
Abstract
Growing evidence supports the hypothesis that type 2 diabetes (T2D) increases the risk of developing dementia. Experimental evidence from mouse models demonstrates that the induction of T2D/insulin resistance (IR) can promote the accumulation of Alzheimer’s disease (AD) pathological features. However, the association of T2D with pathological and clinical phenotypes in humans is unclear. Here we investigate the relationship of indices of IR (HOMA-IR) and pancreatic β-cell function (HOMA-B) with cognitive performance across several domains (Verbal/Visual Episodic Memory, Executive Function, Language and a measure of Global cognition) and AD biomarkers (CSF Aβ42, T-tau/P-tau, hippocampal volume and neocortical Aβ-amyloid burden). We reveal that HOMA-IR (p < 0.001) incrementally increases across diagnostic groups, becoming significantly elevated in the AD group compared with cognitively normal (CN) adults. In CN adults, higher HOMA-IR was associated with poorer performance on measures of verbal episodic memory (p = 0.010), executive function (p = 0.046) and global cognition (p = 0.007), as well as with higher CSF T-tau (p = 0.008) and P-tau (p = 0.014) levels. No association was observed with CSF Aβ or imaging modalities. Together our data suggest that IR may contribute to reduced cognitive performance and the accumulation of CSF tau biomarkers in cognitively normal adults.
Similar content being viewed by others


Introduction
Epidemiological studies indicate that Type 2 diabetes (T2D) is associated with an increased risk of dementia1,2,3,4,5. Clinical studies using cross-sectional designs support this association by showing that cognition is worse in patients with T2D as compared to matched controls without T2D6, 7. Furthermore, studies of structural magnetic resonance imaging (MRI) show that in T2D cognitive impairment is associated with greater levels of vascular lesions as well as with brain atrophy6,7,8,9,10. Prospective MRI studies also show that in T2D brain atrophy occurs at faster rates than in normal ageing11, 12, suggesting that T2D accelerates neurodegeneration.
Animal studies provide additional evidence to show that inducing T2D/insulin resistance (IR) can promote the pathological changes characteristic of Alzheimer’s disease (AD), specifically accumulation of amyloid-beta (Aβ) and tau (see review ref. 13). These studies also implicate common inflammatory or oxidative stress pathways that link these two chronic diseases of ageing (see review ref. 14). However, the association between cognition and AD pathology in human studies and the stage of AD progression where IR has greater impact remains unclear. Evidence to date using Positron emission tomography (PET) studies have so far been inconclusive.
In AD, PET studies of cerebral glucose metabolism (18F-deoxyglucose PET: FDG-PET) and Aβ deposition (e.g. C11-Pittsburg compound B-PET: PiB-PET) show that reduced neuronal glucose metabolism and increased levels of neocortical Aβ accumulation are features that occur early in the disease (see review ref. 15). A small number of cohort studies have investigated the relationship between these imaging markers of AD and T2D. A cross-sectional study in the population-based Mayo Clinic Study of Ageing assessed cerebral glucose metabolism (FDG-PET) and Aβ deposition (PiB-PET) in healthy older and cognitively normal (CN) adults and older people with T2D. The study showed that compared to the controls, those with T2D displayed cerebral hypometabolism, particularly in those regions severely affected in AD, but no differences in neocortical amyloid load16. In the Baltimore, longitudinal study of Ageing (BLSA), no association was observed between measures of peripheral IR or glucose tolerance and neocortical Aβ load (in a PiB-PET scanned cohort) or other pathological features of AD in post-mortem brain17. More recently in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) study, no association was shown betweenT2D and accumulation of neocortical Aβ load (PiB-PET) or increases in CSF Aβ4210. Instead, T2D was associated with lower cortical thickness an increase in CSF total tau (T-Tau) and phosphorylated tau (P-tau). Together, these findings suggest that IR/T2D is not associated with cerebral accumulation of Aβ but with other hallmarks of the disease. However, in a recent cross-sectional study, Aβ deposition was associated with a higher Homeostatic Model Assessment of IR (HOMA-IR) in late-middle aged, normoglycaemic cognitively normal participants18. The conflicting results in the current literature may be in part due to demographic differences in populations (e.g. age, clinical staging of disease and disease progression) and study design. Further, recent meta-analyses suggest that sex can also mediate T2D associations with dementias and associated co-morbidities, such as stroke19,20,21. Thus, the relationship between IR and clinical and pathological features of the early stages of AD, and sex specific effects, requires further study, particularly in normoglycaemic individuals and prior to the onset of cognitive impairment.
In addition to IR, β-cell hyperactivity and dysfunction and subsequent hyperinsulinemia also contribute to hyperglycaemia and T2D (see review ref. 22). Further, recent evidence indicates β-cell dysfunction in AD rodent models23, 24 and that Aβ and Tau have been shown to accumulate in human post-mortem pancreatic tissue in T2D25, possibly contributing to β-cell dysfunction. Despite this evidence, there is a lack of literature investigating pancreatic β-cell activity (HOMA-B) on cognition and AD related pathology.
The overall aim of this study was to investigate if assessments of IR (HOMA-IR) or pancreatic β-cell function (HOMA-B) are altered across diagnostic groups and ascertain their associations with pathological and clinical expressions of AD in the well characterised Australian Imaging Biomarker and Lifestyle (AIBL) study. We hypothesised that HOMA-IR and HOMA-B are altered across diagnostic groups and are associated with poorer cognitive performance and higher burdens of neuroimaging/CSF biomarker load in cognitively normal participants.
Results
Clinical and cognitive descriptive data for the diagnostic groups [cognitively normal adults (CN), mild cognitive impairment (MCI) and AD] are presented in Tables 1 and 2, respectively. A significant difference in age and frequency of the Apolipoprotein E (APOE) ε4 allele was observed across all diagnostic groups (Table 1), with AD and MCI being older and having a significantly higher APOE ε4 frequency compared to CN (p < 0.001). Significant differences between groups for pathological features and cognitive measures confirmed a clear differentiation between diagnostic groups (Table 2).
