
Abstract
Marcha and thiat are traditionally prepared amylolytic starters use for production of various ethnic alcoholic beverages in Sikkim and Meghalaya states in India. In the present study we have tried to investigate the bacterial and fungal community composition of marcha and thiat by using high throughput sequencing. Characterization of bacterial community depicts phylum Proteobacteria is the most dominant in both marcha (91.4%) and thiat (53.8%), followed by Firmicutes, and Actinobacteria. Estimates of fungal community composition showed Ascomycota as the dominant phylum. Presence of Zygomycota in marcha distinguishes it from the thiat. The results of NGS analysis revealed dominance of yeasts in marcha whereas molds out numbers in case of thiat. This is the first report on microbial communities of traditionally prepared amylolytic starters of India using high throughput sequencing.
Similar content being viewed by others
Introduction
Traditional practice of sub-culturing by back-sloping and preservation of essential native microbiota consisting of consortia of yeasts, molds and bacteria, in the form of dry, flattened, or round balls, for alcoholic beverages production in South-East Asia including the Himalayan regions of India, Nepal, Bhutan, and China is the worth wisdom of the ethnic people for centuries1. Some common and uncommon amylolytic starters in Asia are marcha of India, Nepal, and Bhutan, hamei, humao, thiat, phab of India, men of Vietnam, bubod of the Philippines, chiu/chu of China and Taiwan, loogpang of Thailand, ragi of Indonesia, nuruk of Korea, mae/dombae/buh/puhin Cambodia, etc.2,3,4,5,6,7 Traditionally prepared Asian amylolytic starters have consortia of mixed micrbiota representing filamentous molds, yeast and bacteria1,2,3, hence many researchers have studied the fungal, yeast and bacterial populations in Asian starter cultures, commonly based on culture-dependent techniques including phenotypic and 16S rRNA sequencing, and isolated and identified filamentous molds Absidia corymbifera, Amylomyces rouxii, Botryobasidium subcoronatum, Mucor circinelloides forma circinelloides, Mucor hiemalis, Rhizopus oryzae, Rhi. microsporus, Rhi. chinensis, and Rhi. stolonifer, Xeromyces bisporus5, 8, 9; yeasts Candida glabrata, C. tropicalis, Clavispora lusitaniae, Issatchenkia sp., Pichia anomala, P. ranongensis, P. burtonii, Saccharomycopsis fibuligera, Sm. capsularis, Saccharomyces cerevisiae, Sacch. Bayanus 5, 9,10,11,12,13; and bacteria Acetobacter orientalis, A. pasteurianus, Bacillus amyloliquefaciens, B. circulans, B. sporothermodurans, B. subtilis, Pediococcus pentosaceus, Lactobacillus bifermentans, Lb. brevis, Lb. plantarum, Weissella confusa, W. paramesenteroides 5, 14,15,16.
Introduction of culture-independent methods and it’s applicability in food microbiology7, 17, has been a motivation for few researchers to profile the microbial community structure of some Asian starter cultures using PCR-DGGE, pyrosequencing, etc. which is suggestive to provide more insight into the microbial diversity of ethnic starters3, 5, 18,19,20,21,22. Rapid evolution in next generation sequencing (NGS) technologies has enabled researchers to have increased accuracy, throughput, with reasonably low cost and in relatively short period of time17, 23. However, there are still a limited number of studies, characterizing the microbial community composition of fermented foods such as cheese24,25,26, kefir grains27, some ethnic Asian fermented foods28,29,30,31. Furthermore, the information on the community composition of Asian starter culture is rudimentary and needs in depth exploration using cutting edge technologies7.
