
Abstract
Tetrameric ion channels have either swapped or non-swapped arrangements of the S1–S4 and pore domains. Here we show that mutations in the transmembrane domain of TRPV6 can result in conversion from a domain-swapped to non-swapped fold. These results reveal structural determinants of domain swapping and raise the possibility that a single ion channel subtype can fold into either arrangement in vivo, affecting its function in normal or disease states.
Similar content being viewed by others


Introduction
Transient receptor potential (TRP) channels comprise a large and functionally versatile superfamily of cation permeable ion channels that respond to a myriad of sensory modalities1. Structurally, TRP channels are assemblies of four identical subunits arranged symmetrically around the pore axis2,3,4,5,6. The transmembrane domain of TRP channels includes six transmembrane helices (S1–S6) and a pore loop (P-loop) between S5 and S6. The first four helices (S1–S4 domain) form a bundle that plays the role of voltage sensing domain in voltage-gated potassium channels7. S5, P-loop and S6 form the KcsA channel-like pore domain, with the ion conduction pathway lined by the S6 helices and extracellular loops connecting the P-loop helix to S5 and S6. In the cryo-EM reconstructions of the vanilloid subtype TRP channels TRPV12 and TRPV23, 6, the S1–S4 domain of one subunit is packed against the pore domain of the neighboring subunit, producing a domain-swapped arrangement similar to the voltage-gated potassium channel chimera Kv1.2–2.17, 8.
TRPV6 and its close relative TRPV5 are TRP channels that are distinctly Ca2+-selective and play an important role in calcium homeostasis9, 10. Altered expression of TRPV6 is associated with numerous human diseases, including various forms of cancer11. Accordingly, TRPV6 has emerged as an important target for drug design12. We recently solved the structure of a TRPV6 construct engineered to produce well diffracting crystals5. On the basis of electron density and cysteine crosslinking experiments, we built a model of TRPV6 in which the S1–S4 domain of one subunit is packed against the pore domain of the same subunit, creating a non-swapped architecture distinct from TRPV12 and TRPV23, 6. Subsequent studies of HCN13, CNG14, Slo115, Slo2.216 and Eag117 have revealed the prevalence of non-swapped transmembrane domain architecture in tetrameric ion channels.
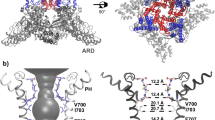
Given the domain-swapped architectures of TRPV12 and TRPV23, 6, and relatively high sequence conservation in the transmembrane region (Fig. 1), the absence of domain swapping in TRPV6 is surprising and warranted further study. We initially wondered whether the mutations present in the TRPV6 crystallization construct5 (TRPV6cryst) affected the domain arrangement. Compared to wild type, TRPV6cryst has a 59-residue C-terminal truncation and four point mutations, including L495Q, which resides in the S5 transmembrane helix (Fig. 1). This L495Q substitution, which was a spontaneous mutation that occurred during gene synthesis, was originally included in TRPV6cryst because it expressed at 3-fold higher levels (Fig. 2) than the same construct with the substitution reverted to the native leucine (TRPV6*). To examine whether the distinct non-swapped arrangement of TRPV6cryst resulted from the L495Q mutation, we were compelled to solve the structure of TRPV6*.

Topology and sequence alignment of TRPV subunits. (a) Membrane topology of TRPV subunits. Red circles and red line indicate four point mutations and C-terminal deletion in TRPV6cryst construct, respectively. (b) Sequence alignment of the transmembrane domain regions in rat TRPV subtypes. Helices are indicated by cylinders above the sequence. L495 (open red box) and the selectivity filter in TRPV6 (thick red line) are highlighted. ¥ marks the N-linked glycosylation site in the extracellular loop connecting S1 and S2 conserved in TRPV6 (and TRPV5) channels.

FSEC traces for crudely solubilized TRPV6cryst and TRPV6*. FSEC traces were recorded using Superose 6 size-exclusion chromatography column monitoring GFP fluorescence. Note the difference in the peak height, indicating approximately 3 fold greater expression levels of TRPV6cryst compared to TRPV6*.
To crystallize TRPV6*, we used the same expression system, purification protocol and crystallization conditions as for TRPV6cryst 5. TRPV6* crystallized in a nearly identical crystal lattice to TRPV6cryst (Table 1) and the overall shape of the molecule was very similar (Fig. 3). Despite these similarities, to our great surprise, the structure of TRPV6* exhibited the domain swapped fold observed in other TRP channels of known structure. Attempts to use the “non-swapped” TRPV6cryst model for molecular replacement resulted in massive negative and positive signals in the Fo-Fc density maps. Similar inconsistency between the crystallographic data and the model was observed when we attempted to use the “swapped” molecular replacement model of TRPV6* to solve the structure of TRPV6cryst. Therefore, the crystallographic data unambiguously demonstrates that the TRPV6* structure is domain-swapped. Apart from the distinct connectivity between S4 and S5 as well as S6 and TRP helix, the domain-swapped TRPV6* and non-swapped TRPV6cryst structures were almost identical. Superposition of TRPV6* and TRPV6cryst excluding regions of S4–S5 (residues 464–497) and S6-TRP helix (residues 577–589) gave a root mean square deviation (RMSD) of 0.865 Å.