Relationships between T2D markers and clinical diagnosis
Serum glucose levels were increased significantly in AD and MCI compared with CN (p = 0.014) (AD > MCI > CN), and a non-significant trend was observed for serum insulin HOMA-IR, but not for HOMA-B (Table 1). These group differences in the HOMA indices became statistically significant after co-varying for BMI, sex, diabetes, use of diabetes medication, smoking, age and APOE genotype. HOMA-IR (Fig. 1A) was significantly increased across diagnostic groups (ANCOVA, F = 8.656, p < 0.001) with post-hoc analysis showing a significant increase in HOMA-IR in the AD group (Bonferroni p < 0.001) compared to CN. A clinical group difference was also observed for HOMA-B (Fig. 1B; F = 4.564, p = 0.011), though between the MCI and CN groups (Bonferroni p = 0.028). In both cases, significant differences were only observed in females (HOMA-IR, F = 6.989 p = 0.001; HOMA-B, F = 5.575 p = 0.004) but not males (HOMA-IR, F = 2.603 p = 0.075; HOMA-B, F = 0.911 p = 0.403). In females HOMA-IR was still only observed to be different in the AD group (Bonferroni p = 0.001) compared to CN, whilst HOMA-B, was significantly lower in CN compared to both the MCI (Bonferroni p = 0.041) and AD (Bonferroni p = 0.017) groups.

HOMA-IR (A) and HOMA-B (B) at baseline within the clinical classifications of AIBL: *HOMA-IR and HOMA-B represented as Estimated Marginal Means ( ± SEM) of Box-Cox transformed raw data. Univariate analysis was performed covarying for BMI, sex, %diabetes, %diabetes medication, %hypertension, smoking, age and APOE ε4 (HOMA-IR, F = 8.656, p < 0.001; HOMA-B, F = 4.564, p = 0.011). Presented p-values are calculated using Post-hoc Bonferoni analysis.
HOMA-IR is associated with cognitive performance and CSF biomarkers
A significant inverse relationships were observed (Table 3) between HOMA-IR and the cognitive composites, verbal episodic memory (β = −0.65, p = 0.010), executive functioning (β = −0.48, p = 0.046) and global composite (β = −0.68, p = 0.007). Significant positive relationships were also observed with CSF T-tau (β = 830.2, p = 0.008) and P-tau (β = 95.9, p = 0.014). Increases in HOMA-B were only observed to be associated with reductions in executive functioning (β = −0.095, p = 0.044) and the global composite (β = −0.011, p = 0.043). Stratification by sex (Table 4, female; Table 5
, male) revealed that prior significant associations held between HOMA-IR and verbal episodic memory (β = −0.63, p = 0.046), executive function (β = −0.61, p = 0.042), the global cognitive composite (β = −0.79, p = 0.014) and both CSF T-tau (β = 639.5, p = 0.048) and P-Tau (β = 93.4, p = 0.031) in females. No significant associations were observed for either HOMA-IR or HOMA-B in males.
Discussion
Our findings demonstrate modest yet significant differences, in both HOMA-IR and HOMA-B, between the clinical classifications of AD, MCI and CN within the AIBL cohort, after covarying for potential confounding variables. Within clinical classifications, compared to the CN group, the AD group had higher HOMA-IR. These findings are consistent with previous reports showing that the prevalence of IR is greater in MCI/AD patients than controls26,27,28. However, in the current study HOMA-B was increased only in the MCI group, suggesting an increase in β-cell function/insulin secretion in this group, an observation consistent with the observed trend towards increasing absolute insulin levels across groups. This may represent a response to control increasing glucose levels during disease progression29, which was also observed in this study. Overall, these initial findings suggested that increased β -cell function/insulin secretion is associated with cognitive impairment, at least in non-demented adults. However, in the absence of overt cognitive impairment (i.e. CN group), compared to HOMA-IR, HOMA-B had no or weak associations with functioning of cognitive domains. This suggests that changes in insulin sensitivity is the stronger, earlier contributor to impairments in cognition. Longitudinal analysis in the cognitively normal that do or do not show clinical disease progression may provide further support for this notion.
We also observed sex differences in levels of HOMA-IR and HOMA-B between diagnostic groups, where upon stratification by sex, the increases observed in the MCI or AD groups were only observed in females. These findings are consistent with outcomes from meta-analyses which indicate women with T2D are at higher risk of stroke and dementia compared to men with T2D19,20,21 and may be a consequence of several factors. For example, studies in different ethnic groups have suggested that age related increases in the prevalence of metabolic syndrome are greater in women then in men (see review ref. 30). Similarly, impaired glucose tolerance has been reported to be more prevalent in older women than men, although impaired fasting glycaemia more prevalent in men31. Further, changes in hormonal status also contribute to an age-associated increased prevalence of metabolic syndrome in women32 and may be driven by androgen/oestrogen imbalances during the peri-menopausal period33. For example reduction in the neuroprotective effects of oestrogen or increases in gonadotropins may act to promote AD related pathological changes (see review ref. 34). With longitudinal study, the interactions between biological markers of IR and indices of AD related biology, and its clinical expression, would provide evidence to confirm our findings that these differences are stronger in woman than men, whilst also providing an indication of the potential contributions of hormones.
Having shown that increases in IR were associated with clinical stages of AD, we wished to ascertain the associations of HOMA-IR and HOMA-B with pathological and clinical expressions of AD prior to the onset of clinical cognitive impairment. In the CN group, greater HOMA-IR was associated with reductions in global cognition and in two of the four cognitive domains assessed, namely verbal episodic memory and executive functioning. A weaker association was observed between HOMA-B and executive function and global cognition. Our findings are in agreement with previous studies showing similar associations of cognitive functioning with HOMA-IR in cognitively normal individuals from community-based volunteer35, population based36 or family based37 studies. Episodic memory is one of the earliest cognitive domains that show changes in the pre-clinical stages of AD. Previous studies have observed episodic memory changes occurring 4–8 years prior to executive function and up to 10-years prior to changes in other cognitive domains38,39,40. Compared to non-diabetics, verbal memory and executive functioning are also strongly affected in T2D41, 42. The association of IR with episodic memory suggests that it plays a role at early preclinical stages of the disease process.
