
Abstract
Cell division cycle 42 (CDC42) plays important roles in polarity establishment and maintenance as well as cell cycle progression and cell division. Although disruption of cell polarity is a prerequisite in epithelial tumor initiation, the roles of CDC42 in tumorigenesis are still poorly understood. Here we find that Cdc42 deficiency inhibits the Kras G12D-induced lung alveoli tumor formation, while conversely promotes bronchiole tumor formation in mice. Bronchial Cdc42 loss destroys contact inhibition potentially through cell polarity disruption, and results in increased tumor formation. In contrast, deletion of Cdc42 in alveoli cells prevents Kras G12D-induced cell proliferation, which leads to reduced tumor formation. Further analyses of clinical specimens uncover a significant positive correlation between CDC42 and type II alveolar epithelial cells marker SP-A, indicating the potential importance of CDC42 in this specific subset of lung cancer. Collectively, we identify the lineage-specific function of CDC42 in lung tumorigenesis potentially through the regulation of cell polarity integrity.
Similar content being viewed by others


Introduction
The Rho GTPase family member CDC42 plays an essential role in regulating multiple cellular processes including cell proliferation, division, migration, morphogenesis, and especially epithelial polarity establishment. In its GTP-bound form, CDC42 initiates actin polymerization, filopodia formation by binding to PAK family of serine/threonine kinases and WASP, and formation of epithelial junctions by binding to PAR6, causing followed phosphorylation of aPKC targets1.
Deregulation of CDC42 may result in multiple cellular defects and significantly contribute to cancer formation1, 2. CDC42 was initially thought to be oncogenic in several cancer types via contributing to cell proliferation, survival, migration and invasion2,3,4. While gene mutations have not been detected in human cancers5, CDC42 has been reported to be overexpressed in several types of human cancers including colorectal adenocarcinoma6, breast cancer7, testicular cancer8 and non-small cell lung cancer9. Elevated CDC42 level may also be detrimental to patient survival as overexpression of CDC42 in melanoma positively correlated with prognostic indicators10. Constitutive activated CDC42 is capable of inducing immortalized fibroblasts transformation11 and functional CDC42 is required for HRas V12 induced colony formation of NIH-3T3 cells in soft agar12.
Interestingly, recent studies based on Cdc42 conditional knock-out mice have uncovered a tumor suppressive role of Cdc42, showing that hepatocyte-specific deletion of Cdc42 results in chronic liver damage, hepatomegaly and development of hepatacellular carcinoma13. Also, induced gene targeting of Cdc42 in murine bone marrow hematopoietic stem/progenitor cells results in a loss of hematopoietic stem cell quiescence and hyperproliferation of blood progenitors14. Consistently, neuroblastomas with N-myc amplification display deletions of the short arm of chromosome 1 containing the Cdc42 gene in 90–95% of cases, and one copy of Cdc42 is consistently lost in this type of cancer15. These data suggest that the role of Cdc42 as oncogene or tumor suppressor might be lineage dependent16.
Lung cancer is one of the most devastating diseases worldwide with different subtypes derived from trachea, bronchiole or peripheral alveoli. Previous studies have detected high CDC42 expression in human lung cancer samples9 and cell lines17 and demonstrate its contribution to cancer cell migration. Moreover, down-regulation of CDC42 is found to inhibit lung cancer cell growth18 and invasiveness17, 19,20,21,22. CDC42 also promotes trans-endothelial migration of lung cancer cells through β1 integrin23. These observation are consistent with oncogenic role of CDC42.
Here through detailed studies of Cdc42 deletion in distinct cell types using lineage specific promoter driven CRE in Kras G12D driven lung cancer mouse model, we have identified both tumor-promoting and tumor-suppressive function of CDC42 in type II alveolar epithelial cells and Club cells, respectively. Our data further show that CDC42 prevents lung bronchiole tumor formation potentially through regulation of cell polarity integrity. In accordance with its tumor promoting role in alveolar tumor formation, CDC42 expression is positively correlated with alveolar marker surfactant protein A1 (SP-A) expression in human lung adenocarcinoma patients.