Non-swapped domain arrangement as a result of single residue substitution. (a-b), Extracellular views of TRPV6* (a) and TRPV6cryst (b) with four subunits (A–D) each shown in different color. Note, the peripheral S1–S4 domains are adjacent to the S5–S6 pore domains of the same subunit in TRPV6cryst, while the adjacent domains are from neighboring subunits in TRPV6*. (c,d) Side views of one S1–S4 domain and two S5–S6 pore domains illustrating S4–S5 and S6-TRP helix connectivity unique to swapped (c) and non-swapped (d) architectures. (e,f) Close-up views of the regions surrounding L495 in TRPV6* (e) and Q495 in TRPV6cryst (f).
We reproduced crystals of TRPV6cryst and confirmed its non-swapped conformation. Notably, we were able to collect new crystallographic data, where both the S4–S5 and S6-TRP helix connectivity in TRPV6cryst is unambiguously resolved in the 2Fo-Fc and Fo-Fc omit maps (Fig. 4), in stark contrast to our previous data5, where density for thirteen residues in S4–S5 linker (F471-Q483) was missing. Similarly, both the 2Fo-Fc and robust Fo-Fc omit maps for TRPV6* demonstrated completely different connectivity of transmembrane domains (Fig. 4) compared to TRPV6cryst. These structural data therefore suggest that the single leucine L495 to glutamine substitution in S5 is responsible for the absence of domain swapping in TRPV6cryst. Because the sequence of TRPV6* is closer to wild-type, the domain-swapped architecture of TRPV6 likely represents the physiologically relevant domain arrangement.

Dramatic change in TRPV6 transmembrane domain connectivity as a result of single-residue substitution in S5. (a–d) Transmembrane portions of TRPV6* (leucine at position 495, left) and TRPV6cryst (glutamine at position 495, right) viewed parallel to the membrane. S4–S5 (upper row) and S6-TRP helix (bottom row) connectivity is illustrated by 2Fo-Fc (blue mesh, 1σ) and omit Fo-Fc (yellow mesh, 2.5σ) maps.
The presence or absence of transmembrane domain swapping could impact the functional behavior of TRPV6 considerably. Nonetheless, ratiometric fluorescence measurements of cells loaded with the Ca2+ sensitive dye Fura-2 showed that the three constructs, TRPV6*, TRPV6cryst and wild type TRPV6, demonstrate pore permeability to Ca2+ and ion channels block by Gd3+ (Fig. 5). Indeed, the pore architectures of TRPV6cryst and TRPV6* are indistinguishable (Fig. 6a,b), and anomalous signals for Ca2+ and Gd3+ indicated that the positions of the pore cation binding sites are similar in TRPV6* (Fig. 6c–f) and TRPV6cryst 5. Thus, the structural and functional integrity of the TRPV6 pore is overall preserved in both domain-swapped and non-swapped folds. Despite these gross similarities, kinetic analysis indicated the rate of increase in Fura-2 signal is faster in TRPV6* compared to TRPV6cryst (Fig. 7). Therefore, the Ca2+ entry function is somewhat compromised in the non-swapped TRPV6cryst construct, presumably due to differences in allosteric communication between the pore domain and the rest of the protein.

Ca2+ permeability and Gd3+ block are preserved in TRPV6 constructs with distinct domain arrangements. (a,b) Representative ratiometric fluorescence measurements for HEK cells expressing TRPV6*. Arrows indicate the time at which the corresponding ion was added. After resuspending the cells in nominally calcium-free buffer, addition of Ca2+ resulted in robust concentration-dependent increase in Fura-2 signal (a). In contrast, pre-incubation of cells with increasing concentrations of Gd3+ resulted in concentration-dependent reduction in Fura-2 signal (b). (c,d) Normalized changes in F340/F380 at varying concentrations of Ca2+ (c) and Gd3+ (d) calculated for wild type TRPV6 (blue), TRPV6cryst (red) and TRPV6* (green). In (d), ΔF340/F380 was measured after preincubation with Gd3+ and subsequent addition of 2 mM Ca2+.

