
Abstract
Canine parvovirus (CPV) outbreaks can have a devastating effect in communities with dense dog populations. The interior region of Alaska experienced a CPV outbreak in the winter of 2016 leading to the further investigation of the virus due to reports of increased morbidity and mortality occurring at dog mushing kennels in the area. Twelve rectal-swab specimens from dogs displaying clinical signs consistent with parvoviral-associated disease were processed using next-generation sequencing (NGS) methodologies by targeting RNA transcripts, and therefore detecting only replicating virus. All twelve specimens demonstrated the presence of the CPV transcriptome, with read depths ranging from 2.2X – 12,381X, genome coverage ranging from 44.8–96.5%, and representation of CPV sequencing reads to those of the metagenome background ranging from 0.0015–6.7%. Using the data generated by NGS, the presence of newly evolved, yet known, strains of both CPV-2a and CPV-2b were identified and grouped geographically. Deep-sequencing data provided additional diagnostic information in terms of investigating novel CPV in this outbreak. NGS data in addition to limited serological data provided strong diagnostic evidence that this outbreak most likely arose from unvaccinated or under-vaccinated canines, not from a novel CPV strain incapable of being neutralized by current vaccination efforts.
Similar content being viewed by others

Introduction
Canine parvovirus type 2 (CPV-2) is a non-enveloped, single-stranded DNA virus that causes fatal gastroenteritis in young dogs1. The CPV-2 genome is 5323 nucleotide long and possesses at least 2 major open reading frames (ORFs)2. CPV-2 infections are associated with significant morbidity and mortality which can reach 91% in untreated pups3. Three variants of CPV type 2 are known, CPV-2a, CPV-2b, and CPV-2c, and are highly contagious due to suspected low infective dose requirements combined with high titers of transmissible virus in stools of affected dogs4, 5. The virus is highly resistant to environmental conditions and can remain viable outside of its host for at least a year6.
CPV-2 infections are one of the most common causes of disease outbreaks in dense canine environments such as kennels or shelters, and timely diagnosis is important in order to control the number of affected individuals7. CPV is commonly diagnosed in veterinary clinics using rapid fecal enzyme-linked immunosorbent assays (ELISA) that target viral antigen. These tests have high specificity but poor sensitivity when compared to PCR or immune-electron microscopy8. Although PCR assays are more sensitive, they can cause difficulty in terms of result interpretation since they can detect live attenuated vaccine strains or produce positive results from dogs showing no symptoms of gastroenteritis. This requires veterinarians to associate PCR results with other clinical signs of CPV infection, the animal’s history, and other laboratory parameters such as leukopenia8, 9. The performance of any of these antigen-targeting methods are highly variable due to the known phenomenon of intermittent shedding of CPV during the earlier and later stages of disease10. Despite these diagnostic challenges, it has been shown that current ELISA and PCR methods are capable of detecting all three variants of CPV in spite of antigenic differences11.
Between January and April 2016, the interior of Alaska experienced an increased number of CPV cases12,13,14. Outbreaks of canine infectious disease in Alaska can be socially and economically detrimental due to the dense dog population needed to support the state sport, dog mushing. The interior of Alaska, in particular, has a concentrated population of dogs due to the increased presence of professional and recreational mushers operating various sized kennels of 10–100+ sled dogs each. Each winter, Alaska hosts many visiting mushers, national and international, who come to the area to train and race their own dogs. Additionally, Alaska hosts several high-profile international dog mushing races, including the Iditarod and the Yukon Quest. These large races frequently include groups of dogs ranging from 350–1350 animals in number, who utilize the same trails, rests stops and parking areas, allowing for extensive comingling and a high potential for disease transmission. Sick dogs can spread the virus through defecation on common trails, where other teams run and transport the virus back to their own kennels. In 2016, the interior of Alaska experienced a mild winter with less than the usual amount of snowfall, leading to accumulation of uncovered fecal material on common mushing trails. In response to the CPV-associated outbreak of disease, mushers were provided notifications throughout the 2016 race season, and were asked to not bring potentially infected dogs to races in order to help slow the progression of the outbreak. Recommendations were also made to isolate sick animals in individual kennels. Due to the perceived increased virulence of the CPV strain or strains associated with the outbreak, additional testing to further characterize the virus was pursued.