Results
Cdc42 loss promotes bronchiole tumor formation but inhibits alveoli tumor formation in Kras mouse model
To investigate the potential role of CDC42 in lung tumorigenesis, we crossed the conditional Cdc42 L/L allele with Lox-Stop-Lox Kras G12D (hereafter named as Kras) allele to get Lox-Stop-Lox Kras G12D; Cdc42 L/L allele (hereafter named as Kras/Cdc42)24, 25. Mice were then inoculated with Cre-expressing adenovirus (Ad-Cre) by nasal inhalation as previously described25, and analyzed at a series of time points (Fig. 1a). In this model, it remains possible that multiple pulmonary cell lineages are infected with Ad-Cre and neoplastic lesions arise from both bronchiole and alveoli epithelial cells25. We initially confirmed the Cdc42 deletion in lung tumors derived from Kras/Cdc42 mouse model (Fig. 1b, Supplementary Figs S1–2). As the control, deletion of Cdc42 alone did not result in any tumor formation over 70 weeks post Ad-Cre treatment (Fig. 1c). Consistent with the essential role of CDC42 in promoting cell division and neoplastic transformation2, 26, Cdc42 loss significantly decreased the lesion number and percentage of alveolar tumors in Kras/Cdc42 mice (Fig. 1d–f). Surprisingly, we observed a significant increase of the lesion number and percentage of bronchiolar tumors in this model (Fig. 1d–f), featured with the papillae protrusion into airway lumens (Fig. 1d). These bronchiolar lesions in Kras/Cdc42 model exhibit a high cell proliferating index (presented by KI67 staining) compared with those in Kras model (Fig. 1g,h). This analysis demonstrated that Cdc42 loss increased formation of bronchial and bronchiolar epithelial tumors, but decreased Kras-induced tumor formation in alveoli.

Cdc42 loss promotes bronchiole tumor formation but inhibits alveoli tumor formation in Kras mouse model. (a) Mouse number analyzed for 3 strains in indicated time points. (b) Up: PCR analysis of conditional Cdc42 allele recombination in tumors from Kras and Kras/Cdc42 mice; Bottom: Western blot of CDC42 expression in tumors from Kras and Kras/Cdc42 mice. Histone 3 (H3) serves as a loading control. The cropped blots are used in the figure. The membranes were cut prior to exposure so that only the portion of gel containing desired bands would be visualized. (c) Representative histology of lung tumors from WT mice and Cdc42 L/L mice at 80 weeks post Ad-cre treatment. Scale bar = 500 μm. (d) Representative pathology of lung tumors from Kras and Kras/Cdc42 mice at 16 weeks post Ad-Cre treatment. The areas in the boxes of left photos were amplified on the right. Scale bar (left) = 500 μm, Scale bar (right) = 100 μm (e,f) Statistical analyses of the number of alveolar and bronchiolar tumors (e) and the percentage of bronchiolar tumors (f) in Kras and Kras/Cdc42 mice at 16 weeks post Ad-Cre treatment. Al: alveolar; Br: Bronchiolar. Data were shown as mean ± s.e.m. *P < 0.01***P < 0.001. (g) Representative immunostaining of KI-67 in alveolar and bronchiolar tumors from Kras and Kras/Cdc42 mice. Scale bar = 50 μm. (h) Statistical analyses of proliferative index by KI-67 immunostraining in bronchiolar and alveolar tumor lesions from Kras and Kras/Cdc42 mice. More than 200 high-power fields (HPF) per mouse were counted. Data were shown as mean ± s.e.m. ***P < 0.001.
Cdc42 loss disrupts bronchiole cell polarity
We then asked how Cdc42 loss promoted the bronchiole tumor formation. Normal bronchioles are lined by pseudostratified or single layer epithelia which potentially contribute to contact inhibition and act as the important barrier for neoplastic transformation27, 28. Since CDC42 plays a central role in establishing and maintaining epithelial polarity which is frequently disrupted during tumor progression, we first analyzed the subcellular localization of a series of polarity proteins in Kras or Kras/Cdc42 bronchioles at three weeks post Ad-Cre treatment. Our data showed a subcellular dislocation of both ZO1 and Phalloidin, markers for tight junction and F-ACTIN assembling respectively, in Kras/Cdc42 mice bronchioles at three weeks post Ad-Cre treatment (Fig. 2a). Similar dislocation of other polarity components including PAR6 and Occludin was also detectable in Kras/Cdc42 mice (Fig. 2b). Moreover, our data from electron microscopy analyses demonstrated that the ultrastructure of tight junction in bronchioles was disrupted in Kras/Cdc42 mice but not in Kras mice (Fig. 2c).

Cdc42 loss disrupts bronchiole cell polarity. (a) Immunofluorescent co-staining of ZO1 (Green), Phalloidin (Red) and 4′,6-diamidino-2-phenylindole (DAPI, blue) in Kras and Kras/Cdc42 mice at 3 weeks post Ad-Cre treatment. The arrows indicate tight junctions at apical surface of bronchiole epithelial cells, and the dashed lines indicate basal surface. Scale bar = 3.5 μm. (b) Immunofluorescent staining of Occludin and PAR6 in Kras and Kras/Cdc42 mice at 3 weeks post Ad-cre treatment. The arrows indicate apical surface of bronchiole epithelial cells. Scale bar = 50 μm. (c) Ultrastructural analysis of tight junction between bronchiole cells in WT, Kras, Cdc42 (dashed line) and Kras/Cdc42 (arrows) mice at 3 weeks post Ad-Cre treatment. Scale bar = 500 nm.