TRPV6 structures with swapped and non-swapped transmembrane domains have identical ion channel pores. (a,b) Superposition of the pore domains in TRPV6* (cyan) and TRPV6cryst (yellow) viewed parallel to the membrane (a) or extracellularly (b), with residues important for cation binding shown in stick representation. Calcium atom is shown as a green sphere. (c–f) Side (c,e) and top (d,f) views of the TRPV6* pore. Front and back subunits in c and e are removed for clarity. Green and pink mesh shows anomalous difference electron density for Ca2+ (c,d, 3σ) and Gd3+ (e,f, 3.5σ), respectively, and ions are shown as spheres of the corresponding color. Positions of cation binding sites are similar to TRPV6cryst 5.

Different kinetics of ratiometric curves for TRPV6* or TRPV6cryst at physiological extracellular calcium concentration. (a-b), Representative ratiometric fluorescence measurements for HEK cells expressing TRPV6* (a) or TRPV6cryst (b). Arrows indicate the time at which 2 mM Ca2+ was added. The curves were fitted with one (red) or two (blue) exponentials. Note, TRPV6* kinetics was well described by the double but not the single exponential. Over three measurements, the time constants of the single exponential fit (τsingle) were 14.5 ± 2.7 s for TRPV6* and 110 ± 16 s for TRPV6cryst, τfast = 2.2 ± 1.0 s, τslow = 54 ± 11 s and Afast = 0.57 ± 0.01 for TRPV6* and τfast = 5.4 ± 2.8 s, τslow = 135 ± 23 s and Afast = 0.10 ± 0.03 for TRPV6cryst. At n = 3, the values of τsingle and Afast for TRPV6* and TRPV6cryst were statistically different (t-test, P < 0.05).
How does the L495Q mutation result in the non-swapped domain arrangement in TRPV6? In TRPV6*, L495 projects its side chain into a hydrophobic pocket formed by residues of S5 and S6 of one subunit and residues at the S4–S5 elbow of the adjacent subunit (Fig. 3e). By contrast, in TRPV6cryst, Q495 is rotated by ~120° with respect to the orientation of L495 in TRPV6* and forms polar interactions with the hydrophilic pocket formed by S4–S6 and the TRP helix of the same subunit (Fig. 3f). These observations suggest that hydrophobic burial of the L495 residue plays an important role in driving the domain-swapped fold of TRPV6. Disruption of these hydrophobic interactions leads to the non-swapped fold. In other members of TRPV family, either leucine or valine occupy the position homologous to L495 in TRPV6 (Fig. 1b). Further, a hydrophobic pocket similar to TRPV6 is also present in the structures of TRPV1/22, 3, 6, suggesting a conserved role for this region in determining the domain arrangement in TRPV channels.
In the case of non-swapped cation channels, the absence of domain swapping was attributed to shorter S4–S5 linkers13,14,15,16,17. We tested whether TRPV6 domain arrangement could be biased toward a non-swapped conformation by shortening of the S4–S5 linker, which has a well-defined helical structure in the domain-swapped TRPV6* but lacks secondary structure in TRPV6cryst. Remarkably, TRPV6* constructs with up to ten residues deleted in the S4–S5 linker expressed robustly and could be isolated as a monodisperse species with gel filtration elution time corresponding to the tetrameric assembly (Fig. 8a,b). We determined the crystal structure of TRPV6* with four residues deleted in the S4–S5 linker (TRPV6*-del1) and confirmed its non-swapped architecture (Fig. 8c,d). Therefore, the length of the S4–S5 linker is indeed a critical determinant of domain swapping. Interestingly, ratiometric fluorescence measurements did not detect measurable Ca2+ influx in HEK cells expressing TRPV6*-del1, suggesting that while the tetrameric expression and assembly in this mutant is preserved, gating function is significantly perturbed.