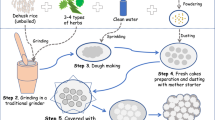
In present study, we attempted to profile the microbial community composition of marcha and thiat, traditionally prepared ethnic starter cultures of India using targeted amplicon sequencing. We selected two different traditionally prepared amylolytic starter cultures from two regions in India, marcha (Fig. 1a) from Sikkim (www.sikkim.gov.in) and thiat (Fig. 1b) from Meghalaya (www.megtourism.gov.in). Marcha is prepared from soaked rice with some wild herbs ((Plumbago zeylanica, Buddleja asiatica and Vernonia cinerea), ginger and red dry chili, 1–2 % of previously prepared marcha powder as an inoculum, crushed in a wooden mortal by wooden pestle, mised and dough are made into round to flatted cakes of different size and shape. Cakes are covered with fern fronds (Glaphylopteriolopsis erubeseens), fermented at room temperature for 24 h, sun dried for 3–5 days and are used as amylolytic starters for production of cereal-based ethnic fermented beverages such as kodo ko jaanr, bhaati jaanr, raksi, etc.2 During thiat preparation, soaked glutinous rice is grinded with leaves and roots of wild plant Amomum aromaticum, 1–2% of old thiat, mixed and made into a dough by adding water. Flat to round balls are made and fermented for 1–3 days. The freshly prepared thiat balls are sun dried for 3–5 days. It is used to ferment alcoholic beverage locally called kiad in Meghalaya2. Fermentation process involved in preparation of these starters is unconditional and may harbor both bacterial and fungal communities as consortia. Therefore, we aimed to explore the bacterial and fungal (filamentous molds and yeasts) communities in marcha and thiat. This is the first report on complete microbial community profile of traditionally prepared amylolytic starters of India using NGS technique.

Traditionally prepared amylolytic starter cultures (a) Marcha and (b) Thiat.
Results
Characterizing microbial diversity
High throughput sequencing and quality trimming of 16S rRNA and ITS gene yielded ~0.85and ~0.29 million quality reads in both marcha and thiat, respectively, which was used for subsequent analysis. Taxonomic assignment of sequences with the reference database resulted into 5,015 operational taxonomic units (OTUs). The average Good’s coverage of both the samples of marcha and thiat for 16Sr RNA amplicon sequencing was found to be 99.08% ± 0.1% (mean ± SD) whereas for ITS region was recorded as 87.5% ± 17.6% (mean ± SD) indicating majority of the diversity was captured.
The estimates of alpha diversity indices revealed significant differences between marcha and thiat when computed for both the bacterial and fungal diversity (Table 1a and b). The bacterial species richness was found to be higher in thiat (4256.83) than marcha (1520.92), in contrast, fungal species richness depicts higher in marcha (5.25) over thiat (5.0). Significant variations were also noticed in non-parametric shannon index for bacterial communities in thiat (5.48) and marcha (4.01). Shannon index for fungal communities follow the reverse trend with marcha (2.25) and thiat (1.80). This observation is suggestive of higher bacterial diversity in thiat while marcha showed higher fungal diversity.
Bacterial community profile of thiat and marcha
16S rRNA gene amplicon sequencing yielded 15 bacterial phyla in thiat and marcha, respectively (Fig. 2a). In thiat bacterial phyla distributions were Proteobacteria (91.4%), Actinobacteria (4%), Firmicutes (4%) and the rest (0.6%) constituted the minor phyla Cyanobacteria, Bacteroidetes, Verrucomicrobia, Fusobacteria, Planctomycetes, Deinococcus-Thermus, Chloroflexi, Synergistetes, Acidobacteria, Saccharibacteria, Gemmatimonadetes, Armatimonadetes. In marcha the phyla distributions of bacteria were Proteobacteria (53.8%), Firmicutes (45.4%) and other minor phyla were 0.8% constituting Actinobacteria, Cyanobacteria, Bacteroidetes, Verrucomicrobia, Fusobacteria, Planctomycetes, Deinococcus-Thermus, Chloroflexi, Synergistetes, Acidobacteria, Saccharibacteria, Gemmatimonadetes, and Armatimonadetes. The abundance of thirteen minor phyla was very less hence percentage of composition was not shown in Fig. 2a. Bacterial phylum Proteobacteria was found to outnumber other bacterial phyla in thiat whereas marcha was found to constitute Proteobacteria and Firmicutes as major phyla.

Taxa distributions of phylum and family at different phylogenetic level in thiat and marcha. (a) bacterial phyla; (b) bacterial family; (c), fungal phyla and (d) fungal family.
At family level, OTUs with ≥1% abundance were filtered which differed quantitatively between thiat and marcha (Fig. 2b). The family level distributions of bacteria in thiat were Enterobacteriaceae (84.6%), Microbacteriaceae (3.24%), Enterococcaceae (2.47%), Clostridiaceae (1.13%) Neisseriaceae (0.87%) and Oxalobacteraceae (0.59%) (Fig. 2b). Whereas the family level of bacterial distributions in marcha were Acetobacteraceae (50.6%), Leuconostocaceae (25.5%), Streptococcaceae (10.5%), Lactobacillaceae (8.38%), Burkholderiaceae (2.13%) and Staphylococcaceae (0.54%) (Fig. 2b).