Cdc42 loss promotes the proliferation of KRAS-activated polarized cells potentially through disruption of cell-cell contact inhibition
To test if Cdc42 loss indeed promotes lung bronchiolar tumorigenesis through disruption of polarity, we utilized two types of cells with distinct polarities including mouse trachea epithelial cells (MTEC) and mouse embryonic fibroblasts (MEF) to analyze the functional consequence of Cdc42 deletion. In comparison with KRAS activation alone, deletion of CDC42 together with KRAS activation almost completely disrupted the tight junction in MTEC, indicated by ZO1 staining (Fig. 3a), and dramatically increased cell growth (Fig. 3b). On the contrary, in MEF cells which did not form tight junction, Cdc42 deletion abrogated the promotive effect of KRAS upon cell proliferation (Fig. 3c,d).

Cdc42 loss promotes the proliferation of KRAS activated cells potentially through disruption of cell-cell contact inhibition. (a) Immunofluorescent staining of ZO1 (Green) and DAPI (blue) in Ad-cre treated MTEC derived from Kras and Kras/Cdc42 mice. Scale bar = 50 μm. (b) Crystal violet staining of MTEC derived from Kras and Kras/Cdc42 mice at 72 h with or without Ad-Cre treatment. (c) Immunofluorescent staining of ZO1 (Green) and DAPI (blue) in Ad-cre treated MEF derived from Kras and Kras/Cdc42 mice. Scale bar = 50 μm. (d) Growth of MEF derived from indicated mice determined by MTT assay during 6 continuous days of cell culture. (e) MTT assay indicated growth of MDCK cell line transfected with indicated vector at 72 h (low density) and 120 h (high density) relative to control (12 h). ***P < 0.001. (f) Quantitative PCR showing the knockdown efficiency of indicated vector in MCDK. (g) Immunofluorescent double staining of Phalloidin (Red) and DAPI (blue) in 3D cultured MDCK expressing KRAS-copGFP (top) or KRAS-shCdc42-copGFP (bottom). Scale bar = 20 μm.
We further took advantage of Madin-Darby canine kidney (MDCK) cells, which is widely used for polarity study. Our data showed that Cdc42 knockdown remarkably inhibited KRAS-activated MDCK cell growth at low cell density but the cell growth was significantly enhanced after reaching 100% confluence (Fig. 3e,f). These data clearly demonstrated that Cdc42 loss plays opposite roles in cell proliferation, depending on whether cell-cell junction exists or not.
The three-dimensional (3D) system is an excellent model of epithelial morphogenesis in which cells embedded in matrigel form acini, a spherical monolayer enclosing a central lumen. To study the potential role of CDC42 in restricting neoplastic transformation, we infected pre-formed MDCK acini (day 6) with a low dose of lenti-virus expressing Kras G12D with or without Cdc42 knockdown. Consistent with previous study, KRAS activated single cell remained quiescent in growth-arrested acini, with apical location of ZO1 and Phalloidin (Fig. 3g, upper panel). In contrast, single cell with Cdc42 knockdown in context of KRAS activation initiated inner-growing cell mass from the epithelial layer with disordered ZO1 and Phalloidin localization (Fig. 3g, bottom panel), which morphologically resembled papillae protruding into the airway lumens in Kras/Cdc42 mice bronchiole (Fig. 1d). These results substantiated the key role of CDC42 and polarity integrity in inhibiting tumor formation.
Lineage-specific deletion of Cdc42 in Kras-driven lung cancer mouse model
Mouse bronchiolar and alveolar tumors are considered to be derived from Club cells and type II alveolar epithelial cells (AECII) respectively29, 30. CCSP and the surfactant protein-C (SP-C) are commonly-used markers to distinguish Club cell and AECII25. We found that the bronchiolar tumors from Kras/Cdc42 model were stained positive for CCSP but negative for SP-C, whereas the alveolar tumors were opposite (Fig. 4a). Based on these data, we hypothesize that CDC42 might play a cell type-specific role in lung tumorigenesis.