Deletions in S4–S5 preserve TRPV6 tetrameric assembly and generate non-swapped topology. (a) Sequence alignment of TRPV6* and mutants with deletions in the S4–S5 linker. Leucine L495 is indicated by the red circle. The sequence corresponding to the resolved regions in TRPV6*-Del1 structure is highlighted by yellow. (b) FSEC traces for purified TRPV6* without and with deletions in S4–S5 following tryptophan fluorescence. Note, all samples demonstrate a single monodisperse tetrameric peak. (c) Transmembrane domain of a single TRPV6*-Del1 subunit demonstrates a non-swapped architecture with the characteristic S6-TRP helix connectivity. Blue mesh shows 2Fo-Fc map (1σ). (d) Close-up view of the S4–S5 illustrating boundaries of a missing region (residues 471–485).
In summary, our studies demonstrate that small sequence changes can result in a major alteration of the transmembrane domain arrangement of a tetrameric ion channel. Depending on the nature of the mutation, this structural alteration can lead to strong or weak changes in ion channel function. We speculate that tetrameric ion channels might be able to switch domain arrangements under a variety of conditions, including dynamic rearrangement between distinct conformations, as a result of mutations, post-translational modifications, or other perturbations. The impact of changes in domain swapping on ion channel function may play a role in pathophysiology.
Methods
Constructs
Our previous crystallization construct TRPV6cryst 5 comprised of residues 1–668 of rat TRPV6 and contained the point mutations I62Y, L92N, M96Q and L495Q. We made a new construct TRPV6*, which is identical to TRPV6cryst except the reversal of the single L495Q mutation in S5 to the native leucine residue. To understand the role of S4–S5 linker in domain swapping, TRPV6*-del1, TRPV6*-del2, TRPV6*-del3 and TRPV6*-del4 constructs (Fig. 8a) were prepared by deleting four (477–480), six (475–480) eight (473–480) and ten (471–480) residues in S4–S5 linker, which were disordered in the original TRPV6cryst structure5.
Expression and Purification
TRPV6 constructs described in this paper were expressed and purified as previously described for the TRPV6cryst 5. In brief, TRPV6 constructs were introduced into the pEG BacMam vector with C-terminal thrombin cleavage site (LVPRG) followed by eGFP and streptavidin affinity tag (WSHPQFEK), for the expression in suspension of baculovirus-transduced HEK293 GnTI– cells. 48–72 hours after transduction with P2 virus, cells were harvested, washed with PBS at pH 8.0 and after sonication, cellular membranes were prepared. The protein was extracted from cellular membranes using 20 mM n-dodecyl-β-D-maltopyranoside, and purified using Strep-Tactin affinity chromatography followed by size exclusion chromatography.
Crystallization and structure determination
Optimized crystals of purified TRPV6* or TRPV6*-del1 were grown in the same condition as crystals of TRPV6cryst 5, which included a reservoir solution consisting of 20–24% PEG 350 MME, 50 mM NaCl and 50 mM Tris-HCl pH 8.0–8.5 in hanging drop configuration at 20 °C. Prior to crystallization using hanging drop method, the protein was subjected to ultracentrifugation (Ti100 rotor, 40000 rpm, 40 min, 4 °C) to remove the aggregated protein. Crystals were cryoprotected by serial transfer into the buffer composed of 100 mM NaCl, 100 mM Tris-HCl pH 8.2, 0.5 mM DDM and 50 mM ammonium formate, containing increasing concentration of PEG 350 MME, with maximum concentration of up to 33–36%, and then flash frozen in liquid nitrogen. To obtain crystals with Ca2+ or Gd3+, protein was incubated with 10 mM CaCl2 or 1 mM GdCl3, respectively, for at least 1 hour at 4 °C prior to crystallization.
X-ray diffraction data collected at APS (beamlines 24-ID-C/E), NSLSII (beamline 17-ID) or ALS (beamline 5.0.2) were indexed, integrated and scaled using XDS18 or HKL200019. The initial phase information and structures were solved by molecular replacement using Phaser20 and the structure of rat TRPV6cryst 5 (PDB ID: 5IWK) as a search probe. Most of TRPV6*, including the ankyrin domain, S1–S4, pore module and C-terminal hook, was nearly identical to the TRPV6cryst; the rest of the structure was built using the omit map as a guide. The robust electron density for the S4–S5 linker was evident from the initial phases obtained by molecular replacement, and map features improved further during refinement. The model was refined by alternating cycles of building in COOT21 and automatic refinement in Phenix22 and Refmac23. All structural figures were made using the PyMOL Molecular Graphics System (Version 1.8 Schrödinger, LLC).
Fura 2-AM measurements
The intracellular Ca2+ measurements from HEK cells expressing rat TRPV6 or TRPV6cryst or TRPV6* were performed as described previously5. All Fura2-AM-based fluorescence measurements were conducted using spectrofluorometer QuantaMasterTM 40 (Photon Technology International) at room temperature in a quartz cuvette under constant stirring.