At the genus level, OTUs with ≥1% abundance were filtered (Fig. 3a,b), which retained 18 differentially abundant genera in both samples of marcha and thiat. Distribution of bacterial genera in marcha were Acetobacter (52.6%), Fructobacillus (21.1%), Lactococcus (10.3%), Lactobacillus (8.4%), Leuconostoc (4.0%) (Fig. 3a), Burkholderia (2.1%) and Gluconacetobacter (1.4%). Genera in thiat were Pantoea (32.4%), Cronobacter (21.4%), Escherichia-Shigella (15.5%), Enterobacter (13.1%), Citrobacter (4.2%) (Fig. 3b), Salmonella (3.2%), Serratia (2.8%), Enterococcus (2.5%), Curtobacterium (2.2%), Kluyvera (1.6%) and Clostridium (1.1%). The composition percentage of bacterial genera which was less than 3.9% was not shown in Fig. 3a,b.

Taxa distributions of genus at different phylogenetic level. (a) bacterial genera in marcha; (b) bacterial genera in thiat; (c) fungal genera in marcha and (d) fungal genera in thiat.
Fungal (filamentous molds and yeasts) composition in thiat and marcha
Fungal ITS gene sequencing and taxonomic analysis demonstrated the predominance of yeast phylum Ascomycota (98.6%) in thiat, whereas the distribution of filamentous phyla Zygomycota was only 1.4% (Fig. 2c). However, in marcha only yeast phylum Ascomycota constituted the fungal diversity (Fig. 2c). Filamentous mold phylum was not detected in marcha. Distributions of fungi (filamentous molds and yeasts) at the family level in thiat were Trichocomaceae (15.7%), Dothioraceae (3.94%), Mucoraceae (2.63%) and unidentified fungi (77.73%). Whereas the distributions of yeasts at the order/family level in marcha were Saccharomycetales (50%), Saccharomycetaceae (37.5%) and Amphisphaeriaceae (12.5%). (Fig. 2d). Distributions of yeasts genera in marcha were Wickerhamomyces (25%), Candida (25%), Kazachstania (25%), Saccharomyces (12.5%) and Pestalotiopsis (12.5%) (Fig. 3c). The filamentous mold genera distribution in thiat were Aspergillus (15.7%), Aureobasidium (3.9%) and Mucor (2.7%) and unidentified genera (77.7%) (Fig. 3d). The unidentified genera represented the yeast phylum Ascomycota in thiat. The sequence reads showed the species of filamentous molds were Aspergillus penicillioides, Aureobasidium pullulans and Mucor circinelloides, whereas the yeasts species were Wickerhamomyces anomalus, Candida quercitrus and Kazachstania exigua (data not shown).
Discussion
Our study provides comprehensive microbial diversity analysis using deep sequencing approach of ethnic amylolytic starter from India. Quantitative differences were noted for the presence of bacterial and fungal taxa among marcha and thiat; which could be the consequence of differences in the preparation, incubation period and most importantly the type of preservations. Alpha diversity estimation of amylolytic starters marcha and thiat using species richness and non-parametric Shannon index suggested higher bacterial diversity in thiat while marcha shows the higher assemblage of fungal diversity with dominance of yeast phylum Ascomycota. Persistence of higher fungal diversity in marcha is determinant factor suggesting the higher acidic conditions of marcha; in contrast, higher bacterial diversity of thiat depicts the faster turnover from acidic to alkali with the presence of acid neutralizing bacterial taxa32.
Acetobacter, Fructobacillus, Lactococcus, Lactobacillus, Leuconostoc, Burkholderia, and Gluconacetobacter were the predominant bacterial genera in marcha. Higher proportion of Acetobacter was possibly due to its retention and enrichment during fermentation. We observed relatively lower proportion of Streptococcus and Lactococcus than Lactobacilli; as Lactobacilli have high acid tolerance over former two33. Though some species of Lactococcus have low acid tolerance, however, they could be isolated from raw milk and were found flourishing during the early stage of fermentation24. This supports the lower abundance of Lactococcus than Lactobacillus as seen in our samples. Another interesting observation was absence of Pediococcus in bacterial community profile which was otherwise present as a one of the dominant genus in earlier report by culture dependent methods in marcha 10, 16. Furthermore, since there is no earlier report on microbial composition based on culture dependent or culture-independent methods of thiat the present study describe microbial diversity of thiat using NGS method as its first report. Pantoea, Cronobacter, Escherichia, Shigella, Enterobacter, Citrobacter, Salmonella, Serratia, and Enterococcus depicts most dominant bacterial genera of thiat each comprised over 0.1% of total bacterial sequences. Significantly varied microbial composition among thiat and marcha is a clear indication of differences in amylolytic starters. Genus Enterobacter was also detected in Mexican alcoholic beverages speculated to originate from the bacterial contamination in raw milk and they subsequently decreased during the fermentation process34. The lactic acid bacterium such as Lb. plantarum seemed to be one factor for the good quality of the alcoholic beverages, as it can perform malolactic fermentation to decrease wine acidity32 and also produces bacteriocins35.