Stratification by sex revealed these associations were again present in females only. In previous studies, there have been varied findings with respect to the effect of sex on the association between HOMA-IR and cognitive functioning. In a cross-sectional study of cognitively healthy elderly community volunteers, higher HOMA-IR was associated with lower verbal fluency performance and reduced grey matter in the temporal lobe of both men and women35. However, in a large, family based Dutch study of 1898 participants, higher HOMA-IR was associated with poorer executive functioning in women only37. A similar association with verbal fluency in women only was shown recently in a population based study of ~6000 adults36. This association was independent of a marker of glucose levels (glycated haemoglobin; HbA1c), indicating that IR may have early effects on cognition and maybe detected prior to development of significant hyperglycaemia and T2D36. Further, in the same study HOMA-IR was associated with poorer verbal fluency in APOE ε4 non-carriers only. Several factors could account for these mixed results including age, cohort characteristics (family vs population vs community based) and the size of the cohort. For example, those studies that show associations in women only36, 37 the analysis was undertaken in larger family or population based cohorts or were younger (~45 yrs. and 53 yrs., respectively) compared to our study and that of Benedict and colleagues35 (71 and 73 yrs., respectively).
IR was also associated with biomarkers of AD pathology within the cognitively normal group where increases in HOMA-IR were associated with increases in CSF T-tau and P-tau. No such relationships were observed with neocortical Aβ burden or CSF-Aβ42 in these groups. These findings suggest that, at earlier stages of the disease process there is a stronger relationship with tau rather than Aβ. Cross-sectional cohort studies evaluating imaging modalities (structural MRI, PET) or AD biomarkers have shown similar results. In these studies, lower cortical thickness and cerebral hypometabolism, but not changes in Aβ were associated with T2D8, 16, 17. More recently, in the ADNI study, T2D-related reduction in cortical thickness was associated with increases in CSF P-tau, but not with neocortical Aβ burden or CSF Aβ42, across diagnostic groups of CN, MCI, and AD10. Our study was not powered to similarly stratify for T2D as only a small proportion (~8%) of the total cohort had the condition. The association of HOMA-IR with increases in CSF tau, even after controlling for T2D, suggests that IR is an early contributor to this process. However, it is acknowledged that this is a cross-sectional analysis and a longitudinal follow-up is required to further establish IR as a determinant of cognitive decline and accumulation of AD pathological features.
Numerous studies in various animal models have shown that diet (HF diet/sucrose/fructose) or chemical (administration of the β-cell toxin streptozotocin (STZ)) induced diabetes promote, tau phosphorylation, and synaptic/neurodegeneration23, 43,44,45,46,47. The potential mechanisms may involve neuroinflammatory and oxidative stress mechanisms14. In addition, impaired cerebral insulin signalling, promoted by IR and T2D, plays a key role in this process and in triggering tau hyper phosphorylation through increased activity of GSK-3β, a major protein kinase that phosphorylates tau (see review ref. 13). These in vivo, animal studies also support a role for T2D/IR in promoting Aβ build up in the brain, where availability of Aβ degrading enzymes such as the insulin degrading enzyme is reduced, inflammatory and oxidative processes promoting Aβ accumulation and formation of toxic oligomers further exacerbating synaptic degeneration (see review ref. 13). As discussed above the findings from the current study and others10, 16, 17 do not fully support this notion in human studies, but do not rule out the possibility that IR is associated with early deposition of Aβ.
Two recent studies have shown increases in HOMA-IR associated with increases in Aβ18, 48. The study by Willette et al.18, showed that higher HOMA-IR was associated with higher amyloid load (as assessed by PiB-PET) in normoglycaemic participants recruited from the Wisconsin Registry for Alzheimer’s disease Prevention (WRAP). The study performed analysis of HOMA-IR with PiB-PET and did not include CSF Aβ42, or CSF T-tau/P-tau levels. The more recent study by Hoscheidt et al.48 assessed participants with a parental history of dementia due to AD recruited from a similar cohort, the Wisconsin Alzheimer’s Disease Research Centre (ADRC), Investigating memory in people at risk, causes and treatments (IMPACT). The study investigated the association of HOMA-IR with CSF AD related biomarkers including Aβ42, CSF sAPPα/sAPPβ and CSF P-tau and showed that increases in HOMA-IR was associated with increased CSF levels of sAPPβ (not sAPPα), and modest association with CSF Aβ42 only. These findings were consistent with IR promoting amyloidogenic processing of APP (i.e. increased sAPPβ) and thus Aβ42 formation. Whilst associations between HOMA-IR and neocortical Aβ burden were not assessed in the latter study, the findings were suggestive of being consistent with that previously observed18.
There are number of factors that may account for the differences in the findings in our cohort and the Wisconsin cohorts. Age may have a major role in determining where in the progression of AD pathology, IR has the greatest impact. The participants in the Wisconsin studies were younger (57.7 yrs49, and 60 yrs18) compared to our study cohort (CN average age 70 yrs.) and other cohorts where no associations with Aβ-amyloid was reported (~75 yrs.10 and ~79 yrs.17). Therefore, it is possible that an earlier relationship occurs between IR and Aβ-amyloid burden, but in older and presymptomatic individuals a relationship with Tau exists. Longitudinal studies that incorporate Aβ-amyloid imaging, tau imaging and CSF Aβ/tau biomarkers are required to map out where IR fits into the progression of the disease. However, both relationships may be useful in the possible use of IR as a factor that contributes to predicting the conversion to cognitive impairment from pre-symptomatic stages.