Lineage-specific deletion of Cdc42 in Club cells and AECII in Kras mouse model. (a) Immunohistochemical staining for SP-C and CCSP in lung tumors from Kras and Kras/Cdc42 mice. The dashed line indicated the bronchiole. Br: Bronchiole. Scale bar = 50 μm. (b) Representative photos of CCSP-CreERT2/Rosa26R -LacZ (upper panel) or SPC-CreERT2/Rosa26R-LacZ (lower panel) mice lung sections at 4 weeks post tamoxifen administration. β-galactosidase staining (blue) indicates targeted cell. The upper panel is co-stained with CCSP and the bottom is co-stained with SP-C. Scale bar = 50 μm. (c) A brief scheme of mouse crossing of SPC-CreERT2 and CCSP-CreERT2 allele with Kras or Kras/Cdc42 mice. (d) Representative pathology and immunohistochemical staining for SP-C and CCSP of lung tumors derived from SPC-Cre-ERT2/Kras (SK), SPC-Cre-ERT2/Kras/Cdc42 (SKC), CCSP-Cre-ERT2/Kras (CK) and CCSP-Cre-ERT2/Kras /Cdc42 (CKC) mice at 8 weeks post Ad-Cre treatment. Br: Bronchiole; Al: Alveoli. Scale bar = 500 μm for H&E, Scale bar = 50 μm for immunostaining. (e) Statistical analyses of the number of alveolar and bronchiolar tumors found in SK and SKC mice. Data were shown as mean ± s.e.m. *P < 0.05. (f,g) Statistical analyses of the number of alveolar and bronchiolar tumors (f) and the percentage of bronchiolar tumors (g) found in CK and CKC mice. Data were shown as mean ± s.e.m. ***P < 0.001.
We then utilized lineage specific ablation of Cdc42 to dissect the potential roles of Cdc42 in bronchiolar and alveolar tumor formation. For this, we first constructed the SPC-Cre-ERT2 (mainly targeting AECII cells) and CCSP-Cre-ERT2 transgenic alleles (mainly targeting Club cells in bronchiolar epithelia31) (Fig. 4b). Given that the CCSP-Cre-ERT2 strain was novelly constructed, we crossed these two alleles to Rosa26R-LacZ reporter mouse strain32 and found that tamoxifen-induced LacZ expression from Rosa26 locus was mainly restricted to Club cells that located at bronchioles, and AECII cells located at alveoli, respectively (Fig. 4b). These data are consistant with our previous study33. We then crossed SPC-Cre-ERT2 or CCSP-CreERT2 allele with either Kras or Kras/Cdc42 mice to generate SPC-Cre-ERT2/Kras (SK)34, SPC-Cre-ERT2/Kras/Cdc42 (SKC), CCSP-Cre-ERT2/Kras (CK) and CCSP-Cre-ERT2/Kras/Cdc42 (CKC) mice and performed the detailed tumor analyses after tamoxifen treatment (Fig. 4c). As expected, the majority of tumors in the SK model were located at alveoli; SKC mice demonstrated significantly decreased tumorigenesis (alveolar tumors) compared to the SK group (Fig. 4d,e). On the other hand, we found that the majority of lung tumors in CKC mice were located at bronchioles, and stained positive for CCSP but negative for SP-C (Fig. 4d). Interestingly, a dramatic increase of tumor formation mainly in bronchiole was detected in CKC mice compared to CK mice (Fig. 4d,f,g). Less than 20% bronchioles tumor was found in CK mice, suggesting that KRASG12D activation alone is difficult to transform the bronchiole epithelium cells with intact cell polarity.
Collectively, these data support that CDC42 functions oppositely in different lineage tumor formation: it promotes tumor growth in alveoli while prevents tumor formation in bronchioles.
High CDC42 level correlates with SP-A level in human lung adenocarcinoma
To further study the potential clinical relevance of our findings from mouse model, we analyzed a set of 84 lung adenocarcinoma for expression patterns of CDC42 and SP-A, a marker for human AECII35. Immunohistochemistry staining were performed and blindly scored according to Allred score system (Fig. 5a). Consistent with previous study9, we found that 57.5% of tumors were positive for CDC42 expression. Pearson correlation analysis showed that CDC42 level positively correlated with SP-A level (Fig. 5b,c; r = 0.3436, P = 0.0014). Although SP-A is a biomarker for normal human AECII, human lung cancer frequently lost the expression of lineage biomarkers. Thus, our clinical data analyses only provide an implication that CDC42 expression might be specifically high in those lung cancer derived from AECII.