Exploration of fungal diversity of ethnic amylolytic starters suggested higher abundance of yeast in marcha and thiat constitutes for 32.33-fold yeast to the filamentous molds. This observation was in coherence with the earlier report of culture-dependent studies showing the dominance of Mucor and Rhizopus genera of Mucorales in marcha 8. Interestingly no filamentous molds were detected in marcha using the applied high throughout sequencing method; the exact reasons for the observed variation in the microbiota have not been identified. This may be due to lower abundance of molds, limited sample size and/or age of the sample and finally also due to inadequate cell lysis which may prevent the release of nucleases36. Our study was in accordance to the previous reports describing the exposure of cheese to different external environments such as manufacturing process; geographical region, etc have varied impact on the microbial composition of the end products28. Thus, we speculate that the factor of geographic environment including altitudes and climate play a more significant role over the manufacturing process in resulting in the different microbial compositions of the starter culture under study. Some other crucial factors that may affect the composition of microbial communities in fermented amylolytic starters are level of hygiene, quality of the glutinous rice, water, as well as the back slopping technique. In this study three dominant yeasts in marcha were Wickerhamomyces anomalus, Candida quercitrus and Kazachstania exigua, followed by Saccharomyces and Pestalotiopsis which also accompany the findings of ref. 21 by PCR-DGGE method. ITS gene sequences analysis of the thiat revealed the existence of Aspergillus penicillioides, Aureobasidium pullulans and Mucor circinelloides as the most dominant filamentous molds in thiat. At family level thiat shows Trichocomaceae, Dothioraceaeand Mucoraceaeas the major constituents of fungal community composition emphasizing the significant differences between thiat and marcha viz differences in starter substrates, preparations, inoculums, consortia, geography, hygiene, preservation technique, caloric values etc.
In the present study Ascomycota was dominant in starter cultures of India like in Korean and Chinese starters cultures, which was also reported earlier, based on NGS tools, in Korean alcoholic beverages3 and in Chinese liquors37. We could also expect similar observation in case of marcha as it has higher abundance of lactic acid bacteria. Aspergullus oryzae has strong secretion of amylases including alpha-amylase, which may accelerate the degradation of grains and provide more nutrients for microbes in alcoholic fermentation38.
Amylolytic starter culture-making technology reflects the traditional knowledge of the ethnic Indian people on sub-culturing desirable inocula from previous batch to new culture using rice as base substrates by back-sloping method. This technique preserves the consortia of microbial community ranging from filamentous molds, yeasts and bacteria which were co-existed in traditionally prepared amylolytic and alcohol producing starters7, and also preserves vast biological genetic resources, otherwise, which may be forced to disappear. Fermented beverages produced by using amylolytic starters in India are generally mild-alcoholic (4–5%), sweet taste with several health benefits to the local consumers as high source of calories, some vitamins and minerals2. Ethnic fermented beverages and alcoholic drinks have the potential to grow beverage industry if proper scientific and technical support are applied to the existing indigenous practices of home based alcoholic fermentation.
Materials and Methods
Sample collection
Samples of sun-dried amylolytic starters marcha and thiat were collected immediately after the preparation from local people of Gangtok and Shillong in Sikkim and Meghalaya states of India, respectively. Dry samples were transferred to sterile containers, sealed, and stored at desiccator at room temperature for the further analysis.