It is also worth noting that the Wisconsin cohorts had a high percentage of participants with a family history of dementia due to AD, and in the Hoescheidt et al. study49, analysis was only performed on those with a family history. In addition, 47% of the cohort were APOE ε4 carriers compared to 26.8% in this study and CSF sAPPβ and P-tau/Aβ42 were higher in APOE ε4 carriers. The Willette et al.18 study also had a higher percentage of APOE ε4 carriers (38.7%). The carriage of APOE ε4 is associated with greater CSF Aβ42 and Aβ-amyloid plaque load50,51,52,53, a strong predictor of AD pathology and cognitive decline54, 55. The apoE4 isoform is also thought to have a greater impact on Aβ accumulation, through impairing clearance/degradation56,57,58,59,60, but also has been associated with the dysregulation of APP processing to promote Aβ production61, 62. These characteristics of the Wisconsin cohorts may also explain the greater association of HOMA-IR with Aβ-amyloid pathology compared to what we observe in the AIBL cohort.
Our findings, in CN adults, indicate that increases in IR are associated with reductions in cognition function and elevations in CSF tau. This and other studies suggest that IR may contribute to AD progression, although it is possible that it is AD pathology that gives rise to metabolic dysfunction. For example, evidence from AD post-mortem studies shows brain regions involved in regulating brain and systemic energy metabolism, such as the hypothalamus contain Aβ plaques and Tau tangles (see review ref. 63). Further, FDG-PET studies show lowered hypothalamic glucose metabolism in MCI or AD than in age matched controls. Additionally, animal studies suggest that peripheral Aβ may drive IR and glucose dysregulation and that tau may be important in maintaining glucose homeostasis (see review ref. 64). Our findings that IR is associated with preclinical reductions in cognitive performance and increased CSF tau levels contribute to this understanding of the relationship between IR and AD. These data are also consistent with those from a recent cross-sectional study from the ADNI cohort which observed that T2D-related brain atrophy was associated with increases in CSF P-tau in CN, MCI and AD groups and which led to the conclusion that T2D may promote neurodegeneration independent of AD dementia diagnosis10. Our observed associations of increasing IR with reductions in cognition and increases in CSF T-tau and P-tau in CN adults suggests IR may also lead directly to changes in cognition and tau accumulation in early AD. Longitudinal analysis, particularly in controls/MCI participants that convert may provide some insight into the direction of this association. Ideally, these studies would be complemented with PET imaging of relevant brain regions including the hypothalamus. Although, FDG-PET studies have shown reduced glucose utilisation in the AD/MCI hypothalamus65, 66 studies with Aβ or tau tracers have not been forthcoming, potentially due to the small size of the hypothalamus, limiting spatial resolution, and off-target binding, which may increase the difficulty in assessing this brain region.
There are some limitations to our findings. Firstly, there is the potential that observed findings are currently sample dependent for several reasons; participants were volunteers and not randomly selected from the community, therefore the findings of this study may only be applicable to similar cohorts and the study used cognitive composites derived from specific cognitive assessments, and the use of similar composites derived from different cognitive assessments may result in different findings. Second, the analysis is cross-sectional and informs that there is an association of HOMA indices with cognitive functioning. The findings merit longitudinal analysis to determine if; (1) insulin resistance is associated with the progression of AD pathology and at what stages this relationship is observed (i.e. amyloid at early stages/ tau at later stages) and (2) the contribution of IR/T2D to predicting cognitive impairment. The HOMA indices are calculated from fasting glucose and insulin levels. Although commonly used in association studies, particularly large cohort studies, a closer measure of insulin sensitivity is utilising the euglycaemic-hyperinsulinaemic clamp to quantify insulin secretion and assess metabolism of glucose. Given that this method requires to be done in a clinical setting it would not be feasible in large cohort studies, but once confirmation of a role for IR in the progression of the disease/conversion to cognitive impairment/AD pathology, studies could be undertaken in a small subset to tease out potential mechanisms. As the focus was associations with IR, our study variables did not include cardiovascular or atherosclerotic factors (i.e. cholesterol, triglycerides), however recent studies investigating the relationship of IR with atherosclerotic factors in AD/MCI patients show that increased insulin/decreased response to insulin are independent predictors for AD and MCI67.
We have demonstrated that in the AIBL cohort increases in HOMA-IR are associated with reductions in cognitive domains that show early changes in AD with concomitant increases in CSF tau levels. Longitudinal analysis in this and additional cohorts that incorporate tau imaging would be requried to confirm this association with accumnulation of pathological changes. A similar analysis in individuals at risk will also determine if there are earlier associations with Aβ burden and if T2D contributes to the conversion of pre-sympotmatic to early cogntive impairment (MCI) or MCI to AD. As early IR, prior to significant hyperglycaemia, is modifiable this work has implications in identifying and assessing strategies to reduce/prevent the onset cognitive decline and pathology associated with T2D.
Materials and Methods
Participants
The study reports on data collected from the AIBL study, a prospective longitudinal study of ageing. This cross-sectional study presents baseline data from 1264 participants (905 cognitively normal (CN), 156 mild cognitive impaired (MCI), 203 Alzheimer’s disease (AD)). Information regarding AIBL study design, enrolment process, neuropsychological assessments and diagnostic criteria has been previously described68. Ethics approval for the AIBL study and all experimental protocols was provided by the ethics committees of Austin Health, St Vincent’s Health, Hollywood Private Hospital and Edith Cowan University. All experiments and methods were carried out in accordance with the approved guidelines and regulations and all volunteers gave written informed consent before participating in the study.