Sequence analysis of the VP2 gene, the most abundant and immunogenic protein produced for construction of the viral particle capsid, is used to help subtype and further characterize wildtype CPV15,16,17,18. Surveillance of this particular protein is critical for assessing the potential efficacy of the current vaccination strategy and can also be used to relate individual infections in outbreak situations19,20,21,22. Beyond VP2 investigation, deep-sequencing of the whole genome has been shown to be useful in order to better understand the true nature of CPV molecular diversity and discover new variants23, 24.
As a clinical laboratory, it is our approach to target RNA molecules upon initial assessment of a clinical specimen to account for common RNA viruses as well as DNA viruses being actively transcribed while simultaneously reducing background genomic DNAs. In this experiment, we use next-generation sequencing (NGS) to detect and characterize actively replicating CPV in rectal swabs of canines associated with a suspected outbreak in the interior of Alaska between January - April 2016 by targeting RNA transcripts.
Materials and Methods
Specimens
Twelve rectal swab specimens representing two communities, A (n = 5) & B (n = 7), were collected for the sole purpose of disease diagnosis. Communities involved are 258 kilometers (approximately 160 miles) apart. The University of Alaska Fairbanks Institutional Animal Care and Use Committee (IACUC) has determined that this research project did not require IACUC review. Protocol review is not required for diagnostics performed during the course of a disease investigation. All methods were carried out in accordance with relevant guidelines and regulations.
Referral Testing (serology, PCR, and genotyping)
Specimens were sent to Cornell University, Ithaca, NY for evaluation of CPV IgG and IgM antibodies using hemagglutination inhibition (HAI), nucleic acid using PCR, and genotyping (Table 1). Total antibody was evaluated, as well as the IgM to IgG ratio upon application of 2-mercaptoethanol to dissociate IgM antibody molecules. Laboratory interpretation guidelines suggest that a 4-fold or greater decrease in titer after 2-mercaptoethanol treatment is evidence of recent parvovirus exposure. Post-vaccination levels of total antibody can range from 80 to 2,560 HAI, with ranges around 80 demonstrating need for booster vaccination. In addition to serotyping, canine parvovirus PCR was used to rule-in specimens containing CPV nucleic acid and two specimens were genotyped.
Nucleic acid isolation in preparation for sequencing
Flocked swabs were used to collect specimens from the rectum of expired canines presumed to be related to the outbreak (n = 12). Confirmation of parvoviral infection was made by rapid fecal enzyme-linked immunosorbent assays (ELISA), necropsy findings consistent with acute parvoviral illness (hemorrhagic enteritis with fibrinous serositis), or both. Swabs were immediately placed in viral transport medium to stabilize viral particles. Representative aliquots (~1 mL) of each specimen were centrifuged for 10 minutes at 15,000 rpm to pellet and discard debris as well as the majority of the host and bacterial cells. The supernatant was transferred into a new centrifuge tube and a portion of the supernatant (~500uL) was used as the starting material for nucleic acid isolation using phenol/chloroform followed by ethanol precipitation as previously described25, 26. DNAse I (New England Biolabs, Inc.) was added to the isolated total nucleic acid and purified again using phenol/chloroform followed by ethanol precipitation to reduce the overall contaminating genomic DNAs.
Library preparation for sequencing
NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Inc) was used to construct sequencing library from a starting quantity of 10–200ng of total RNA. Individual indexes were used to barcode the fragments and allow for specimen pooling. Fragment sizes of ~300 bp and larger were selected during AMPure bead cleanup.
