
Abstract
Alzheimer’s disease (AD) is characterized by deposition of amyloid beta (Aβ) peptides into senile plaques in the brain. While most familial mutations are associated with early-onset AD, recent studies report the AD-protective nature of two genetic human Aβ variants, i.e. A2T and A2V, in the heterozygous state. The mixture of A2V Aβ1-6 (Aβ6) hexapeptide and WT Aβ1–42 (Αβ42) is also found neuroprotective. Motivated by these findings, in this study we investigate the effects of WT, A2V, and A2T Aβ6 hexapeptide binding on the monomeric WT Aβ42 landscape. For this purpose, we have performed extensive atomistic Replica Exchange Molecular Dynamics simulations, elucidating preferential binding of Aβ42 with the A2V and A2T hexapeptides compared to WT Aβ6. A notable reorganization of the Aβ42 landscape is revealed due to hexapeptide association, as manifested by lowering of transient interactions between the central and C-terminal hydrophobic patches. Concurrently, Aβ6-bound Aβ42 monomer exhibits alternative structural features that are strongly dependent on the hexapeptide sequence. For example, a central helix is more frequently populated within the A2T-bound monomer, while A2V-bound Aβ42 is often enhanced in overall disorder. Taken together, the present simulations offer novel molecular insights onto the effect of the N-terminal hexapeptide binding on the Aβ42 monomer structure, which might help in explaining their reported amyloid inhibition properties.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD), presently accounting for 60–70% of the 47.5 million dementia cases worldwide1, is symptomatic of progressive and irreversible loss of cognitive brain functions including memory, language skills, and spatiotemporal perception. AD is neuro-pathologically characterized by the aggregation of amyloid beta (Aβ) peptides into extracellular senile plaques, formation of intracellular neurofibrillary tangles, and subsequent neuronal degeneration2, 3. The causative Aβ peptide exists in varying lengths, Aβ40 and Aβ42 being the most abundant isoforms. Of these, Aβ42 is more aggregation-prone and toxic in nature4.
Solid-state (ss) NMR studies reveal that both Aβ40 and Aβ42 fibrils are composed of intermolecular stack of β-strands, termed as cross-β conformation, formed via self-complementary side-chain interfaces – so-called dry steric zipper5. In both Aβ40 and Aβ42 fibrils, residues around the central hydrophobic cluster (CHC) and the hydrophobic C-terminal region (CTR) form two parallel, in-register β-strands separated by a hydrophilic turn region6, 7. Structural polymorphism has been reported within Aβ fibrils, revealing alternative β-sheet arrangements, such as a triple β-motif8 or a double horse-shoe like structure composed of five β-strands9.
Aβ aggregation is initiated by a rate-limiting primary nucleation process, in which monomers self-associate into oligomeric ‘paranuclei’, followed by fibril assembly10. Oligomers can also be formed via a fibril-catalyzed secondary nucleation pathway from the monomeric peptides11. Soluble Aβ oligomers are reportedly more toxic than the fibrils and monomers, and are considered as pathological target for the treatment of AD12, 13. The neurotoxicity of those oligomers is correlated to their β-sheet content14 that increases with the order of polymerization15. Possible mechanism for Aβ oligomer toxicity includes direct association with membrane, resulting in membrane disruption and/or pore formation16.
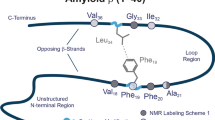
The monomer to oligomer transition is thought to be initiated by transient misfolding of the monomer to an aggregation-competent, hairpin-like conformation17,18,19,20,21,22. This structure is topologically similar to the peptide monomer conformation in fibrils, in which CHC and CTR are in direct contact, and residues 22–29 form a turn-like structure19, 23, 24 (hereafter referred as the turn region, see Fig. 1a). Designed Aβ sequences that fold into β-hairpin monomer conformation reportedly stabilize toxic, β-sheet rich oligomers and inhibit amyloid fibril formation20. However, translating the structural knowledge of the ordered amyloid fibrils to the Aβ monomer and pre-fibrillar, soluble oligomers remains non-trivial using conventional experiments, as those species are highly metastable, structurally heterogeneous, and disordered in nature25, 26. Aβ peptide belongs to the class of Intrinsically Disordered Peptides (IDP)27 that lack unique 3-dimentional structure, and instead exist as a dynamic ensemble of interconverting conformations. Molecular simulations at various resolutions28,29,30,31,32,33 have served as a useful means, particularly when combined with experiments, to characterize structure and binding of the disordered peptides.

Simulation of Aβ42 with WT, A2V, or A2T Aβ6. (a) Amino acid sequence of Aβ42. Residues 1–16, 17–21, 22–29, and 30–42 of Aβ42 are denoted as NTR, CHC, turn and CTR, respectively. Color-code used to describe different regions of Aβ42 is also shown. Aβ6 derivatives are shown inside the black rectangle. Amino acid at position 2 of the hexapeptide is depicted in red. (b) The initial structure of the WT Aβ42 monomer and the A2T Aβ6 fragment immersed in water. Peptides are shown using cartoon representation. Aβ42 chain is color-coded according to Fig. 1a. Aβ6 is shown in red, and residue 2 is represented in orange using van der Waals (vdW) spheres. Water molecules (in brown) and sodium ions (in green) are shown as points and spheres respectively. (c) Backbone RMSD (in nm) of Aβ42, with respect to the initial peptide structure, as a function of simulation time (in ns) at 308.4 K. Moving averages of 1 ns are shown. The raw data (WT-bound: light brown, A2V-bound: light green, A2T-bound: light blue) are also plotted. (d) Residue-wise turn propensity (in %) at 308.4 K, using two different time-windows, 60–130 ns (solid line) and 60–200 ns (dashed line).
The Aβ N-terminal region (NTR), that is primarily disordered in fibril structures6, 7, has long been relatively neglected in terms of its role in downstream events of AD. However, a number of familial mutations leading to aberrant monomer misfolding, aggregation, and related toxicity, have been identified at the NTR, such as the English (H6R) and the Tottori (D7N)34. Interestingly, a familial A2V mutation, while causing dementia in homozygous carriers, has been reported to provide protection in heterozygous carriers35. In a seminal whole-genome sequencing study on an Icelandic population, the rare A2T mutation was found to be AD protective and also offered protection from age-related cognitive decline in non-AD patients36. Experimental37,38,39,40,41 and simulation studies42,43,44 have revealed that, both A2V and A2T mutations differentially affect the Aβ monomer and oligomer structures, peptide aggregation kinetics, and associated toxicity. The protective nature of the WT/A2V(T) Aβ cross-interactions thus provides a natural path towards design of mechanism-based AD therapeutics. Along this line, Di Fede et al.45 recently showed that Aβ1-6A2V in D-form directly interacts with WT Aβ40/42 and prevents amyloid fibril formation in vivo. Further testing of the hexapeptide tagged with the TAT sequence [Aβ1-6A2VTAT(D)] in a mouse model revealed alteration of oligomer size distribution, aggregation inhibition, and cerebral amyloid deposition46. However, little is known about the molecular mechanism underlying the inhibitory effect of those short NTR peptide fragments.