Cognitive Measures
The neuropsychological test battery administered in the AIBL study has been described in detail previously68. This included the Mini-Mental State examination (MMSE), Clock drawing test, California Verbal Learning Test- Second edition (CVLT-II), Logical Memory I and II (LMI; LMI; Story A only), D-KEFS verbal fluency, a 30-item version of the Boston Naming Test (BNT), Wechsler Test of Adult Reading (WTAR), Digit Span and Digit Symbol-Coding subtests of the Wechsler Adult Intelligence Scale-Third edition (WAIS-III), the Stroop task (Victoria version), and the Rey Complex Figure Test (RCFT). Cognitive measures are presented as five domain specific composite scores, derived from data collected from the neuropsychological battery as previously published by69. The five domain composites included in this study were; Verbal Episodic Memory (VEM; CDR sum of boxes (CDRSB), LMII CVLT false positives (CVLTFP) and long delay free recall (CVLTLDFR)), Visual Episodic Memory (ViEM; CDRSB, RCFT; 3 and 30 minute delayed recall, and recognition hits (RCFT3DR, RCFT30DR and RCFTHits)), Executive Functioning (EF; CDRSB, Stroop, Verbal Fluency Task (FAS), Category Switching Total (CatSwTot)), Language (LANG; CDRSB, Category Fluency (CatFl), BNT) and a statistically driven global composite (CDRSB, MMSE, LMII, CVLTFP and Clock), the latter aimed as a sensitive measure for longitudinal decline in individuals predisposed to AD69. All composites were calculated with a correction for age, sex, years of education, premorbid IQ (WAIS-III Full Scale Intelligence Quotient (FSIQ) and depressive symptoms (Geriatric Depression Scale (GDS))69.
Biochemistry, Genotyping and HOMA estimations
Baseline fasted blood samples were taken and fractionated, with one aliquot sent to clinical pathology laboratories in Perth and Melbourne, as described previously68. As part of the clinical pathology assessment, fasting plasma insulin (FPI) and fasting plasma glucose levels (FPG) were assessed. The stated reference ranges are the ranges established in the clinical pathology laboratory in accordance with the national guidelines (http://www.nata.asn.au/, http://www.health.gov.au/npaac). The FPI and FPG were used to calculate values of Homeostatic modelling assessment (HOMA). The HOMA model is often used in cross-sectional and longitudinal studies to estimate insulin sensitivity and pancreatic β-cell functioning as alternatives to more direct assessments such as glucose clamping or acute insulin response, which are not practical in large cohort studies (see review ref. 70). Initially, comparisons of two methods of HOMA was performed; the HOMA1 (“original method”) using equations developed by Mathews and colleagues71 and the HOMA2 “the computer model”) developed by Levy and colleagues72. These models and the differences between them have been extensively discussed elsewhere70. HOMA1 was calculated using the following: HOMA-IR = (FPI x FPG)/22.5; HOMA-B (%) = (20x FPI)/(FPG = 3.5) to estimate IR and β-cell functioning respectively. HOMA2 was calculated from a Microsoft Excel macro accessed via the Oxford University website (www.dtu.ox.ac.uk/homacalculator/). Pearson’s correlation analysis of the data generated from HOMA1 and HOMA2 analysis revealed a strong significant correlation between these indices (HOMA1-IR vs HOMA2-IR, r = 0.976, p < 0.001; HOMA1-B vs HOMA2 B, r = 0.717, p < 0.0001), indicating suitability of both methods for this data set. However, HOMA1 was utilised as it is the most commonly used method in large cohort cross-sectional or longitudinal studies. All reference to HOMA in this report reflects HOMA1 calculated indices.
DNA was extracted and APOE genotype determined previously described73. Briefly, QIAamp DNA Blood Maxi Kits (Qiagen, Hilden, Germany) were used per manufacturer’s instructions to extract from whole blood. APOE genotype was determined from two separate TaqMan® (Thermo Fisher Scientific, Waltham, MA) genotyping assays for the single nucleotide polymoprhisms rs7412 (assay ID: C____904973_10) and rs429358 (assay ID: C___3084793_20). TaqMan® genotyping assays were performed on a QuantStudio 12 K Flex™ Real-Time-PCR systems (Thermo Fisher Scientific, Waltham, MA) using the TaqMan® GTXpress™ Master Mix (Thermo Fisher Scientific, Waltham, MA) methodology as per manufacturer instructions. APOE carrier status was defined by the presence (1 or 2 copies; ε4 + ) or absence (0 copies; ε4−) of the APOE ε4 allele.
CSF collection and Aβ42, T-tau, and P-tau181P quantitation were performed as previously described74. Briefly, approximately 10–14 ml of CSF was collected in the morning by routine lumbar puncture after overnight fasting directly into one 15 ml polypropylene tube (Greiner Bio-One188271), employing a protocol like that recommended by the Alzheimer’s Biomarkers Standardization Initiative75. All CSF samples for analysis were taken from aliquots prepared and stored as previously described74 and thawed at time of assay. All CSF samples were analyzed in duplicate using the enzyme-linked immunosorbent assay (ELISA): INNOTEST Aβ-amyloid (1–42; Aβ42), INNOTEST hTAU Ag (T-tau), and INNOTEST Phospho-tau (P-tau; 181 P; P-tau181P) (Innogenetics, Ghent, Belgium) per published standard methods. Mean intra-assay coefficients of variation for these assays are as previously published74. This study reports on data from 66 study participants from whom CSF was taken at the baseline time point of the AIBL study.
Brain Imaging
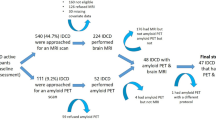
Data was available at baseline from a total of 379 AIBL participants (262 CN, 69 MCI, 48 AD) who underwent Aβ-amyloid imaging with positron emission tomography using either 11C-Pittsburgh Compound B (PiB), 18F-florebetapir or 18F-Flutemetamol as described elsewhere76,77,78. PET standardized uptake value ratios (SUVR) were determined for all tracers using CapAIBL, a web based freely availably MR-less methodology79. Briefly, SUVs were summed and normalized to either the cerebellar cortex SUV (PiB), whole cerebellum SUV (florbetapir, FBP) or pons SUV (flutemetamol, FLUTE) to yield the target-region to reference-region SUVR. To allow for the analysis of tracer specific SUVRs as a single continuous variable, a linear regression transformation, termed the “Before the Centiloid Kernel Transformation” (BeCKeT) scale, was applied to FBP and FLUTE SUVR to generate PiB-like SUVR units80.
Hippocampal Volume data was available for 319 (229 CN, 52 MCI, 38 AD) participants at baseline. Hippocampal volumes were determined through MRI, parameters of which have been previously described81. Briefly, participants underwent T1 weighted MRI using the ADNI 3-dimensional (3D) Magnetization Prepared Rapid Gradient Echo (MPRAGE) sequence on 1.5 T or 3 T scanners. Hippocampal volume was calculated after correcting for age in years and intracranial volume (sum of grey matter, white matter and cerebrospinal fluid volumes), as previously described82.