Community DNA Extraction
The total community DNA was extracted using ProMega DNA kit (ProMega). 1g of amylolytic starter culture sample was suspended in lysis solution and incubated at 65 °C for 15 min. Subsequently, the RNA was eliminated from the cellular lysate by administering the RNase solution following incubation at 35 °C for 15 min. The residual proteins were removed by adding protein precipitation solution and centrifuged at max speed. Finally, the DNA was precipitated by adding isopropanol, which was purified with two washes of 70% ethanol. The quality of DNA was checked on 0.8% agarose gel and concentration was measured using Nano-DropND-1000 spectrophotometer (Nano Drop technologies, Willington, USA) as described by ref. 39. The DNA was stored at −20 °C until further processing.
Amplicon sequencing
Amongst the nine hypervariable regions of bacterial 16S rRNA gene, we have targeted V4 hyper-variable region40 to investigate bacterial diversity of marcha and thiat. The universal 16S rRNA gene primer sets F515 and 806R41 was used for the amplification of V4 hyper-variable region. Similarly, fungal Internal Transcribed Spacer (ITS) II region was targeted for taxonomic profiling amylolytic starters, which was subjected to amplification using ITS1 and ITS2 primers. The library preparation of both the 16S rRNA and ITS gene amplicons were in accordance with the protocols of Illumina (USA). These amplicon libraries were further processed for sequencing using AMPure XT beads (Beckman Coulter Genomics, Danvers, MA, USA). The resultant product was screened with the LabChip GX (Perkin Elmer, Waltham, MA, USA) and with the Library Quantification Kit for Illumina (Kapa Biosciences, Woburn, MA, USA). Subsequently, the 16S rRNA and ITS gene library were sequenced on the Illumina MiSeq platform using 2x 250bp chemistry. The sequences obtained from high throughput sequencing effort were submitted to National Center for Biotechnology Information (NCBI) which are available under BioProject ID PRJNA376467.
Bioinformatics analysis
The raw sequences generated from MiSeq platform was assembled using FLASH tool (Fast Length Adjustment of Short reads) a Paired end assembler for DNA sequences42. The assembled reads were subjected to quality filtering using via Quantitative Insights into Microbial Ecology (QIIME) 1.842. Sequence reads were assigned to bacterial and fungal operational taxonomic units (OTUs) by a closed reference-based OTU picking approach by using SILVA and UNITE reference databases, respectively. The OTU picking was carried out using UCLUST method with similarity threshold of 97%43. Taxonomic assignments were performed using RDP naïve bayesian classifier44. Alpha diversity indices like Chao, Shannon and Simpson were calculated via QIIME after rarefying all samples to the same sequencing depth45, 46.
Data availability
The sequences obtained from high throughput sequencing effort, was submitted to NCBI which are available under Bio Project ID PRJNA376467.
References
Tamang, J. P. & Kailasapathy, K. Fermented foods and beverages of the world. CRC Press (Eds.), Taylor and Francis Group, New York (2010).
Tamang, J. P. Himalayan fermented foods: microbiology, nutrition and ethnic value, CRC Press, Taylor and Francis Group, New York (2010).
Jung, M. J., Nam, Y. D., Roh, S. W. & Bae, J. W. Unexpected convergence of fungal and bacterial communities during fermentation of traditional Korean alcoholic beverages inoculated with various natural starters. Food Microbiol. 30, 112–23 (2012).
Sujaya, I. et al. Identification and characterization of yeasts in brem, a traditional Balinese rice wine. World J. Microbiol. Biotechnol. 20, 143–150 (2004).
Thanh, V. N., Mai, L. T. & Tuan, D. A. Microbial diversity of traditional Vietnamese alcohol fermentation starters (banh men) as determined by PCR-mediated DGGE. Int. J. Food Microbiol. 128, 268–273 (2008).
Yamamoto, S. & Matsumoto, T. Rice fermentation starters in Cambodia: cultural importance and traditional methods of production. Southeast Asian Studies. 49, 192–213 (2011).
Tamang, J. P., Watanabe, K. & Holzapfel, W. H. Review: Diversity of microorganisms in global fermented foods and beverages. Front. Microbiol. 7, 377, doi:10.3389/fmicb.2016.00377 (2016).
Tamang, J. P., Sarkar, P. K. & Hesseltine, C. W. Traditional fermented foods and beverages of Darjeeling and Sikkim - a review. J. Sci. Food Agric. 44, 375–385 (1988).
Dung, N. T. P., Rombouts, F. M. & Nout, M. J. R. Functionality of selected strains of moulds and yeasts from Vietnamese rice wine starters. Food Microbiol. 23, 331–340 (2006).