Given the importance of monomer misfolding in the amyloidogenesis pathway, it can be expected that binding of these Aβ-derived short inhibitor peptides results in remodeling of the full-length Aβ monomer landscape, thereby interfering with the subsequent aggregation. In this study, we take the first steps towards quantifying the effects of the binding of three different Aβ NTR hexapeptide (Aβ6) variants on the Aβ42 monomeric landscape. The hexapeptide variants studied are WT, A2V, and A2T Aβ6. For this purpose, we have performed large-scale, atomistically-detailed Replica Exchange Molecular Dynamics (REMD) simulations of the full-length peptide and hexapeptide variant in explicit water. In recent years, REMD has been extensively used as a tool to successfully sample conformational ensembles of amyloid proteins43, 44, 47,48,49. Our simulations reveal that these NTR-derivatives cause a substantial and sequence-dependent reconfiguration of the Aβ42 landscape. Taken together, this simulation study illustrates the potential of the NTR-based hexapeptides toward mechanism-based therapeutic design and calls for further extensive structural investigation, both computational and experimental.
Results
Simulating Aβ42 monomer in presence of WT, A2V, or A2T Aβ6 hexapeptide variant
The amino acid sequence of the Aβ42 monomer and the three Aβ6 NTR-derivatives studied here are shown in Fig. 1a. For brevity, we will refer these three Aβ42 + Aβ6 systems as WT-bound (or + WT), A2V-bound (or + A2V), and A2T-bound (or + A2T) henceforth. The simulated system comprising of the solvated full-length peptide monomer and hexapeptide variant is represented in Fig. 1b. To quantify the effect of Aβ6 binding on Aβ42 landscape, we compared the simulations of Aβ42 in presence of Aβ6 with earlier reported simulations of the free Aβ42 monomer42.
To ensure that the choice of the initial peptide conformation did not bias the results, we computed the root-mean-square deviation (RMSD) of the Aβ42 backbone from the initial peptide structure as a function of simulation time. Figure 1c depicts the backbone RMSD evolution of the 308.4 K replica as an example. The backbone RMSD reaches a value of 1.00 nm around 20 ns, then quickly equilibrates to ~1.20 nm by 50 ns, and fluctuates around this value (with a standard deviation of 0.12 nm) for the rest of the simulation. Based on this result, we consider that the REMD trajectories reached equilibration by around 50 ns, and the 60–200 ns portion of the trajectories was regarded as the production run. The production ensemble consists of structures extracted every 50 ps from the twelve replicas (276.0–308.4 K), resulting into 33,600 structures.
We further confirmed sampling convergence by comparing the residue-wise turn propensity of Aβ42 over the time intervals of 60–130 ns and 60–200 ns (Fig. 1d, also see Table S1). The overall turn propensity shows a mean value of ~50% for the hexapeptide-bound Aβ42 monomer, and is almost indistinguishable over the two time-intervals. Equilibration and convergence of the simulations were also tested in terms of additional structural properties (see Supplementary Information, Fig. S1, and Table S1). These results suggest that the production ensemble is not biased towards the initial conformation and has reasonably converged to a quasi-equilibrium state.
The radius of gyration (Rg) distribution of the Aβ42 monomer, as estimated from the production ensemble (see Supplementary Fig. S2), reveals a mean value around 1.05 nm for the free Aβ42 and is unchanged upon hexapeptide binding. This value agrees well with the value of the hydrodynamic radius (0.9 nm) reported in fluorescence correlation spectroscopy measurements50 and Rg values reported in earlier REMD simulation studies47, 51. The estimated Rg value indicates a collapsed structure, as Rg scales as N1/3 (where N = 42)52, 53. We have also quantified the extent of disorder within the production ensemble by performing an RMSD-based clustering (see Supplementary Information). The results, as shown in Supplementary Fig. S2, imply that the top 50 clusters cumulatively represent ~90% of the total population for all systems. Thus, the Aβ42 peptide remains intrinsically disordered in presence of the hexapeptide variant, to the same extent as it is in its free state.
Hexapeptide interaction modifies Aβ42 secondary structure profile
Figure 2 shows the ensemble-averaged, residue-wise population of the secondary structure elements within the free Aβ42 ensemble. The standard error values were obtained from standard deviations estimated by dividing the simulation data into four 35 ns long, non-overlapping blocks between 60–200 ns. The calculated standard error values are negligible, indicating statistical significance of the values reported (see Model and Methods in Supplementary Information). A notable feature of the secondary structure profile of free Aβ42, is a >20% β-strand propensity around the CHC (residues 17–21) and the CTR (residues 30–41). The residue-wise secondary structure distributions of hexapeptide-bound Aβ42 can be found in Supplementary Fig. S3. The overall disordered nature of Aβ42 in all systems is illustrated, as coil and turn population together account for 70–75% of secondary structure. Hexapeptide binding in general results in considerable lowering of the CHC and CTR β-strand propensity (at least by 10% in majority of those residues, see Fig. 2b–d), when compared to free Aβ42. The β-strand tendency reduction is most prominent in the CHC residues. In contrast, the preCHC region (residues 11–16) exhibits enhanced β-strand tendency in all three hexapeptide-bound Aβ42 systems. β-strand formation is also noticed around residues 6 and 29 in WT-bound Aβ42 and residues 28–30 in A2V-bound Aβ42. The first 17 residues appear more helix-rich in the WT-bound and A2T-bound peptide (Fig. 2b,d). On the other hand, the helicity becomes almost negligible due to A2V hexapeptide binding.