Statistical analysis
All statistical analysis was conducted using IBM SPSS Statistics (version 23; IBM Corp, Armonk, NY, USA) with the level of significance set to α = 0.05 (two-tailed). All variables were assessed for conformation to a normal distribution. Box-Cox transformations was used to correct variables departing from a normal distribution83. For all variables, except HOMA indices, the calculated lambda (λ) equated to no transformation. HOMA indices underwent transformations (T(Y)) prior to analysis, specifically: T(Y) = ((Y + 1)−0.079−1)/−0.079 and T(Y) = (Y0.04−1)/0.04 for HOMA-IR and HOMA-B, respectively (with Y representing the HOMA index). Analysis of demographic variables was undertaken using a One-way Analysis of Variance (ANOVA) to determine differences in continuous variables across clinical classifications differences in categorical variables determined using the χ2-test. Differences in HOMA indices between clinical classifications were assessed using an analysis of covariance (ANCOVA) via a General Linear Model (GLM) with Bonferroni correction. Associations between HOMA indices and cognitive composites and pathological brain changes in CN were assessed using linear regression analysis. Both ANCOVA and linear regression analyses, for all dependent variables, covaried for body mass index (BMI), diabetes (yes/no), diabetes medication (yes/no), hypertension (yes/no), smoking (yes/no), and APOE genotype (ε4+/ε4−), with sex and age only covaried for with biomarker dependent variables.
Data availability
All data and samples used in this study are derived from the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study of Ageing. All AIBL data, and that specific to this study, is publically accessible to all interested parties through an Expression of Interest procedure and is governed by the AIBL Data Use Agreement, for more information please see https://aibl.csiro.au/awd/.
References
Leibson, C. L. et al. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann NY Acad Sci 826, 422–427 (1997).
Arvanitakis, Z. et al. Diabetes mellitus and progression of rigidity and gait disturbance in older persons. Neurology 63, 996–1001 (2004).
Ott, A. et al. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53, 1937–1942 (1999).
Peila, R., Rodriguez, B. L., Launer, L. J. & Honolulu-Asia Aging, S. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 51, 1256–1262 (2002).
Xu, W. L., Qiu, C. X., Wahlin, A., Winblad, B. & Fratiglioni, L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology 63, 1181–1186 (2004).
Manschot, S. M. et al. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 55, 1106–1113 (2006).
van Harten, B., Oosterman, J. M., Potter van Loon, B. J., Scheltens, P. & Weinstein, H. C. Brain lesions on MRI in elderly patients with type 2 diabetes mellitus. Eur Neurol 57, 70–74, doi:10.1159/000098054 (2007).
Moran, C. et al. Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care 36, 4036–4042, doi:10.2337/dc13-0143 (2013).
Biessels, G. J. & Reijmer, Y. D. Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 63, 2244–2252, doi:10.2337/db14-0348 (2014).
Moran, C. et al. Type 2 diabetes, skin autofluorescence, and brain atrophy. Diabetes 64, 279–283, doi:10.2337/db14-0506 (2015).
Kooistra, M. et al. Diabetes mellitus and progression of vascular brain lesions and brain atrophy in patients with symptomatic atherosclerotic disease. The SMART-MR study. J Neurol Sci 332, 69–74, doi:10.1016/j.jns.2013.06.019 (2013).
van Elderen, S. G. et al. Progression of brain atrophy and cognitive decline in diabetes mellitus: a 3-year follow-up. Neurology 75, 997–1002, doi:10.1212/WNL.0b013e3181f25f06 (2010).
Verdile, G., Fuller, S. J. & Martins, R. N. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis 84, 22–38, doi:10.1016/j.nbd.2015.04.008 (2015).
Verdile, G. et al. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediators Inflamm 2015, 105828, doi:10.1155/2015/105828 (2015).
Cohen, A. D. & Klunk, W. E. Early detection of Alzheimer’s disease using PiB and FDG PET. Neurobiol Dis 72 Pt A, 117–122, doi:10.1016/j.nbd.2014.05.001 (2014).
Roberts, R. O. et al. Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation. J Nucl Med 55, 759–764, doi:10.2967/jnumed.113.132647 (2014).
Thambisetty, M. et al. Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol 70, 1167–1172, doi:10.1001/jamaneurol.2013.284 (2013).
Willette, A. A. et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement 11, 504–510, e501, doi:10.1016/j.jalz.2014.03.011 (2015).
Huxley, R., Barzi, F. & Woodward, M. Excess risk of fatal coronary heart disease associated with diabetes in men and women: meta-analysis of 37 prospective cohort studies. BMJ 332, 73–78, doi:10.1136/bmj.38678.389583.7C (2006).
Chatterjee, S. et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared With Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 39, 300–307, doi:10.2337/dc15-1588 (2016).
Peters, S. A., Huxley, R. R. & Woodward, M. Diabetes as a risk factor for stroke in women compared with men: a systematic review and meta-analysis of 64 cohorts, including 775,385 individuals and 12,539 strokes. Lancet 383, 1973–1980, doi:10.1016/S0140-6736(14)60040-4 (2014).
Keane, K. N., Cruzat, V. F., Carlessi, R., de Bittencourt, P. I. Jr. & Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and beta-Cell Dysfunction. Oxid Med Cell Longev 2015, 181643, doi:10.1155/2015/181643 (2015).
Vandal, M. et al. Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes 63, 4291–4301, doi:10.2337/db14-0375 (2014).
Park, S., Kim, D. S., Kang, S. & Moon, N. R. beta-Amyloid-induced cognitive dysfunction impairs glucose homeostasis by increasing insulin resistance and decreasing beta-cell mass in non-diabetic and diabetic rats. Metabolism 62, 1749–1760, doi:10.1016/j.metabol.2013.08.007 (2013).