Sequencing and analysis
Libraries were pooled and sequenced using the Illumina MiSeq system and the MiSeq Reagent Kit v2 (Illumina) 500-cycle sequencing kit. Paired-end sequencing was performed (2 × 251 bp) to achieve all base reads available in the 500-cycle sequencing format. Read files generated by the sequencer were analyzed by GSNAP reference sequence alignment using NCBI Accession NC_001539.1 as the reference genome for parvovirus.
Results
Serological data was difficult to obtain since affected animals would expire before blood draws could be acquired. Only three dogs in this data set were tested for canine parvovirus antibodies (Parvo6, 7, and 12) each demonstrating low to medium levels of acceptable antibody (Table 1). PCR was performed on most specimens in this dataset unanimously ruling-in the presence of CPV nucleic acid. Three specimens were genotyped, Parvo1 and Parvo2 (representing the same animal and therefore an opportunity to assess diagnostic reproducibility) were typed as a 2/2a virus, and Parvo10 as a 2b virus.
Table 2 describes the general sequencing metrics for each specimen tested. The number of reads aligning to the canine parvovirus reference sequence (NC_001539.1) ranged from 47 reads for Parvo11 to 263,625 reads for Parvo9. This type of range can be expected when blindly testing clinical specimens and is dependent on sample collection and handling as well as the stage of disease presenting in the canine at the time of specimen collection. Read depth ranged from 2.2X to 12,381X and genome coverage ranged from 44.8% to 96.5% when aligned to the reference genome. Canine parvovirus sequences were heavily masked amongst other sequence data representing an average of 0.95% (range 0.0015% to 6.7%) of the metagenome across all specimens sequenced.
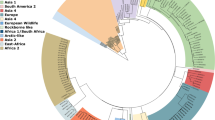
Whole genome phylogenetic analysis was performed using CLC Workbench 8 (Fig. 1). Parvo1 and Parvo11 samples were not included in this analysis due their lower than optimum read depths (<30X). Whole genome sequences from the remaining 10 specimens were compared to 13 reference genomes from NCBI representing many variations of subtype 2a and 2b canine parvoviruses as well as two versions of the attenuated vaccine strain. Phylogenetic analysis shows that there were two distinct groups of viruses circulating in the outbreak and these groups were directly associated with the geographical location of the animal when it became ill. Parvo2, 3, 6, &7 formed a group of somewhat older origin and represented animals from Community A. The other grouping of viruses sequenced (Parvo4, 5, 8, 9, 10, & 12) shows more recent evolution and were collected from animals approximately 260 kilometers away in Community B. Interestingly, Parvo6 was a dog that had been transferred from Community B to Community A; however, the sequence data would suggest that the parvovirus infection was actually caused by a virus picked up in Community A, not brought in from Community B.

Phylogram of clinical specimen isolates in relation to reference sequences of canine parvovirus 2a, 2b, and vaccine candidate genomes. Parentheses next to 10 of the 12 specimens analyzed indicate the age of the canine and kennel origination. Two specimens, Parvo1 and Parvo11, are not included in this analysis due to inadequate read depth and subsequent insufficient sequence availability. Two major groupings are recognized as distinctly 2a viruses (Parvo2, 7, 3, 6) and 2b viruses (Parvo10, 9, 5, 4, 8, 12).
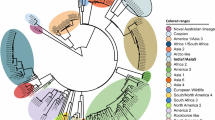
Further analysis of the VP2 protein of each one of these groupings was performed (Fig. 2). Parvo6 represented the Community A grouping and Parvo9 represented the Community B grouping due to their high genome coverage percentages and read depths (>30X). The nucleic acid VP2 sequences for these two viruses were 99.4% similar differing by 10 bases. Five (50%) of the base differences resulted in four amino acid sequence differences in the VP2 protein at residues 267, 324, 426, and 440. The amino acid difference at residue 426 is a common codon affected by polymorphism and is used to diagnostically differentiate CPV-2a and CPV-2b viruses with the use of specific sequence-based probes. The amino acid difference at position 426 for Parvo6 was asparagine, specific to CPV-2a viruses, and for Parvo9 was aspartic acid which is specific to CPV-2b viruses. The amino acid residue at position 297 was alanine for both Parvo6 and Parvo9 suggesting they are new strains of CPV-2a and −2b, as described by Decaro et al.27.