Secondary structural content of bound and free Aβ42. (a) Residue-wise population distribution (in %) of the secondary structural elements in free Αβ42 monomer. The values were estimated from our earlier published REMD study42. Standard errors of mean were estimated by dividing the production portion of replicas into four 35 ns long, non-overlapping blocks (see Methods). (b–d) Differences (in percentage) in residue-wise β-strand, 310-helix, and α-helix propensities between bound and free Aβ42. Positive values indicate higher occurrence in the hexapeptide-bound system, and vice versa. (b) WT-bound, (c) A2V-bound, and (d) A2T-bound.
The short peptide mainly populates coil structure (≥60%) in all three variant forms (Supplementary Fig. S4). Interestingly, the A2V hexapeptide shows notable (~15%) propensity to form β-strand around residues 2 and 3, when compared to the two other variants.
Wild-type CHC-CTR interaction is reduced due to Aβ6 binding
Next, we compare the intramolecular contact map of the bound Aβ42 with that of the free peptide, to reveal the effect of short peptide binding on the Aβ42 tertiary structure (see Fig. 3 and Supplementary Fig. S5). It should be noted that all long-range (|i-j| > 8) contacts observed are transiently populated (with a ~12% average probability of formation) within the unbound Aβ42 ensemble, in accordance with its intrinsically disordered nature. Noticeable long-range interactions are: (i) a set of anti-parallel, hairpin interactions between the CHC and CTR residues, (ii) a second, less-extensive set of hairpin interactions within CTR residues 30–41, and (iii) contacts between two termini. The observed β-strand proclivity of the CHC and CTR residues, the dominant (>70%) turn tendency of residues 24–28, and the CHC-CTR tertiary interactions, together suggest transient population of a CHC-CTR β-hairpin monomer. Such transiently formed, extended hairpin-like monomer structures are believed to be aggregation-prone in nature, therefore triggering the formation of soluble, toxic oligomers and subsequent polymerization20, 23, 24.

Characterization of Aβ42 tertiary structure. (a) Ensemble-averaged intramolecular Cα contact map of the free Aβ42 monomer42 (upper triangle). Only non-sequential contacts, i.e., |i–j| ≥ 3, are considered. Arithmetic difference between the contact probabilities of hexapeptide-bound and free Aβ42 are also plotted (a, lower triangle: WT-bound; b, upper triangle: A2V-bound; b, lower triangle: A2T-bound). Color-scales used for contact probability and contact probability differences (bound minus free) are shown on top of the figures.
Interestingly, the Aβ42 tertiary contact map reveals significant changes due to hexapeptide binding. Only the contacts showing a probability difference value higher than 0.06 were considered for this analysis. First of all, hexapeptide binding induces a marked ≥20% reduction in the CHC-CTR interaction (Fig. 3). At the same time, the D23-K28 salt-bridge frequency decreases from 13% in free Aβ42 to 8–9% in the bound peptide. These findings together suggest that hexapeptide binding reduces the transient folding into an aggregation-prone β-hairpin structure, of which CHC-CTR contacts and D23-K28 salt-bridge are key features17, 18, 21, 54. Additionally, interactions of extreme NTR with rest of the protein generally decrease in the bound Aβ42. On the other hand, enhanced interaction between the preCHC residues 12–16 and the CTR residues 30–35 is observed, following the order: A2T-bound (12%) <WT-bound (16%) <A2V-bound (26%). Resembling what was seen in secondary structure analysis (Fig. 2), the tertiary structural profile of the WT-bound and A2T-bound Aβ42 appear similar to each other. For example, the hairpin contacts within CTR are consistently present in the WT-bound and A2T-bound Aβ42 (to the same or increased extent as found in free Aβ42), but decreases upon A2V hexapeptide binding. Enhanced binding between the CHC region and residues 24–30 is noticed in the A2V-bound system (Fig. 3b). The strong β-strand propensity in those residues combined with enhanced turn tendency of residues 24–27 consistently suggest formation of a register-shifted β-hairpin in the A2V-bound Aβ42 (see Fig. 2 and Supplementary Fig. S3).
A2V and A2T substitutions favor Aβ42-Aβ6 binding
Binding free energy (∆Gbind) between Aβ6 and Aβ42 was calculated by using the Molecular Mechanics - Poisson-Boltzmann Surface Area (MM-PBSA)55 method (Fig. 4a). Both electrostatic (∆Eelec) and Van der Waals (∆EVdW) terms equally contribute to the WT Aβ6 – Aβ42 binding energy. Using WT-hexapeptide binding as reference, it was observed that binding is preferred upon A2V and A2T mutation. While ∆EVdW remains the same (~−27 kcal/mol), ∆Eelec decreases from −25 kcal/mol to −37 kcal/mol in A2V-bound and −43 kcal/mol in A2T-bound Aβ42, indicating a strengthening of the interpeptide electrostatic interactions. Interestingly, SPR experiments also indicated more favorable binding of the 1–6 A2V peptide with Aβ40, when compared to the WT hexapeptide45. A comprehensive breakdown of the constituent MM-PBSA energy terms is given in Supplementary Table S2.

Intermolecular binding analysis. (a) Interpeptide binding free energy estimation, as estimated using the MM-PBSA method. Van der Waals, electrostatic, and total binding energy values (±standard errors of mean) are reported in kcal/mol. (b) Average number of Aβ42 heavy-atom contacts per residue across the hexapeptide sequence. Standard error values were estimated by dividing the production data into four 35 ns long blocks (see Methods). (c–e) Quaternary heavy-atom contact probability maps: (c) WT-bound, (d) A2V-bound, and (e) A2T-bound systems.
Figure 4b illustrates the average number of heavy-atom contacts with the full-length peptide across the hexapeptide residues. This analysis suggests average ~3.3 interpeptide heavy-atom contact formation per hexapeptide residue for all three Aβ6 variants. Figure 4b further indicates that, a valine or threonine at position 2 of Aβ6 interacts more frequently than an alanine. This frequency increase is stemming from the preferential interaction with Aβ42 residues 12–22, 28, and 40, as suggested by the interpeptide contact maps shown in Fig. 4c–e. A threonine at positon 2 of Aβ6 also allows E3 to be more interacting, especially with the preCHC residues 11–16. On the other hand, the probability of contact formation with aromatic F4 residue from Aβ6 is enhanced, when the hexapeptide variant is more hydrophobic in nature (WT or A2V). In those scenarios, the contacting Aβ42 residues are 3–6, 14–17, and 24–27 (Fig. 4c and d). The primary mode of hexapeptide binding involves NTR Aβ42 residues, irrespective of the Aβ6 variant sequence studied here (Fig. 4c–e). Residues 1–7 of Aβ42 exhibit ~12% probability of contact formation with A2V and A2T Aβ6, and ~17% with WT hexapeptide (Fig. 4c). Additional contacting Aβ42 residues are residues 12–17 and 34 with WT Aβ6 (Fig. 4c), CHC residues with A2V Aβ6 (Fig. 4d), and preCHC residues 11–16 with A2T Aβ6 (Fig. 4e). Overall, the A2V and A2T variants are more efficient in sequestering the central hydrophobic core and binding to K28, when compared to WT Aβ6. Thus, A2V and A2T Aβ6 appear stronger candidates for disrupting the CHC-CTR interaction and central salt bridge formation17, 54, which are crucial components of the β-hairpin structure found in oligomers and fibrils.