References
Venkatachalam, K. & Montell, C. TRP channels. Annual review of biochemistry 76, 387–417, doi:10.1146/annurev.biochem.75.103004.142819 (2007).
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112, doi:10.1038/nature12822 (2013).
Zubcevic, L. et al. Cryo-electron microscopy structure of the TRPV2 ion channel. Nature structural & molecular biology 23, 180–186, doi:10.1038/nsmb.3159 (2016).
Paulsen, C. E., Armache, J. P., Gao, Y., Cheng, Y. & Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517, doi:10.1038/nature14871 (2015).
Saotome, K., Singh, A. K., Yelshanskaya, M. V. & Sobolevsky, A. I. Crystal structure of the epithelial calcium channel TRPV6. Nature. nature17975 [pii] 534, 506–511, doi:10.1038/nature17975 (2016).
Huynh, K. W. et al. Structure of the full-length TRPV2 channel by cryo-EM. Nature communications 7, 11130, doi:10.1038/ncomms11130 (2016).
Doyle, D. A. et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77 (1998).
Long, S. B., Tao, X., Campbell, E. B. & MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382 (2007).
Voets, T. et al. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. The Journal of biological chemistry 276, 47767–47770, doi:10.1074/jbc.C100607200 (2001).
van Goor, M. K. C., Hoenderop, J. G. J. & van der Wijst, J. TRP channels in calcium homeostasis: from hormonal control to structure-function relationship of TRPV5 and TRPV6. Biochimica et biophysica acta 1864, 883–893, doi:10.1016/j.bbamcr.2016.11.027 (2017).
Wissenbach, U. et al. TRPV6 and prostate cancer: cancer growth beyond the prostate correlates with increased TRPV6 Ca2+ channel expression. Biochem Bioph Res Co 322, 1359–1363, doi:10.1016/j.bbrc.2004.08.042 (2004).
Peters, A. A. et al. Calcium Channel TRPV6 as a Potential Therapeutic Target in Estrogen Receptor-Negative Breast Cancer. Mol Cancer Ther 11, 2158–2168, doi:10.1158/1535-7163.MCT-11-0965 (2012).
Lee, C. H. & MacKinnon, R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell 168, 111–120 e111, doi:10.1016/j.cell.2016.12.023 (2017).
Li, M. et al. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature 542, 60–65, doi:10.1038/nature20819 (2017).
Tao, X., Hite, R. K. & MacKinnon, R. Cryo-EM structure of the open high-conductance Ca2+ -activated K+ channel. Nature 541, 46–51, doi:10.1038/nature20608 (2017).
Hite, R. K. & MacKinnon, R. Structural Titration of Slo2.2, a Na+ -Dependent K+ Channel. Cell 168, 390–399 e311, doi:10.1016/j.cell.2016.12.030 (2017).
Whicher, J. R. & MacKinnon, R. Structure of the voltage-gated K(+) channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669, doi:10.1126/science.aaf8070 (2016).
Kabsch, W. Xds. Acta Crystallogr D. Biol Crystallogr 66, 125–132, doi:10.1107/S0907444909047337 (2010).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol 276, 307–326, doi:10.1016/S0076-6879(97)76066-X (1997).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D 63, 32–41, doi:10.1107/S0907444906045975 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66, 213–221, doi:10.1107/S0907444909052925 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D 67, 355–367, doi:10.1107/S0907444911001314 (2011).
Acknowledgements
We thank members of the E.C. Greene Lab for assistance with their fluorimeter. We also thank the personnel at beamlines 24-ID-C/E of APS, AMX of NSLSII and 5.0.2 of ALS. This work was supported by the NIH grants R01 NS083660, R01 CA206573 (A.I.S) and T32 GM008281 (K.S.), by the Pew Scholar Award in Biomedical Sciences and the Irma T. Hirschl Career Scientist Award (A.I.S.).
Author information
Authors and Affiliations
Contributions
A.K.S., K.S. and A.I.S. designed the project. A.K.S. and K.S. performed the experiments. A.K.S., K.S. and A.I.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Accession codes. Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 5WO6, 5WO7, 5WO8, 5WO9 and 5WOA (see Table 1 for detail).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, A.K., Saotome, K. & Sobolevsky, A.I. Swapping of transmembrane domains in the epithelial calcium channel TRPV6. Sci Rep 7, 10669 (2017). https://doi.org/10.1038/s41598-017-10993-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10993-9
This article is cited by
-
A pentameric TRPV3 channel with a dilated pore
Nature (2023)
-
Molecular pathway and structural mechanism of human oncochannel TRPV6 inhibition by the phytocannabinoid tetrahydrocannabivarin
Nature Communications (2023)
-
Structural mechanism of human oncochannel TRPV6 inhibition by the natural phytoestrogen genistein
Nature Communications (2023)
-
Evolutionary analyses reveal independent origins of gene repertoires and structural motifs associated to fast inactivation in calcium-selective TRPV channels
Scientific Reports (2020)
-
Structural insights on TRPV5 gating by endogenous modulators
Nature Communications (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