Tamang, J. P. & Sarkar, P. K. Microflora of murcha: an amylolytic fermentation starter. Microbios. 81, 115–122 (1995).
Tsuyoshi, N. et al. Identification of yeast strains isolated from marcha in Sikkim, a microbial starter for amylolytic fermentation. Int. J. Food Microbiol. 99, 135–146 (2005).
Jeyaram, K., Mohendro Singh, W., Capece, A. & Romano, P. Molecular identification of yeast species associated with ‘Hamei”- a traditional starter used for rice wine production in Manipur, India. Int. J. Food Microbiol. 124, 115–125 (2008).
Jeyaram, K., Tamang, J. P., Capece, A. & Romano, P. P. Geographical markers for Saccharomyces cerevisiae strains with similar technological origins domesticated for rice-based ethnic fermented beverages production in North East India. Antonie van Leeuwenhoek. 100, 569–578 (2011).
Dung, N. T. P., Rombouts, F. M. & Nout, M. J. R. Characteristics of some traditional Vietnamese starch-based rice wine starters (Men). LWT-Food Sci. Technol. 40, 130–135 (2007).
Hesseltine, C. W. & Ray, M. L. Lactic acid bacteria in murcha and ragi. J. Appl. Bacteriol. 64, 395–401 (1988).
Tamang, J. P. et al. Lactic acid bacteria in Hamei and Marcha of North East India. Indian J. Microbiol. 47, 119–125 (2007).
Cocolin, L., Alessandria, V., Dolci, P., Gorra, R. & Rantsiou, R. Culture independent methods to assess the diversity and dynamics of microbiota during food fermentation. Int. J. Food Microbiol. 167, 29–43 (2013).
Chao, S. H. et al. Microbial diversity analysis of fermented mung beans (Lu-Doh-Huang) by using pyrosequencing and culture methods. PLOS ONE. 8(5), e63816 (2013).
Chen, B., Wu, Q. & Xu, Y. Filamentous fungal diversity and community structure associated with the solid state fermentation of Chinese Maotai-flavor liquor. Int. J. Food Microbiol. 179, 80–84 (2014).
Lv, X. C., Huang, X. L., Zhang, W., Rao, P. F. & Ni, L. Yeast diversity of traditional alcohol fermentation starters for Hong Qu glutinous rice wine brewing, revealed by culture-dependent and culture-independent methods. Food Control. 34, 183–190 (2013).
Sha, S. P., Anupma, A., Pradhan, P., Prasad, G. S. & Tamang, J. P. Identification of yeasts by PCR-mediated DGGE in marcha, an ethnic amylolytic starter of India. J. Ethnic Foods. 3, 292–296 (2016).
Zhu, L. et al. BOX-PCR and PCR-DGGE analysis for bacterial diversity of a naturally fermented functional food (Enzyme). Food Bioscience. 5, 115–122 (2014).
Mayo, B. et al. Impact of next generation sequencing techniques in food microbiology. Curr. Genomics. 15(4), 293–309 (2014).
Alegría, Á., Szczesny, P., Mayo, B., Bardowski, J. & Kowalczyk, M. Biodiversity in Oscypek, a traditional Polish cheese, determined by culture-dependent and-independent approaches. Appl. Environ. Microbiol. 78, 1890–8 (2012).
Quigley, L. et al. High-throughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses. Appl. Environ. Microbiol. 78, 5717–23 (2012).
Ercolini, D., De Filippis, F., La Storia, A. & Iacono, M. A “remake” of the microbiota involved in the production of water buffalo mozzarella cheese by high throughput sequencing. Appl. Environ. Microbiol. 78, 8142–8145 (2012).
Bourrie, B. C. T., Willing, B. P. & Cotter, P. D. The microbiota and health promoting characteristics of the fermented beverage kefir. Front. Microbiol. 7, 647 (2016).
Nam, Y. D., Lee, S. Y. & Lim, S. I. Microbial community analysis of Korean soy- bean pastes by next-generation sequencing. Int. J. Food Microbiol. 155, 36–42 (2012).
Romi, W., Ahmed, G. & Jeyaram, K. Three-phase succession of autochthonous lactic acid bacteria to reach a stable ecosystem within 7 days of natural bamboo shoot fermentation as revealed by different molecular approaches. Mol. Ecol. 24, 3372–3389 (2015).
Keisam, S., Romi, W., Ahmed, G. & Jeyaram, K. Quantifying the biases in metagenome mining for realistic assessment of microbial ecology of naturally fermented foods. Sci. Rep. 6, 34155 (2016).