Alternative structures are stabilized on the hexapeptide-bound Aβ42 landscape
To further disseminate the changes in the Aβ42 monomeric landscape due to hexapeptide binding, we have estimated a two-dimensional Potential of Mean Force (PMF, see Methods and Fig. 5). For this purpose, the following two reaction coordinates were defined: (i) the number of residues in CHC and CTR region that are in β-strand conformation, NβCHC+CTR; and (ii) the number of Cα contacts between CHC and CTR, NCCHC-CTR; both normalized to one. This choice was motivated by the secondary and tertiary structural changes resulting from short peptide binding (Figs 2 and 3). Interestingly, while the full-length monomer in all four systems (three Aβ6-bound and free Aβ42) sampled similar regions of the energy landscape, the population distribution was substantially affected upon hexapeptide binding (Fig. 5). This observation led to further investigation of six regions on the PMF plot, S1 to S6 (see Fig. 5, Table S3 and Methods). The population distribution within these regions is shown in Table 1 for all systems, suggesting that the S1–S6 populations together represent 80–90% of the total ensemble in each case. The free Aβ42 monomer frequently (~47%) visits the S6 region that corresponds to high values of NCCHC-CTR and NβCHC+CTR, indicating possible CHC-CTR β-hairpin population. However, a RMSD-based clustering of the S6 sub-population reveals high structural diversity even within this region. Only those structures with NβCHC+CTR > 0.6 consistently show a single or a double hairpin motif comprised of CHC and CTR residues (Supplementary Fig. S6). Binding to Aβ6 variant dramatically reduces S6 population, e.g. to 21% in WT-bound, to 22% in A2V-bound, and to 13% in A2T-bound Aβ42 (Table 1), indicating that Aβ6 binding inhibits CHC-CTR hairpin interaction that is thought to be crucial for aggregation nucleation and toxicity18, 20.

Bound and free Aβ42 monomeric landscapes. (a–d) 2D potential of mean force (PMF, in kcal/mol) plots as a function of the number of residues in CHC and CTR region that are in β-strand conformation (NβCHC+CTR), and the number of Cα contacts between CHC and CTR (NCCHC-CTR), both normalized to one. (a) free monomer, (b) WT-bound, (c) A2V-bound, and (d) A2T-bound systems. Solid black rectangles denote the S1–S5 states that are more frequently populated upon hexapeptide binding. The S6 sub-population (dashed black rectangle) corresponds to the CHC-CTR hairpin-rich region.
Concurrently, alternative conformations emerge on the hexapeptide-bound Aβ42 monomeric landscape, which substantially depend on the Aβ6 sequence (Fig. 5). Those alternative structures were further investigated in detail by performing a RMSD-based clustering on the S1–S5 sub-populations. Regions representing at least 10% of total population were only considered (Table 1). Representative structures are shown in Supplementary Fig. S10. Visual inspection of the representative structures, as extracted from clustering, revealed emergence of four main structural features within the alternative Aβ42 monomeric conformations: (i) an overall unstructured population visited mainly by the A2V-bound Aβ42, (ii) a preCHC/CHC helix populated in WT and A2T-bound monomer, (iii) a CTR β-hairpin, found in WT and A2T-bound monomer, and (iv) NTR-CTR β-sheet seen within WT and A2V-bound Aβ42 (see Fig. 6 and Supplementary Fig. S7). In the following, we discuss these four different classes of alternative structures in detail. Secondary structure and tertiary interaction profiles for those alternative populations are illustrated in Supplementary Fig. S8. A small, but non-negligible, 11% population consisting of a non-native register-shifted β-hairpin is seen in A2V-bound S5 structures, which is further discussed in the Supplementary Information (see Supplementary Fig. S9).

Alternative Aβ42 structures are increasingly populated due to hexapeptide binding. (a) Highly unstructured (within A2V-bound S1 and S2). (b) preCHC/CHC helix (within A2T-bound S1 and S3). (c) CTR-hairpin (within WT-bound and A2T-bound S4). (d) NTR-CTR β-sheet (within WT-bound S5 and A2V-bound S4). An RMSD-based clustering with 0.3 nm pairwise Cα-RMSD cut-off was performed for each sub-population. Representative structures from the largest clusters are displayed. Relative population (in %) of the corresponding cluster within each state is reported in parenthesis. Peptides are displayed using cartoon representation. The Aβ42 peptide is colored according to the color scheme shown in Fig. 1a, while the Aβ6 peptide is colored in red. Different orientations of Aβ6 within each cluster are shown. Aβ42 residues that are within 0.4 nm (heavy atoms only) of the hexapeptide are displayed in licorice representation.
Disordered structures
The A2V-bound S1 and S2 states are consistent with an overall disordered structure (Fig. 6a and Supplementary Fig. S7), and together account for about 28% of the total population. Those Aβ42 structures mainly sample turn or coil conformation. The hexapeptide extensively engages with the CHC, 22–29 turn, or CTR residues, which directly competes with the CHC-CTR hairpin interactions needed to nucleate Aβ aggregation. V2 from the short peptide is frequently involved in these interactions.
Central helix structures
The preCHC/CHC helix is primarily seen in the A2T-bound S1, A2T-bound S3, and in WT-bound S2 states (Fig. 6b and Supplementary Fig. S7). This helical feature is most dominant in A2T-bound Aβ42, accounting for about 34% of the total ensemble (Table 1). Additional features, such as β-strand at the Aβ42 NTR or within the short peptide, are also seen (see Supplementary Fig. S7). In these structures, the hexapeptide mainly binds to the NTR, preCHC and the turn (residues 22–29) regions of Aβ42. H6 from the hexapeptide dominates the quaternary binding interface (Table 1) and contacts with NTR or preCHC residues.