CDC42 expression positively correlates with alveolar marker SP-A expression in human lung adenocarcinoma. (a) Representative group (low, medium, high) of immunohistochemical staining for SP-C and CDC42 in 84 human lung adenocarcinoma samples. Scale bar = 50 μm. (b) Expression level of CDC42 or SP-A in 84 human lung adenocarcinoma samples. Scale bar = 50 μm. (c) Pearson correlation analysis showed a significant positive correlation between CDC42 expression and SP-A expression (P < 0.01, r = 0.3436) in 84 human lung adenocarcinoma. Full line represents linear regression of data.
Discussion
In this study, we have identified an unexpected cell type-dependent role of CDC42 in lung tumorigenesis. In AECII, Cdc42 loss strongly prevents Kras-driven neoplastic transformation, establishing the tumor-promotive role of CDC42. These results are consistent with early studies carried out in various mammalian cell lines by overexpressing dominant-negative and/or constitutively active mutants showing the necessity of CDC42 in transformation11, 12, 36. In contrast, our data shows that Cdc42 loss not only disrupted epithelial cell polarity and branching morphogenesis of the developing lung as previously described37, but also promotes KRAS-induced overgrowth and tumor formation in adult Club cells.
Our results further suggest that Cdc42 loss induces Kras-derived bronchiole tumor formation potentially through disruption of cell polarity and cell-cell contact inhibition. Compared with alveolar cells, bronchiolar epithelia cells are close-packed and express polarity-related protein specifically located on apical or basal, which function as a non-cell-autonomous tumor suppressor to restrict KRAS-induced cell proliferation. Since CDC42 plays an essential role in regulation of cell polarity in these cells, its deletion leads to disrupted cell-cell junction, the loss of contact inhibition, eventually triggers cell overgrowth. Together, these data suggest that the cell polarity maintained by CDC42 might serve as an important barrier for bronchiolar tumor formation in lungs. Our findings are consistent with Previous studies showing that the dysfunction of certain polarity genes is associated with tumor initiation or progression38,39,40.The role of CDC42 in cell proliferation (mainly in alveoli) and contact inhibition (mainly in bronchiole) during the tumorigenesis is summarized in Fig. 6.

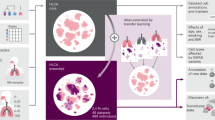
Schematic illustration of the function of CDC42 in lung cancer. In this study, we have identified an unexpected cell type-dependent role of CDC42 in lung tumorigenesis. In type II alveolar epithelial cells (AECII), Cdc42 loss strongly prevents Kras-driven neoplastic transformation, establishing the tumor-promotive role of CDC42. The pseudostratified or single-layer polarized Club cells, are separated by apical junctions which function as physical barrier by providing a tight seal between the membranes of the neighboring cells. These internal physical barriers of the neighboring cells function as a non-cell-autonomous tumor suppressor to restrict Kras-induced cell proliferation and prevent bronchiole tumor formation. Loss of core polarity protein CDC42 disrupted cell polarity and cell-cell contact inhibition, thereby leading to Kras induced bronchiole papillary hyperplasia. Green: normal cells; Pink: genetically mutated cells.
In conclusion, our data show that CDC42 functions in a cell-type specific manner in lung tumorigenesis in context with different cell lineages. Loss of Cdc42 promoted KRAS-induced Club cell tumor formation, underscoring an important tumor initiation function of polarity loss which was previously considered a by-product of abnormal cell accumulation. In addition, studies from mouse model and cell lines can be further applied to human pathological conditions. Of course, more detailed studies are needed to determine the feasibility and therapeutic benefit of targeting CDC42 in human disease.
Materials and Methods
Mouse treatment
Kras G12D, and Rosa26-LacZ mice were originally generously provided by Dr. T. Jacks and Dr. L. Cheng, respectively. Cdc42 L/L mice were generated as described before24, 41. The SPC-CreERT2 allele was made as previously reported34. The CCSP-CreERT2 allele was made using the rat CCSP promoter31 driving the CreERT2 gene expression with similar strategy as previously reported for SPC-CreERT2 allele34. Progeny were screened using southern blot and confirmed by PCR strategy. All mice were housed in a specific pathogen-free environment at the Shanghai Institute of Biochemistry and Cell Biology and treated in strict accordance with protocols approved by the Institutional Animal Care and Use Committee of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. All experiments were performed in accordance with relevant guidelines and regulations. For Kras, Kras/Cdc42 lung cancer mice model, mice were treated with 2 × 106 plague-forming units (PFU) of Ad-Cre (purchased from University of Iowa adenoviral core) or lenti-virus through nasal inhalation at 6~8 weeks of age25. For SPC-Cre-ERT2/Kras (SK), SPC-Cre-ERT2/Kras/Cdc42 (SKC), CCSP-CreERT2/Kras (CK) or CCSP-CreERT2/Kras/Cdc42 (CKC) mouse models, tamoxifen in sunflower oil (40 mg/kg) were given to mice via intraperitoneal injection daily for five continuous doses. Mice were sacrificed at a serial time points for gross inspection, histopathological examination and molecular analyses. The numbers of lung bronchiolar and alveolar tumors were analyzed according to pathology for different mouse cohorts and the percentage of regional tumors was then calculated accordingly.
