1, which are imported via a piggy-backing mechanism. Biochim Biophys Acta 1863, 148–156 (2016).
Gee, R., McGroarty, E., Hsieh, B., Wied, D. M. & Tolbert, N. E. Glycerol phosphate dehydrogenase in mammalian peroxisomes. Arch Biochem Biophys 161, 187–193 (1974).
Opperdoes, F. R. & Borst, P. Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: the glycosome. FEBS Lett 80, 360–364 (1977).
Murphy, P. A., Krahling, J. B., Gee, R., Kirk, J. R. & Tolbert, N. E. Enzyme activities of isolated hepatic peroxisomes from genetically lean and obese male mice. Arch Biochem Biophys 193, 179–185 (1979).
Antonenkov, V. D. Dehydrogenases of the pentose phosphate pathway in rat liver peroxisomes. Eur J Biochem 183, 75–82 (1989).
Al-Saryi, N. A. et al. Pnc1 piggy-back import into peroxisomes relies on Gpd1 homodimerisation. Sci Rep 7, 42579 (2017).
Gould, S. J., Keller, G. A., Hosken, N., Wilkinson, J. & Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol 108, 1657–1664 (1989).
Williamson, D. H., Lund, P. & Krebs, H. A. The redox state of free nicotinamide adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 103, 514–527 (1967).
Baumgart, E., Fahimi, H. D., Stich, A. & Volkl, A. L-lactate dehydrogenase A4- and A3B isoforms are bona fide peroxisomal enzymes in rat liver. Evidence for involvement in intraperoxisomal NADH reoxidation. J Biol Chem 271, 3846–3855 (1996).
McClelland, G. B., Khanna, S., Gonzalez, G. F., Butz, C. E. & Brooks, G. A. Peroxisomal membrane monocarboxylate transporters: evidence for a redox shuttle system? Biochem Biophys Res Commun 304, 130–135 (2003).
Wiese, S. et al. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol Cell Proteomics 6, 2045–2057 (2007).
Gronemeyer, T. et al. The proteome of human liver peroxisomes: identification of five new peroxisomal constituents by a label-free quantitative proteomics survey. PLoS One 8, e57395 (2013).
Ebikeme, C. et al. Ablation of succinate production from glucose metabolism in the procyclic trypanosomes induces metabolic switches to the glycerol 3-phosphate/dihydroxyacetone phosphate shuttle and to proline metabolism. J Biol Chem 285, 32312–32324 (2010).
Guther, M. L., Urbaniak, M. D., Tavendale, A., Prescott, A. & Ferguson, M. A. High-confidence glycosome proteome for procyclic form Trypanosoma brucei by epitope-tag organelle enrichment and SILAC proteomics. J Proteome Res 13, 2796–2806 (2014).
Bakker, B. M. et al. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev 25, 15–37 (2001).
Effelsberg, D., Cruz-Zaragoza, L. D., Tonillo, J., Schliebs, W. & Erdmann, R. Role of Pex21p for Piggyback Import of Gpd1p and Pnc1p into Peroxisomes of Saccharomyces cerevisiae. J Biol Chem 290, 25333–25342 (2015).
Goldstein, A. L. & McCusker, J. H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553 (1999).
Orr-Weaver, T. L., Szostak, J. W. & Rothstein, R. J. Genetic applications of yeast transformation with linear and gapped plasmids. Methods Enzymol 101, 228–245 (1983).
Gietz, R. D. & Sugino, A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 (1988).
Motley, A. M. & Hettema, E. H. Yeast peroxisomes multiply by growth and division. J Cell Biol 178, 399–410 (2007).
Motley, A. M., Galvin, P. C., Ekal, L., Nuttall, J. M. & Hettema, E. H. Reevaluation of the role of Pex1 and dynamin-related proteins in peroxisome membrane biogenesis. J Cell Biol 211, 1041–1056 (2015).
Gao, A. W. et al. A sensitive mass spectrometry platform identifies metabolic changes of life history traits in C. elegans. Sci Rep 7, 2408 (2017).
Acknowledgements
Nadal A. Al-Saryi and Murtakab Y. Al-Hejjaj were funded by a PhD scholarship from the Higher Committee for Education Development in Iraq. Georgia Hulmes was funded by a University of Sheffield PhD studentship and Lakhan Ekal by a University of Sheffield Vice Chancellor’s Indian Scholarship. Chantell Payton was supported by a School of Life Sciences studentship from the University of Lincoln, UK. We also thank Arno van Cruchten for technical assistance.
Author information
Authors and Affiliations
Contributions
N.A.S., M.Y.A., C.v.R., G.H., L.E., C.P. and E.H. performed the experiments. All authors interpreted the results. E.H., C.v.R. and R.W. designed the study. E.H. wrote the manuscript with the input of remaining authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Saryi, N.A., Al-Hejjaj, M.Y., van Roermund, C.W.T. et al. Two NAD-linked redox shuttles maintain the peroxisomal redox balance in Saccharomyces cerevisiae . Sci Rep 7, 11868 (2017). https://doi.org/10.1038/s41598-017-11942-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11942-2
This article is cited by
-
Reprogramming methanol utilization pathways to convert Saccharomyces cerevisiae to a synthetic methylotroph
Nature Catalysis (2023)
-
Comparative metabolomic analysis of furfural stress response in Aspergillus terreus
Cellulose (2019)
-
Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages
Cell Death & Disease (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