CTR hairpin structures
The CTR-hairpin is the main characteristic of the WT-bound and A2T-bound S4 states (Fig. 6c and Supplementary Fig. S7), representing 18% and 20% of the total ensemble, respectively. The hairpin involves residues 30–41, with residues 34–38 generally in a turn conformation. It must be noted that the C-terminal hairpin with a G37/38 hinge has been reported in earlier simulation studies of free wild-type Aβ42 51, 56. In some of those structures, NTR Aβ42 residues directly interact with the CTR hairpin, forming a β-sheet conformation. In some structures, the NTR, preCHC, and/or CHC residues adopt helical form. Within this sub-population, the hexapeptide directly contacts with the CTR hairpin or the CHC residues, again disrupting the CHC-CTR intramolecular interactions. F4 and R5 from Aβ6 dominate the intermolecular binding.
NTR-CTR β-sheet structures
The WT-bound S5 and the A2V-bound S4 states are characterized by an NTR-CTR β-sheet (Fig. 6d, Supplementary Fig. S7d). These sub-populations account for about 16.5% of the WT-bound and 23% of the A2V-bound ensemble (Table 1). The NTR of Aβ42 is mainly found to be in contact with the hexapeptide, whereas F4 from Aβ6 governs the quaternary association (Table 1).
Discussion
In light of the AD-protective nature of familial A2V and A2T mutations in heterozygous carriers, therapeutic design based on the molecular basis of WT/A2V(A2T) Aβ cross-interaction appears promising. In vivo and in vitro studies have further confirmed the striking ability of the A2V and A2T Aβ variants to impede Aβ nucleation, neurotoxicity, and aggregation37, 38, 41, 57, 58 by directly interacting with WT Aβ. Remarkably, this inhibitory effect was found to be retained, even when WT Aβ42 was co-incubated with a short 1–6 WT or A2V Aβ variant, the effect being more prominent for the A2V variant35, 45, 46. Recently, short A2T NTR fragments of varying lengths have also been reported to inhibit fibrillization and rescue from toxicity59. A molecular characterization of the NTR hexapeptide binding with the full-length peptide is therefore crucial towards understanding of the aggregation-inhibitive and neuroprotective properties of those short peptides, which is useful for introducing new AD therapy. To our knowledge, the present simulation study reports for the first time the striking effect of the Aβ NTR hexapeptide binding on the WT Aβ42 monomeric landscape. Our results reveal remarkable differences between the WT, A2V, and A2T hexapeptide variant in terms of Aβ42 binding, and resulting structural changes.
While the overall disordered nature of Aβ42 persists, the transient CHC-CTR β-hairpin interactions that are implicated in Aβ aggregation and toxicity18,19,20 is strikingly reduced upon hexapeptide binding. Consequently, short-lived alternative populations emerge on the monomeric conformational landscape. The good agreement between the conformational landscape of the free and hexapeptide-bound monomer indicates involvement of conformational selection60 in the binding process. However, present simulations do not allow to quantify if, and to what extent, conformational selection governs binding. In the bound state, Aβ6 frequently populates a ‘fuzzy’ cloud of possible orientations around a transiently populated Aβ42 structure (Fig. 6 and Supplementary Fig. S7). In those fuzzy complexes61, the loss in overall binding enthalpy is somewhat compensated by a lower entropic loss. As a result of such dynamic interaction, long range electrostatic and transient physical contacts may increase61, leading to greater number of initial contacts and an increased capture radius62. Being an IDP, Aβ42 does not provide a unique binding groove to the hexapeptide, leading to promiscuous binding63. However, the Aβ42 NTR is often found engaged with the hexapeptide, which is desired, given the emerging pivotal role of NTR in Aβ structure, oligomerization/aggregation, associated toxicity, as well as interactions with anti-amyloid molecules64,65,66. A more flexible NTR is often associated with higher toxicity, as Aβ42 NTR is found more flexible than that of Aβ40 in monomer67 and dimer simulations68.
Four distinct structural features are consistently noticed within the emerging alternative Aβ42 populations due to hexapeptide binding: (i) an overall unstructured population, (ii) a preCHC/CHC helix, (iii) a CTR β-hairpin, and (iv) an NTR-CTR β-sheet. The relative propensity of hexapeptide-bound Aβ42 to visit these alternative sub-populations is strongly dependent on the Aβ6 sequence. For example, a central helix or a CTR β-hairpin is more frequently populated within the A2T-bound monomer, while A2V-bound Aβ42 is often disordered, or forms an intramolecular β-sheet involving both termini.
The emergence of a preCHC/CHC helix in the A2T or WT hexapeptide-bound Aβ42 (Fig. 6b and Supplementary Fig. S7) is of interest, since helix stabilization in this region has been reported to reduce fibril formation69 and counteract toxic oligomer formation70. Mutation such as V18A/F19A/F20A or addition of phospho-L-serine in the CHC region increases helical content, and subsequently reduces aggregation69. Therefore, enhanced helical Aβ42 population resulting from WT or A2T hexapeptide binding may impede Aβ42 aggregation, which is consistent with experimental findings35, 45.
A2V hexapeptide binding favors an overall unstructured population of monomeric Aβ42. Since Aβ monomer misfolding is implicated in formation of β-sheet rich oligomers20, the simulation results are in line with the experimental observation that binding of A2V hexapeptide inhibits β-structure formation in full-length Aβ45. Redirecting IDPs such as Aβ and α-synuclein towards stable, unstructured non-toxic oligomer formation has been associated with the aggregation and toxicity inhibiting effect of resveratrol71 and epigallocatechin gallate (EGCG) molecules72. Taken together, enhanced disorder in A2V Aβ6-bound Aβ42 might prevent toxic oligomer formation.
It is widely accepted that transiently formed β-strand/sheet-rich monomeric structures can promote aggregation and toxic oligomer formation in amyloidogenic proteins73,74,75,76,77, such as huntingtin exon1, α-synuclein and IAPP. Previous studies have also indicated a positive correlation between hydrophobic solvent exposure and aggregation propensity (and related toxicity)14. The hydrophobic solvent accessible surface area is consistently estimated to be smaller in the alternative Aβ42 structures with the C-terminal hairpin (2174 ± 62 Å2) or the NTR-CTR β-sheet (2375 ± 67 Å2), when compared to structures with a CHC-CTR β-hairpin (2567 ± 76 Å2). Thus, enhanced population of these atypical β-strand/sheet-rich conformations may help in lowering Aβ aggregation and toxicity. Commensurate to this speculation, a monomer with a C-terminal hairpin was more frequently seen in simulations of the protective A2T Aβ variant compared to the wild-type variant42. On the other hand, a double β-hairpin was often visited by the causative A2V Aβ monomer.