Visual depiction of sequence alignment to reference genome, VP2 gene location within the canine parvovirus genome and analysis of 18 amino acid positions. Parvo6 and Parvo9 were chosen as representatives of each group of viruses to visualize NGS data and varying sequencing depths for each major protein. The VP2 region of each specimen was analyzed at key amino acid positions reflecting canine parvovirus subtype. Parvovirus specimens are ordered and categorized as they are depicted in Fig. 1 (phylogram). “New” canine parvovirus 2a and 2b stem from emerging viruses showing variability at the 426 aa position27.
Discussion
The NGS methodology proved to be an effective diagnostic tool for further characterizing CPV in rectal swabs of affected canines. Not only was the virus detected, but the RNA for all viral proteins was obtained in many cases suggesting that the virus was undergoing active replication and causing symptoms. Despite only targeting RNA transcripts, near whole genomes were obtained making it possible to construct a phylogenetic tree that demonstrated the presence of 2 distinct subtypes of a CPV virus, CPV-2a present in dogs living in and around Community A and CPV-2b present in dogs living in Community B. Similarity of these 2 CPV virus groups to previously published CPV sequences as well as the limited presence of protective CPV antibodies in tested serum suggest that the outbreak did not involve a novel strain of CPV, but rather exposure in an under-vaccinated on unvaccinated, and therefore immunologically naive, dog population.
Summary Line
Next-generation sequencing was used for diagnosing and further characterizing a canine parvovirus outbreak in Alaska.
References
Hoelzer, K. & Parrish, C. R. The emergence of parvoviruses of carnivores. Veterinary research 41, 39, doi:10.1051/vetres/2010011 (2010).
Reed, A. P., Jones, E. V. & Miller, T. J. Nucleotide sequence and genome organization of canine parvovirus. Journal of virology 62, 266–276 (1988).
Nandi, S. & Kumar, M. Canine Parvovirus: Current Perspective. Indian journal of virology: an official organ of Indian Virological Society 21, 31–44 (2010).
Pollock, R. V. Experimental canine parvovirus infection in dogs. The Cornell veterinarian 72, 103–119 (1982).
Pollock, R. V. & Coyne, M. J. Canine parvovirus. The Veterinary clinics of North America. Small animal practice 23, 555–568 (1993).
Gordon, J. C. & Angrick, E. J. Canine parvovirus: environmental effects on infectivity. Am J Vet Res 47, 1464–1467 (1986).
Crawford, C. Management of Disease Outbreaks in Animal Shelters (2013).
Schmitz, S., Coenen, C., Matthias, K., Heinz-Jürgen, T. & Neiger, R. Comparison of Three Rapid Commercial Canine Parvovirus Antigen Detection Tests with Electron Microscopy and Polymerase Chain Reaction. Journal of Veterinary Diagnostic Investigation 21, 344–345, doi:10.1177/104063870902100306 (2009).
Decaro, N. et al. Long-term viremia and fecal shedding in pups after modified-live canine parvovirus vaccination. Vaccine 32, 3850–3853, doi:10.1016/j.vaccine.2014.04.050 (2014).
Mylonakis, M. E., Kalli, I. & Rallis, T. S. Canine parvoviral enteritis: an update on the clinical diagnosis, treatment, and prevention. Veterinary Medicine: Research and Reports 7, 91–100 (2016).
Markovich, J. E. et al. Effects of canine parvovirus strain variations on diagnostic test results and clinical management of enteritis in dogs. Journal of the American Veterinary Medical Association 241, 66–72, doi:10.2460/javma.241.1.66 (2012).
Morrow, W. Canine parvovirus outbreak hits Interior Alaska. The Daily News Miner February 25 (2016).
Press, T. A. Canine parvovirus outbreak reported in Alaska’s interior. The Juneau Empire February 28 (2016).