Histopathological Analysis
Histopathological analysis was performed as described before25. Briefly, mice were sacrificed and lung tissues were inflated and fixed in PFA, embedded in paraffin or frozen in optimal cutting temperature compound (OCT) and sectioned for hematoxylin and eosin (H&E) staining. Immunohistochemical and immunofluorescence analyses were performed as described34. Antibodies against the following proteins were used: KI-67 (Novocastra Laboratories Ltd), CDC42 (Abcam), ZO1 (33-9100, Zymed); PAR6 (sc-14405, Santa Cruz), Occludin (353197a Zymed), Phalloidin (R415 Invitrogen), CCSP (Santa Cruz), SP-C (AB3786, Chemicon), Beta Galactosidase (ab9631, Abcam), Biotinylated goat anti-rabbit secondary antibody (ZYMED company), Alexa Fluor 555 or 488 conjugated anti-mouse, rat or rabbit IgG secondary antibodies (Invitrogen).
β-galactosidase activity analyses
For β-galactosidase activity assays (X-gal staining), mouse lungs were isolated and immediately incubated for 2 hours in a 20-fold volume of ice-cold fixative (1% formaldehyde, 0.2% gluteraldehyde and 0.02% NP40 in PBS) at 4 °C on a rolling platform. The fixative was removed and the tissues were washed twice in PBS for 20 min at room temperature. The galactosidase substrate (5 mM K3Fe (CN)6, 5 mM K4Fe (CN)6, 2 mM MgCl2, 0.02% NP40, 0.1% sodium deoxycholate and 1 mg/ml X-gal in PBS) was then added and the tissues were incubated in the dark overnight. The stained tissues were transferred to tissue cassettes and paraffin blocks were prepared following standard methods.
Constructs, Cell culture and in vitro Growth Characteristics
The ORF of Kras gene was amplified from human cDNAs, and point mutated at G12D following protocol of STRATAGENE QuikChange ® XL Site-Directed Mutagenesis Kit. The Kras G12D and the shRNAs towards human CDC42 was ligated into expression vectors pCDH-EF1-copGFP (Systems Biosciences). The target sequence is: shCdc42: 5′-GCCTATCACTCCAGAGACT-3′.
Mouse embryonic fibroblasts (MEF)
Embryonic day 13.5 embryos were dissected from Kras/Cdc42 intercrossed females, and MEF were isolated and maintained in DMEM (Hyclone) medium supplemented with 10% fetal bovine serum (FBS, Biochrom, AG), penicillin and streptomycin. MEF (~4 × 105) at 6-well plate were virally infected with Ad-Cre (4 × 106 CFU) overnight and then changed with fresh medium. MDCK and HEK293T cells (ATCC) were cultured in DMEM with 10% FBS. Viral infection of cells was as previously described42.
For in vitro growth assay, cells were seeded in 96-well plates (2500 cells per well), and the proliferation information were obtained by MTT (3–4, 4-dimethylthiazol-2, 5 diphenyl tetrabromide) assay every day as previously described43.
Mouse tracheal epithelial cell culture (MTEC)
MTEC harvest was performed as previously described44. Briefly, 8–10 wk Kras or Kras/Cdc42 mice were sacrificed and immersed in 70% ethanol (avoiding airway submersion), and tracheas were resected from the larynx to the bronchial main branches and collected in ice-cold Ham’s F-12 pen-strep. After overnight digestion of 0.25% trypsin at 4 °C, the suspended trachea cells were collected for incubation in tissue culture plates for 3–4 hrs in 5% CO2 at 37 °C to adhere fibroblasts, and nonadherent cells were collected by centrifugation, resuspended in MTEC/Plus medium, and maintained in collagen coated 24-well plate. After 72 hrs growth, MTEC (~1 × 105) at 24-well plate were virally infected with Ad-Cre (1 × 106 CFU) overnight and then changed with fresh medium. Immuno or crystal violet staining was performed after another 4 days’ growth.