Redirecting Aβ monomers to atypical conformations appears a more feasible and attractive strategy for treating AD, compared to directly targeting Aβ production, as Aβ is essential in modulating synaptic activity, neuronal viability, and has shown potential antioxidative functions78, 79. The alternative conformations stabilized on the hexapeptide-bound Aβ landscape might be off the aggregation pathway, as indicated by the earlier experiments69,70,71,72. However, it has been proposed that the α-helical epitope, while present in a sub-population of monomeric Aβ, may be absent in oligomers and higher order aggregates. In fact, solution NMR in presence of helix promoting agents such as HFIP or SDS suggests helix formation only in Aβ monomers80. Thus, the A2T hexapeptide likely recognizes an epitope that is only present in a monomeric Aβ42 sub-population, resulting into a higher preference for monomer. On the other hand, the unstructured peptide conformation is found in a broad variety of Aβ species including oligomers and is related to lower toxicity71, 72. Therefore, targeting the unstructured epitope, as observed in the A2V hexapeptide binding, seems a more rational approach for AD therapeutic design. Our simulations reveal that the A2V short peptide binding stabilizes the unstructured monomer, subsequently sequestering the central and/or C-terminal hydrophobic regions and targeting the central turn region, thus eliminating the essential structural features needed for toxic Aβ oligomer and fibril formation. The unique population of disordered Aβ42, together with stronger binding affinity of the A2V hexapeptide, may explain its more pronounced effect on inhibiting β-sheet rich oligomer formation and aggregation of the full-length peptide, with respect to what was seen with the WT hexapeptide45.
A number of phenolic compounds, such as EGCG72, Congo Red dye81 and resveratrol71, have been previously identified to prevent Aβ aggregation and toxicity. Short peptide fragments derived from natural Aβ sequence have also been extensively studied as potential inhibitors of AD. The hydrophobic CHC derivatives82, while being effective fibrillation inhibitors, are reportedly less efficient in preventing oligomerization. On the other hand, CTR fragments83, 84 are more successful in mediating both oligomerization and fibrillogenesis through diverse mechanisms. Recent experiments establish the amyloid inhibition properties of the NTR Aβ fragment that is mainly hydrophilic in nature. The present study consistently illuminates the sequence-dependent, differential ability of the NTR hexapeptides to reconfigure the Aβ folding landscape away from disease-implicated structures. It will be intriguing to study the effect of length and sequence variation of the short peptide on the Aβ42 folding landscape, which will be addressed in future. In summary, our intriguing findings unravel the key structural features of the full-length Aβ monomer binding with N-terminal fragment derivatives, which offers novel molecular insights onto their amyloid inhibition properties and can further guide rational design of mechanism-based therapeutics for devastating protein aggregation diseases.
Model and Methods
Description of the REMD protocol, including system setup, simulation parameters, etc. are provided in the Model and Methods section of the Supplementary Information. The Aβ42 monomer and the Aβ6 hexapeptide were placed together in a 5.7 × 5.7 × 5.7 nm3 sized cubic box containing ~5,600 water molecules, and charge-neutralized with Na+ and Cl− atoms. The simulated system thus contains a total of about 17,400 atoms. A combination of the OPLS-AA force field85 and TIP3P water model86 was used for all simulations reported in this study, which has been reported to generate Aβ ensemble consistent with NMR measurements47, 51. During REMD, 64 replicas, each 200 ns long, spanning an exponentially distributed temperature range of 276.0–592.3 K were simulated in parallel and exchanged intermittently, resulting in an aggregate of 12.8 μs of conformational sampling for each system. Details of simulation equilibration and convergence assessment, structural characterization, clustering, binding energy calculations, PMF analysis, and error calculations are also reported in the Supplementary Information.
Data Availability
All simulation data will be available upon request.
References
Dementia, Fact Sheet No 362, WHO Media Centre, (April 2016), http://www.who.int/mediacentre/factsheets/fs362/en/.
Selkoe, D. J. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science 275, 630–631 (1997).
Yankner, B. A. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron 16, 921–932 (1996).
Klein, A. M., Kowall, N. W. & Ferrante, R. J. Neurotoxicity and oxidative damage of beta amyloid 1-42 versus beta amyloid 1-40 in the mouse cerebral cortex. Ann N Y Acad Sci 893, 314–320 (1999).
Nelson, R. & Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr Opin in Struct Biol 16, 260–265 (2006).
Petkova, A. T. et al. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA 99, 16742–16747 (2002).
Luhrs, T. et al. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci USA 102, 17342–17347 (2005).
Xiao, Y. et al. Abeta(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol 22, 499–505 (2015).
Walti, M. A. et al. Atomic-resolution structure of a disease-relevant Abeta(1-42) amyloid fibril. Proc Natl Acad Sci USA 113, E4976–4984 (2016).
Roychaudhuri, R., Yang, M., Hoshi, M. M. & Teplow, D. B. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem 284, 4749–4753 (2009).
Cohen, S. I. et al. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA 110, 9758–9763 (2013).
Larson, M. E. & Lesne, S. E. Soluble Abeta oligomer production and toxicity. J Neurochem, 125–139 (2012).
Cheng, I. H. et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 282, 23818–23828 (2007).
Ladiwala, A. R. A. et al. Conformational differences between two amyloid beta oligomers of similar size and dissimilar toxicity. Journal of Biological Chemistry (2012).
Ono, K., Condron, M. M. & Teplow, D. B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci USA 106, 14745–14750 (2009).
Jang, H. et al. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc Natl Acad Sci USA 107, 6538–6543 (2010).
Reddy, G., Straub, J. E. & Thirumalai, D. Influence of Preformed Asp23−Lys28 Salt Bridge on the Conformational Fluctuations of Monomers and Dimers of Aβ Peptides with Implications for Rates of Fibril Formation. The Journal of Physical Chemistry B 113, 1162–1172 (2009).
Larini, L. & Shea, J.-E. Role of beta-Hairpin Formation in Aggregation: The Self-Assembly of the Amyloid-beta(25–35) Peptide. Biophysical Journal 103, 576–586 (2012).
Hoyer, W., Grönwall, C., Jonsson, A., Ståhl, S. & Härd, T. Stabilization of beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc Natl Acad Sci USA 105, 5099–5104 (2008).