Zhang, J. et al. Metagenomic approach reveals microbial diversity and predictive microbial metabolic pathways in Yucha, a traditional Li fermented food. Sci. Rep. 6, 32524 (2016).
Kosseva, M., Beschkov, V., Kennedy, J. F. & Lloyd, L. L. Malolactic fermentation in Chardonnay wine by immobilized. Lactobacillus casei cells. Process Biochem. 33, 793–797 (1998).
Rogosa, M., Mitchell, J. A. & Wiseman, R. F. A selective medium for the isolation and enumeration of oral and fecal lactobacilli. J. Bacteriol. 62, 132 (1951).
Escalante, A. et al. Analysis of bacterial community during the fermentation of pulque, a traditional Mexican alcoholic beverage, using apolyphasic approach. Int. J. Food Microbiol. 124, 126–34 (2008).
Navarro, L., Zarazaga, M., Ruiz-Larrea, F., Torres, C. & Saenz, J. Bacteriocin production by lactic acid bacteria isolated from Riojared wines. J. Appl. Microbiol. 88, 44–51 (2000).
Dolci, P., Alessandria, V., Rantsiou, K. & Cocolin, L. Advanced methods for the identification, enumeration, and characterization of microorganisms in fermented foods. In Advances in Fermented Foods and Beverages: Improving Quality, Technologies and Health Benefits. Elsevier (2015).
Li, X. R. et al. Bacterial and fungal diversity in the traditional Chinese liquor fermentation process. Int. J. Food Microbiol. 146, 31–7 (2011).
Li, H. Simultaneous saccharification and fermentation of broken rice: an enzymatic extrusion liquefaction pretreatment for Chinese rice wine production. Bioprocess Biosyst. Eng. 36, 1141–1148 (2013).
Kumbhare, S. V. et al. Insights into diversity and imputed metabolic potential of bacterial communities in the continental shelf of Agatti Island. PLoS One. 10, 0129864 (2015).
Bartram, A. K., Lynch, M. D., Stearns, J. C., Moreno-Hagelsieb, G. & Neufeld, J. D. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77, 3846–3852 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336 (2010).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDA seq: paired-end assembler for illumina sequences. BMC Bioinformatics 13, 31 (2012).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 26, 2460–2461 (2010).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Bokulich, N. A., Lucy Joseph, C. M., Allen, G., Benson, A. K. & Mills, D. A. Next-generation sequencing reveals significant bacterial diversity of Botrytized wine. PLoS ONE. 7, e36357 (2012).
Blaalid, R. et al. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 13, 218–224 (2013).
Acknowledgements
Authors gratefully acknowledged the financial support of Department of Biotechnology, New Delhi for research project and DAILAB (DBT-AIST International Laboratory for Advanced Biomedicine), Bioinformatics Centre of DBT.
Author information
Authors and Affiliations
Contributions
S.P.S., A.A. and P.P. contributed to this present work as co-authors that are a part of their research work. K.J. and A.S. helped and assisted in all the molecular work and NGS (Bioinformatics and statistical) analyses. A.S., Y.S. and J.P.T. have framed this research paper along with all the authors involved. All authors critically revised, read and approved the final manuscript with approval of J.P.T.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sha, S.P., Jani, K., Sharma, A. et al. Analysis of bacterial and fungal communities in Marcha and Thiat, traditionally prepared amylolytic starters of India. Sci Rep 7, 10967 (2017). https://doi.org/10.1038/s41598-017-11609-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11609-y
This article is cited by
-
Unveiling the microbial communities and metabolic pathways of Keem, a traditional starter culture, through whole-genome sequencing
Scientific Reports (2024)
-
Analysis of the bacterial and fungal populations in South African sorghum beer (umqombothi) using full-length 16S rRNA amplicon sequencing
World Journal of Microbiology and Biotechnology (2023)
-
Effect of powdery mildew on interleaf microbial communities and leaf antioxidant enzyme systems
Journal of Forestry Research (2023)
-
Microbial Diversity and Functional Potential of Keem: A Traditional Starter Culture for Alcoholic Beverage—Application of Next-Generation Amplicon and Shotgun Metagenome Sequences
Molecular Biotechnology (2023)
-
Advancing Fermented Food Products: Exploring Bioprocess Technologies and Overcoming Challenges
Food and Bioprocess Technology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