Electron microscopy
After the mice were sacrificed, small fragments (~1 mm3) of lung were fixed overnight in a solution containing 4% paraformaldehyde, 2.5% gluteraldehyde in 0.1 M PBS (pH 7.4). Tissue fragments were washed for 20 min in 0.1 M PBS for three times and subsequently treated with 2% osmium tetroxide in PBS for 1.5 hrs. After washing for 5 min × 3 with PBS, specimens were dehydrated in a graded ethanol series and embedded in Epon. For orientation purposes, 1 µm sections were stained with toluidine blue. Ultrathin sections (70–80 nm) were then cut with a Reichert–Jung ultramicrotome, collected on formvar coated nickel grids, stained with uranyl acetate for 10 min and with lead citrate for 7 min. Samples were examined with a FEI Tecnai G2 Spirit Transmission Electron Microscope.
Human Lung Cancer Specimen analyses
A total of 84 NSCLC patient samples were collected with the approval by the institutional review committees of Shanghai Cancer Center, Fudan University. Patients gave written informed consents. The specimens were used for immunostaining of SP-A and CDC42 and analyzed for clinical relevance. All human methods were performed in accordance with the relevant guidelines and regulations.
Statistical Analysis
Data were analyzed by Student’s t test or Pearson correlation test; P < 0.05 was considered significant.
Data availability
All data generated or analysed during this study are included in this published article.
References
Etienne-Manneville, S. Cdc42–the centre of polarity. Journal of cell science 117, 1291–1300 (2004).
Stengel, K. & Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cellular signalling 23, 1415–1423 (2011).
Sahai, E. & Marshall, C. J. RHO-GTPases and cancer. Nature reviews Cancer 2, 133–142 (2002).
Cook, D. R., Rossman, K. L. & Der, C. J. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene 33, 4021–4035 (2014).
Rihet, S. et al. Mutation status of genes encoding RhoA, Rac1, and Cdc42 GTPases in a panel of invasive human colorectal and breast tumors. Journal of cancer research and clinical oncology 127, 733–738 (2001).
Gomez Del Pulgar, T. et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. International journal of oncology 33, 185–193 (2008).
Fritz, G., Brachetti, C., Bahlmann, F., Schmidt, M. & Kaina, B. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. British journal of cancer 87, 635–644 (2002).
Kamai, T. et al. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 10, 4799–4805 (2004).
Liu, Y. et al. Abnormal expression of p120-catenin, E-cadherin, and small GTPases is significantly associated with malignant phenotype of human lung cancer. Lung cancer 63, 375–382 (2009).
Tucci, M. G. et al. Involvement of E-cadherin, beta-catenin, Cdc42 and CXCR4 in the progression and prognosis of cutaneous melanoma. The British journal of dermatology 157, 1212–1216 (2007).
Lin, R., Bagrodia, S., Cerione, R. & Manor, D. A novel Cdc42Hs mutant induces cellular transformation. Current biology: CB 7, 794–797 (1997).
Qiu, R. G., Abo, A., McCormick, F. & Symons, M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Molecular and cellular biology 17, 3449–3458 (1997).
van Hengel, J. et al. Continuous cell injury promotes hepatic tumorigenesis in cdc42-deficient mouse liver. Gastroenterology 134, 781–792 (2008).
Yang, L. et al. Cdc42 critically regulates the balance between myelopoiesis and erythropoiesis. Blood 110, 3853–3861 (2007).
Valentijn, L. J. et al. Inhibition of a new differentiation pathway in neuroblastoma by copy number defects of N-myc, Cdc42, and nm23 genes. Cancer research 65, 3136–3145 (2005).
Lammermann, T. et al. Cdc42-dependent leading edge coordination is essential for interstitial dendritic cell migration. Blood 113, 5703–5710 (2009).
Chen, Q. Y. et al. Expression analysis of Cdc42 in lung cancer and modulation of its expression by curcumin in lung cancer cell lines. International journal of oncology 40, 1561–1568 (2012).
Zhu, X. et al. miR-137 inhibits the proliferation of lung cancer cells by targeting Cdc42 and Cdk6. FEBS letters 587, 73–81 (2013).
Asnaghi, L. et al. E-cadherin negatively regulates neoplastic growth in non-small cell lung cancer: role of Rho GTPases. Oncogene 29, 2760–2771 (2010).
Zhang, S. et al. The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42 recruitment and activity. Cancer research 68, 740–748 (2008).
Chang, J. S. et al. GIT1 promotes lung cancer cell metastasis through modulating Rac1/Cdc42 activity and is associated with poor prognosis. Oncotarget 6, 36278–36291 (2015).
Hua, K. T. et al. N-alpha-acetyltransferase 10 protein suppresses cancer cell metastasis by binding PIX proteins and inhibiting Cdc42/Rac1 activity. Cancer cell 19, 218–231 (2011).
Reymond, N. et al. Cdc42 promotes transendothelial migration of cancer cells through beta1 integrin. The Journal of cell biology 199, 653–668 (2012).