Sandberg, A. et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc Natl Acad Sci USA 107, 15595–15600 (2010).
Hou, L. et al. Solution NMR studies of the A beta(1-40) and A beta(1-42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 126, 1992–2005 (2004).
Mitternacht, S., Staneva, I., Hard, T. & Irback, A. Comparing the folding free-energy landscapes of Abeta42 variants with different aggregation properties. Proteins 78, 2600–2608 (2010).
Lazo, N. D., Grant, M. A., Condron, M. C., Rigby, A. C. & Teplow, D. B. On the nucleation of amyloid beta-protein monomer folding. Protein Sci 14, 1581–1596 (2005).
Ahmed, M. et al. Structural conversion of neurotoxic amyloid-[beta]1-42 oligomers to fibrils. Nat Struct Mol Biol 17, 561–567 (2010).
Fandrich, M. Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J Mol Biol 421, 427–440 (2012).
Nagel-Steger, L., Owen, M. C. & Strodel, B. An Account of Amyloid Oligomers: Facts and Figures Obtained from Experiments and Simulations. Chembiochem 17, 657–676 (2016).
Gall, T. L., Romero, P. R., Cortese, M. S., Uversky, V. N. & Dunker, A. K. Intrinsic disorder in the protein data bank. Journal of Biomolecular structure and dynamics 24, 325–341 (2007).
Lin, Y. S. & Pande, V. S. Effects of familial mutations on the monomer structure of Abeta42. Biophys J 103, L47–49 (2012).
Esteras-Chopo, A. et al. A molecular dynamics study of the interaction of D-peptide amyloid inhibitors with their target sequence reveals a potential inhibitory pharmacophore conformation. J Mol Biol 383, 266–280 (2008).
Ganguly, D. & Chen, J. Modulation of the disordered conformational ensembles of the p53 transactivation domain by cancer-associated mutations. PLoS Comput Biol 11, e1004247 (2015).
Best, R. B., Zheng, W. & Mittal, J. Balanced protein–water interactions improve properties of disordered proteins and non-specific protein association. Journal of chemical theory and computation 10, 5113–5124 (2014).
Das, P., Kang, S.-g., Temple, S. & Belfort, G. Interaction of Amyloid Inhibitor Proteins with Amyloid Beta Peptides: Insight from Molecular Dynamics Simulations. PLoS ONE 9, e113041 (2014).
Vitalis, A. & Caflisch, A. Micelle-like architecture of the monomer ensemble of Alzheimer’s amyloid-beta peptide in aqueous solution and its implications for Abeta aggregation. J Mol Biol 403, 148–165 (2010).
Ono, K., Condron, M. M. & Teplow, D. B. Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid beta-protein assembly and toxicity. Journal of Biological Chemistry 285, 23186–23197 (2010).
Di Fede, G. et al. A Recessive Mutation in the APP Gene with Dominant-Negative Effect on Amyloidogenesis. Science 323, 1473–1477 (2009).
Jonsson, T. et al. A mutation in APP protects against Alzheimer/‘s disease and age-related cognitive decline. Nature 488, 96–99 (2012).
Messa, M. et al. The peculiar role of the A2V mutation in amyloid-beta (Abeta) 1–42 molecular assembly. Journal of Biological Chemistry (2014).
Benilova, I. et al. The Alzheimer Disease Protective Mutation Ala2Thr Modulates Kinetic and Thermodynamic Properties of Abeta Aggregation. Journal of Biological Chemistry (2014).
Zheng, X., Liu, D., Roychaudhuri, R., Teplow, D. B. & Bowers, M. T. Amyloid β-Protein Assembly: Differential Effects of the Protective A2T Mutation and Recessive A2V Familial Alzheimer’s Disease Mutation. ACS chemical neuroscience 6, 1732–1740 (2015).
Meisl, G., Yang, X., Frohm, B., Knowles, T. P. & Linse, S. Quantitative analysis of intrinsic and extrinsic factors in the aggregation mechanism of Alzheimer-associated Abeta-peptide. Sci Rep 6, 18728 (2016).
Murray, B. et al. A2T and A2V Abeta peptides exhibit different aggregation kinetics, primary nucleation, morphology, structure, and LTP inhibition. Proteins 84, 488–500 (2016).
Das, P., Murray, B. & Belfort, G. Alzheimer’s protective A2T mutation changes the conformational landscape of the Abeta1-42 monomer differently than does the A2V mutation. Biophys J 108, 738–747 (2015).
Das, P., Chacko, A. R. & Belfort, G. Alzheimer’s Protective Cross-Interaction between Wild-Type and A2T Variants Alters Aβ42 Dimer Structure. ACS Chemical Neuroscience (2016).
Nguyen, P. H., Sterpone, F., Campanera, J. M., Nasica-Labouze, J. & Derreumaux, P. Impact of the A2V mutation on the Heterozygous and Homozygous Aβ1-40 Dimer Structures from Atomistic Simulations. ACS Chemical Neuroscience (2016).
Di Fede, G. et al. Good gene, bad gene: New APP variant may be both. Progress in neurobiology 99, 281–292 (2012).
Di Fede, G. et al. Tackling amyloidogenesis in Alzheimer’s disease with A2V variants of Amyloid-β. Scientific reports 6 (2016).
Rosenman, D. J., Connors, C. R., Chen, W., Wang, C. & Garcia, A. E. Abeta Monomers Transiently Sample Oligomer and Fibril-Like Configurations: Ensemble Characterization Using a Combined MD/NMR Approach. J Mol Biol 425, 3338–3359 (2013).
Nedumpully-Govindan, P. & Ding, F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci Rep 5, 8240 (2015).
Levine, Z. A., Larini, L., LaPointe, N. E., Feinstein, S. C. & Shea, J. E. Regulation and aggregation of intrinsically disordered peptides. Proc Natl Acad Sci USA 112, 2758–2763 (2015).
Nag, S. et al. Nature of the Amyloid-beta Monomer and the Monomer-Oligomer Equilibrium. Journal of Biological Chemistry 286, 13827–13833 (2011).
Carballo-Pacheco, M. & Strodel, B. Comparison of force fields for Alzheimer’s A beta42: A case study for intrinsically disordered proteins. Protein Sci 26, 174–185 (2017).
Mao, A. H., Lyle, N. & Pappu, R. V. Describing sequence-ensemble relationships for intrinsically disordered proteins. Biochem J 449, 307–318 (2013).