Thomas, M. Outbreak of Parvovirus Confirmed in Alaskan Dog Kennels. The Nome Nugget March 4 (2016).
Soma, T., Taharaguchi, S., Ohinata, T., Ishii, H. & Hara, M. Analysis of the VP2 protein gene of canine parvovirus strains from affected dogs in Japan. Research in veterinary science 94, 368–371, doi:10.1016/j.rvsc.2012.09.013 (2013).
Wang, H.-C., Chen, W.-D., Lin, S.-L., Chan, J. P.-W. & Wong, M.-L. Phylogenetic Analysis of Canine Parvovirus VP2 Gene in Taiwan. Virus genes 31, 171–174, doi:10.1007/s11262-005-1791-0 (2005).
Zhang, R. et al. Phylogenetic analysis of the VP2 gene of canine parvoviruses circulating in China. Virus genes 40, 397–402, doi:10.1007/s11262-010-0466-7 (2010).
Zienius, D., Lelesius, R., Kavaliauskis, H., Stankevicius, A. & Salomskas, A. Phylogenetic characterization of Canine Parvovirus VP2 partial sequences from symptomatic dogs samples. Polish journal of veterinary sciences 19, 187–196, doi:10.1515/pjvs-2016-0023 (2016).
Amrani, N. et al. Molecular epidemiology of canine parvovirus in Morocco. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases 41, 201–206, doi:10.1016/j.meegid.2016.04.005 (2016).
Clegg, S. R. et al. Molecular Epidemiology and Phylogeny Reveal Complex Spatial Dynamics in Areas Where Canine Parvovirus Is Endemic. Journal of Virology 85, 7892–7899, doi:10.1128/jvi.01576-10 (2011).
Kapil, S. et al. Canine Parvovirus Types 2c and 2b Circulating in North American Dogs in 2006 and 2007. Journal of Clinical Microbiology 45, 4044–4047, doi:10.1128/jcm.01300-07 (2007).
Ohshima, T. et al. Chronological analysis of canine parvovirus type 2 isolates in Japan. The Journal of veterinary medical science/the Japanese Society of Veterinary Science 70, 769–775 (2008).
Pérez, R. et al. Phylogenetic and Genome-Wide Deep-Sequencing Analyses of Canine Parvovirus Reveal Co-Infection with Field Variants and Emergence of a Recent Recombinant Strain. PLoS ONE 9 (2014).
Zhu, Y. et al. Genome Sequence of a Canine Parvovirus Strain, CPV-s5, Prevalent in Southern China. Genome Announcements 2, doi:10.1128/genomeA.01141-13 (2014).
Ge, F. et al. Preferential Amplification of Pathogenic Sequences. Scientific reports 5, 11047, doi:10.1038/srep11047 (2015).
Parker, J. & Chen, J. Application of next generation sequencing for the detection of human viral pathogens in clinical specimens. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology 86, 20–26, doi:10.1016/j.jcv.2016.11.010 (2017).
Decaro, N. & Buonavoglia, C. Canine parvovirus–a review of epidemiological and diagnostic aspects, with emphasis on type 2c. Veterinary microbiology 155, 1–12, doi:10.1016/j.vetmic.2011.09.007 (2012).
Acknowledgements
The authors wish to thank the veterinary community in Alaska for contributing specimens for further investigation as well as the laboratorians at the Biological Research and Diagnostics at the University of Alaska Fairbanks for performing initial testing and referral. This research was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103395 and by an equipment grant of Alaska State Public Health Laboratories.
Author information
Authors and Affiliations
Contributions
J.C. designed the study, J.P. and J.C. performed experiments and analyzed the data. J.P. wrote the initial manuscript. M.M. and K.H. provided clinical diagnosis. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parker, J., Murphy, M., Hueffer, K. et al. Investigation of a Canine Parvovirus Outbreak using Next Generation Sequencing. Sci Rep 7, 9633 (2017). https://doi.org/10.1038/s41598-017-10254-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10254-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