Yuan, H. et al. Hepatocyte-specific deletion of Cdc42 results in delayed liver regeneration after partial hepatectomy in mice. Hepatology 49, 240–249 (2009).
Jackson, E. L. et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes & development 15, 3243–3248 (2001).
Shinjo, K. et al. Molecular cloning of the gene for the human placental GTP-binding protein Gp (G25K): identification of this GTP-binding protein as the human homolog of the yeast cell-division-cycle protein CDC42. Proceedings of the National Academy of Sciences of the United States of America 87, 9853–9857 (1990).
Lee, M. & Vasioukhin, V. Cell polarity and cancer–cell and tissue polarity as a non-canonical tumor suppressor. Journal of cell science 121, 1141–1150 (2008).
Royer, C. & Lu, X. Epithelial cell polarity: a major gatekeeper against cancer? Cell death and differentiation 18, 1470–1477 (2011).
Atkinson, J. J., Adair-Kirk, T. L., Kelley, D. G., Demello, D. & Senior, R. M. Clara cell adhesion and migration to extracellular matrix. Respiratory research 9, 1 (2008).
Castranova, V., Rabovsky, J., Tucker, J. H. & Miles, P. R. The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicology and applied pharmacology 93, 472–483 (1988).
Perl, A. K., Tichelaar, J. W. & Whitsett, J. A. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic research 11, 21–29 (2002).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Han, X. et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nature communications 5, 3261 (2014).
Wallace, S. W., Durgan, J., Jin, D. & Hall, A. Cdc42 regulates apical junction formation in human bronchial epithelial cells through PAK4 and Par6B. Molecular biology of the cell 21, 2996–3006 (2010).
Kishore, U. et al. Surfactant proteins SP-A and SP-D in human health and disease. Archivum immunologiae et therapiae experimentalis 53, 399–417 (2005).
Fidyk, N., Wang, J. B. & Cerione, R. A. Influencing cellular transformation by modulating the rates of GTP hydrolysis by Cdc42. Biochemistry 45, 7750–7762 (2006).
Wana, H. et al. CDC42 is required for structural patterning of the lung during development. Developmental Biology 374, 46–57 (2013).
Regala, R. P. et al. A typical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer research 69, 7603–7611 (2009).
Iden, S. et al. Tumor type-dependent function of the par3 polarity protein in skin tumorigenesis. Cancer cell 22, 389–403 (2012).
Ozdamar, B. et al. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 307, 1603–1609 (2005).
Wu, X. et al. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes & development 20, 571–585 (2006).
Naldini, L., Blomer, U., Gage, F. H., Trono, D. & Verma, I. M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proceedings of the National Academy of Sciences of the United States of America 93, 11382–11388 (1996).
Zheng, C., Sun, Y. H., Ye, X. L., Chen, H. Q. & Ji, H. B. Establishment and characterization of primary lung cancer cell lines from Chinese population. Acta pharmacologica Sinica 32, 385–392 (2011).
You, Y., Richer, E. J., Huang, T. & Brody, S. L. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. American journal of physiology Lung cellular and molecular physiology 283, L1315–1321 (2002).
Acknowledgements
We thank Drs Yi Zheng and Boyi Gan for helpful discussion and comments; Dr. Tyler Jacks for providing the Kras mice; J. Cao, Y. Zhou, S. Liu for technical supports. We are extremely grateful to National Centre for Protein Science Shanghai (Shanghai Science Research Center, Protein Expression and Purification system) for their instrument support and technical assistance. This work was supported by the National Basic Research Program of China 2017YFA0505500, the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), the National Natural Science Foundation of China (81325015, 81430066, 31621003, 31370747, 81402276, 81402371, 81401898, 81402498, 81101583, 81372509), Science and Technology Commission of Shanghai Municipality (15XD1504000), and Shanghai Institutes for Biological Sciences (2014KIP304).
Author information
Authors and Affiliations
Contributions
H.J. and C.Z. conceived the project and designed experiments. H.J., C.Z., L.Y. and Y.W. wrote the manuscript. C.Z. and Y.W. carried out most of the experiments and analyzed the data. S.Z., F.L., Y.F., L.Z., Q.Y., X.Z., K.W., Z.W., and Z.C. provided technical supports and helpful comments. Z.C., K.W., L.Z., Y.G. and Q.Y. help revise the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, C., Wang, Y., Yang, L. et al. Cell Division Cycle 42 plays a Cell type-Specific role in Lung Tumorigenesis. Sci Rep 7, 10407 (2017). https://doi.org/10.1038/s41598-017-10891-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10891-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