Dima, R. I. & Thirumalai, D. Asymmetry in the shapes of folded and denatured states of proteins. J Phys Chem B, 6564-6570 (2004).
Ultsch, M. et al. Structure of Crenezumab Complex with Abeta Shows Loss of beta-Hairpin. Sci Rep 6, 39374 (2016).
Kollman, P. A. et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33, 889–897 (2000).
Olubiyi, O. O. & Strodel, B. Structures of the amyloid beta-peptides Abeta1-40 and Abeta1-42 as influenced by pH and a D-peptide. J Phys Chem B 116, 3280–3291 (2012).
Maloney, J. A. et al. Molecular Mechanisms of Alzheimer’s Disease Protection by the A673T Allele of Amyloid Precursor Protein. Journal of Biological Chemistry (2014).
Hashimoto, Y. & Matsuoka, M. A mutation protective against Alzheimer’s disease renders amyloid β precursor protein incapable of mediating neurotoxicity. Journal of Neurochemistry 130, 291–300 (2014).
Lin, T. W. et al. Alzheimer’s amyloid-beta A2T variant and its N-terminal peptides inhibit amyloid-beta fibrillization and rescue the induced cytotoxicity. PLoS One 12, e0174561 (2017).
Boehr, D. D., Nussinov, R. & Wright, P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 5, 789–796 (2009).
Tompa, P. & Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci 33, 2–8 (2008).
Shoemaker, B. A., Portman, J. J. & Wolynes, P. G. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci USA 97, 8868–8873 (2000).
Tompa, P., Szasz, C. & Buday, L. Structural disorder throws new light on moonlighting. Trends Biochem Sci 30, 484–489 (2005).
Zago, W. et al. Neutralization of soluble, synaptotoxic amyloid beta species by antibodies is epitope specific. The Journal of Neuroscience 32, 2696–2702 (2012).
Urbanc, B. et al. Structural Basis for Abeta1-42 Toxicity Inhibition by Abeta C-Terminal Fragments: Discrete Molecular Dynamics Study. Journal of Molecular Biology 410, 316–328 (2011).
Barz, B., Olubiyi, O. O. & Strodel, B. Early amyloid [small beta]-protein aggregation precedes conformational change. Chemical Communications 50, 5373–5375 (2014).
Ball, K. A., Phillips, A. H., Wemmer, D. E. & Head-Gordon, T. Differences in beta-strand Populations of Monomeric A beta 40 and A beta 42. Biophysical Journal 104, 2714–2724 (2013).
Barz, B. & Urbanc, B. Dimer Formation Enhances Structural Differences between Amyloid β-Protein (1–40) and (1–42): An Explicit-Solvent Molecular Dynamics Study. PLoS ONE 7, e34345 (2012).
Paivio, A., Nordling, E., Kallberg, Y., Thyberg, J. & Johansson, J. Stabilization of discordant helices in amyloid fibril-forming proteins. Protein Sci 13, 1251–1259 (2004).
Nerelius, C. et al. Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proc Natl Acad Sci USA 106, 9191–9196 (2009).
Ladiwala, A. R. et al. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J Biol Chem 285, 24228–24237 (2010).
Ehrnhoefer, D. E. et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 15, 558–566 (2008).
Qiao, Q., Bowman, G. R. & Huang, X. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J Am Chem Soc 135, 16092–16101 (2013).
Mirecka, E. A. et al. beta-Hairpin of Islet Amyloid Polypeptide Bound to an Aggregation Inhibitor. Sci Rep 6, 33474 (2016).
Hoop, C. L. et al. Huntingtin exon 1 fibrils feature an interdigitated beta-hairpin-based polyglutamine core. Proc Natl Acad Sci USA 113, 1546–1551 (2016).
Chen, M., Tsai, M., Zheng, W. & Wolynes, P. G. The Aggregation Free Energy Landscapes of Polyglutamine Repeats. J Am Chem Soc 138, 15197–15203 (2016).
Salveson, P. J., Spencer, R. K. & Nowick, J. S. X-ray Crystallographic Structure of Oligomers Formed by a Toxic beta-Hairpin Derived from alpha-Synuclein: Trimers and Higher-Order Oligomers. J Am Chem Soc 138, 4458–4467 (2016).
Plant, L. D., Boyle, J. P., Smith, I. F., Peers, C. & Pearson, H. A. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci 23, 5531–5535 (2003).
Kumar, D. K. et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8, 340ra372 (2016).
Tomaselli, S. et al. The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1-42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding. Chembiochem 7, 257–267 (2006).
Frid, P., Anisimov, S. V. & Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res Rev 53, 135–160 (2007).
Tjernberg, L. O. et al. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J Biol Chem 271, 8545–8548 (1996).
Zheng, X. et al. Mechanism of C-Terminal Fragments of Amyloid beta-Protein as Abeta Inhibitors: Do C-Terminal Interactions Play a Key Role in Their Inhibitory Activity? J Phys Chem B 120, 1615–1623 (2016).
Li, H., Rahimi, F. & Bitan, G. Modulation of Amyloid beta-Protein (Abeta) Assembly by Homologous C-Terminal Fragments as a Strategy for Inhibiting Abeta Toxicity. ACS Chem Neurosci 7, 845–856 (2016).
Jorgensen, W. L., Maxwell, D. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on confromational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79, 926–935 (1983).
Acknowledgements
The authors acknowledge the support from IBM BlueGene Science Program. The authors also thank Anita Chacko for contributions to initial stages of this project.
Author information
Authors and Affiliations
Contributions
P.D. designed study; P.D. performed simulations; S.C. and P.D. analyzed simulations; S.C. and P.D. wrote manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chakraborty, S., Das, P. Emergence of Alternative Structures in Amyloid Beta 1-42 Monomeric Landscape by N-terminal Hexapeptide Amyloid Inhibitors. Sci Rep 7, 9941 (2017). https://doi.org/10.1038/s41598-017-10212-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10212-5
This article is cited by
-
Effect of ambient polycyclic aromatic hydrocarbons and nicotine on the structure of Aβ42 protein
Frontiers of Environmental Science & Engineering (2023)
-
Molecular dynamics simulation-based understanding of the structure and property of amyloid proteins at multiple length scales
JMST Advances (2023)
-
Long-range Regulation of Partially Folded Amyloidogenic Peptides
Scientific Reports (2020)
-
Computer Simulation of Protein Materials at Multiple Length Scales: From Single Proteins to Protein Assemblies
Multiscale Science and Engineering (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