Miklossy, J. et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol Aging 31, 1503–1515, doi:10.1016/j.neurobiolaging.2008.08.019 (2010).
Craft, S. et al. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol Aging 17, 123–130 (1996).
Craft, S. et al. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: relationship to severity of dementia and apolipoprotein E genotype. Neurology 50, 164–168 (1998).
Reger, M. A. et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis 13, 323–331 (2008).
Morris, J. K., Vidoni, E. D., Honea, R. A. & Burns, J. M. & Alzheimer’s Disease Neuroimaging, I. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol Aging 35, 585–589, doi:10.1016/j.neurobiolaging.2013.09.033 (2014).
Pucci, G. et al. Sex- and gender-related prevalence, cardiovascular risk and therapeutic approach in metabolic syndrome: A review of the literature. Pharmacol Res 120, 34–42, doi:10.1016/j.phrs.2017.03.008 (2017).
Unwin, N., Shaw, J., Zimmet, P. & Alberti, K. G. Impaired glucose tolerance and impaired fasting glycaemia: the current status on definition and intervention. Diabet Med 19, 708–723 (2002).
Janssen, I., Powell, L. H., Crawford, S., Lasley, B. & Sutton-Tyrrell, K. Menopause and the metabolic syndrome: the Study of Women’s Health Across the Nation. Arch Intern Med 168, 1568–1575, doi:10.1001/archinte.168.14.1568 (2008).
Lovejoy, J. C., Champagne, C. M., de Jonge, L., Xie, H. & Smith, S. R. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int J Obes (Lond) 32, 949–958, doi:10.1038/ijo.2008.25 (2008).
Pike, C. J. Sex and the development of Alzheimer’s disease. J Neurosci Res 95, 671–680, doi:10.1002/jnr.23827 (2017).
Benedict, C. et al. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes Care 35, 488–494, doi:10.2337/dc11-2075 (2012).
Ekblad, L. L. et al. Insulin resistance is associated with poorer verbal fluency performance in women. Diabetologia 58, 2545–2553, doi:10.1007/s00125-015-3715-4 (2015).
Schuur, M. et al. Insulin-resistance and metabolic syndrome are related to executive function in women in a large family-based study. Eur J Epidemiol 25, 561–568, doi:10.1007/s10654-010-9476-y (2010).
Elias, M. F. et al. The preclinical phase of alzheimer disease: A 22-year prospective study of the Framingham Cohort. Arch Neurol 57, 808–813 (2000).
Grober, E. et al. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. J Int Neuropsychol Soc 14, 266–278, doi:10.1017/S1355617708080302 (2008).
Derby, C. A. et al. Screening for predementia AD: time-dependent operating characteristics of episodic memory tests. Neurology 80, 1307–1314, doi:10.1212/WNL.0b013e31828ab2c9 (2013).
Palta, P., Schneider, A. L., Biessels, G. J., Touradji, P. & Hill-Briggs, F. Magnitude of cognitive dysfunction in adults with type 2 diabetes: a meta-analysis of six cognitive domains and the most frequently reported neuropsychological tests within domains. J Int Neuropsychol Soc 20, 278–291, doi:10.1017/S1355617713001483 (2014).
Vincent, C. & Hall, P. A. Executive Function in Adults With Type 2 Diabetes: A Meta-Analytic Review. Psychosom Med 77, 631–642, doi:10.1097/PSY.0000000000000103 (2015).
Mehla, J., Chauhan, B. C. & Chauhan, N. B. Experimental induction of type 2 diabetes in aging-accelerated mice triggered Alzheimer-like pathology and memory deficits. J Alzheimers Dis 39, 145–162, doi:10.3233/JAD-131238 (2014).
Knight, E. M., Martins, I. V., Gumusgoz, S., Allan, S. M. & Lawrence, C. B. High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging 35, 1821–1832, doi:10.1016/j.neurobiolaging.2014.02.010 (2014).
Takeda, S. et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA 107, 7036–7041, doi:10.1073/pnas.1000645107 (2010).
Wang, Y. et al. Synergistic exacerbation of mitochondrial and synaptic dysfunction and resultant learning and memory deficit in a mouse model of diabetic Alzheimer’s disease. J Alzheimers Dis 43, 451–463, doi:10.3233/JAD-140972 (2015).
Ho, L. et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J 18, 902–904, doi:10.1096/fj.03-0978fje (2004).
Hoscheidt, S. M. et al. Insulin Resistance is Associated with Increased Levels of Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease and Reduced Memory Function in At-Risk Healthy Middle-Aged Adults. J Alzheimers Dis 52, 1373–1383, doi:10.3233/JAD-160110 (2016).
Hoscheidt, S. M. et al. Insulin resistance is associated with lower arterial blood flow and reduced cortical perfusion in cognitively asymptomatic middle-aged adults. J Cereb Blood Flow Metab. doi:10.1177/0271678X16663214 (2016).
Reiman, E. M. et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA 106, 6820–6825, doi:10.1073/pnas.0900345106 (2009).
Castellano, J. M. et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3, 89ra57, doi:10.1126/scitranslmed.3002156 (2011).
Bales, K. R. et al. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci 29, 6771–6779, doi:10.1523/JNEUROSCI.0887-09.2009 (2009).
Morris, J. C. et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67, 122–131, doi:10.1002/ana.21843 (2010).
Lim, Y. Y. et al. Abeta amyloid, cognition, and APOE genotype in healthy older adults. Alzheimer’s & dementia: the journal of the Alzheimer’s Association 9, 538–545, doi:10.1016/j.jalz.2012.07.004 (2013).
Lim, Y. Y. et al. APOE epsilon4 moderates amyloid-related memory decline in preclinical Alzheimer’s disease. Neurobiol Aging 36, 1239–1244, doi:10.1016/j.neurobiolaging.2014.12.008 (2015).
LaDu, M. J. et al. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem 269, 23403–23406 (1994).
Yang, D. S. et al. Apolipoprotein E promotes the binding and uptake of beta-amyloid into Chinese hamster ovary cells in an isoform-specific manner. Neuroscience 90, 1217–1226 (1999).
Hone, E. et al. Alzheimer’s disease amyloid-beta peptide modulates apolipoprotein E isoform specific receptor binding. J Alzheimers Dis 7, 303–314 (2005).
Deane, R. et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest 118, 4002–4013, doi:10.1172/JCI36663 (2008).
Sharman, M. J. et al. APOE genotype results in differential effects on the peripheral clearance of amyloid-beta42 in APOE knock-in and knock-out mice. J Alzheimers Dis 21, 403–409, doi:10.3233/JAD-2010-100141 (2010).
Irizarry, M. C. et al. Apolipoprotein E modulates gamma-secretase cleavage of the amyloid precursor protein. J Neurochem 90, 1132–1143, doi:10.1111/j.1471-4159.2004.02581.x (2004).
Hass, S., Weidemann, A., Utermann, G. & Baier, G. Intracellular apolipoprotein E affects Amyloid Precursor Protein processing and amyloid Abeta production in COS-1 cells. Mol Genet Genomics 265, 791–800 (2001).
Ishii, M. & Iadecola, C. Metabolic and Non-Cognitive Manifestations of Alzheimer’s Disease: The Hypothalamus as Both Culprit and Target of Pathology. Cell Metab 22, 761–776, doi:10.1016/j.cmet.2015.08.016 (2015).
Bharadwaj, P. et al. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-beta, Amylin, and Tau Proteins. J Alzheimers Dis, doi:10.3233/JAD-161192 (2017).
Nestor, P. J., Fryer, T. D., Smielewski, P. & Hodges, J. R. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Ann Neurol 54, 343–351, doi:10.1002/ana.10669 (2003).
Cross, D. J. et al. Loss of olfactory tract integrity affects cortical metabolism in the brain and olfactory regions in aging and mild cognitive impairment. J Nucl Med 54, 1278–1284, doi:10.2967/jnumed.112.116558 (2013).
Macesic, M. et al. Impaired Insulin Sensitivity And Secretion In Patients With Alzheimer’s Disease: The Relationship With Other Atherosclerosis Risk Factors. Curr Vasc Pharmacol (2016).
Ellis, K. A. et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 21, 672–687, doi:10.1017/S1041610209009405 (2009).
Burnham, S. C. et al. Novel Statistically-Derived Composite Measures for Assessing the Efficacy of Disease-Modifying Therapies in Prodromal Alzheimer’s Disease Trials: An AIBL Study. J Alzheimers Dis 46, 1079–1089, doi:10.3233/JAD-143015 (2015).
Wallace, T. M., Levy, J. C. & Matthews, D. R. Use and abuse of HOMA modeling. Diabetes Care 27, 1487–1495 (2004).
Matthews, D. R. et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985).
Levy, J. C., Matthews, D. R. & Hermans, M. P. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21, 2191–2192 (1998).
Brown, B. M. et al. Influence of BDNF Val66Met on the relationship between physical activity and brain volume. Neurology 83, 1345–1352, doi:10.1212/WNL.0000000000000867 (2014).
Li, Q. X. et al. Alzheimer’s Disease Normative Cerebrospinal Fluid Biomarkers Validated in PET Amyloid-beta Characterized Subjects from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study. J Alzheimers Dis 48, 175–187, doi:10.3233/JAD-150247 (2015).
Vanderstichele, H. et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: a consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement 8, 65–73, doi:10.1016/j.jalz.2011.07.004 (2012).
Rowe, C. C. et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31, 1275–1283, doi:10.1016/j.neurobiolaging.2010.04.007 (2010).
Clark, C. M. et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 305, 275–283, doi:10.1001/jama.2010.2008 (2011).
Vandenberghe, R. et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: a phase 2 trial. Ann Neurol 68, 319–329, doi:10.1002/ana.22068 (2010).
Bourgeat, P. et al. Comparison of MR-less PiB SUVR quantification methods. Neurobiol Aging 36(Suppl 1), S159–166, doi:10.1016/j.neurobiolaging.2014.04.033 (2015).
Villemagne, V. et al. En Attendant Centiloid. Advances in Research 2, 723–729 (2014).
Bourgeat, P. et al. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology 74, 121–127, doi:10.1212/WNL.0b013e3181c918b5 (2010).
Dore, V. et al. Cross-sectional and longitudinal analysis of the relationship between Abeta deposition, cortical thickness, and memory in cognitively unimpaired individuals and in Alzheimer disease. JAMA Neurol 70, 903–911, doi:10.1001/jamaneurol.2013.1062 (2013).
Box, G. E. P. & Cox, D. R. An Analysis of Transformations. J Roy Stat Soc B 26, 211–252 (1964).
Acknowledgements
Funding for the AIBL study was provided in part by the study partners [Commonwealth Scientific Industrial and research Organization (CSIRO), Edith Cowan University (ECU), Mental Health Research institute (MHRI), National Ageing Research Institute (NARI), Austin Health, CogState Ltd.]. The AIBL study has also received support from the National Health and Medical Research Council (NHMRC) and the Dementia Collaborative Research Centres program (DCRC2), as well as funding from the Science and Industry Endowment Fund (SIEF) and the Cooperative Research Centre (CRC) for Mental Health – funded through the CRC Program (Grant ID:20100104), an Australian Government Initiative. GV is supported by the Curtin Senior Resarch Fellowship (CRF140196), West Australian Department of Health Merit Award and the National Health and Medical Research Council (APP1105698) awarded to G.V, P.N., P.F. and R.M. We thank all those who took part as subjects in the study for their commitment and dedication to helping advance research into the early detection and causation of AD. We kindly thank all AIBL Research Group members (http://aibl.csiro.au/about/aibl-research-team/).
Author information
Authors and Affiliations
Contributions
S.M.L., G.V. designed the study, collated, analysed and interpreted data and drafted the manuscript; S.G., A.W. helped collate data and draft the manuscript; T.P., V.D., Q.X.L., helped collate data and reviewed the manuscript; S.B. provided data analysis and interpretation and reviewed the manuscript; V.L.V. helped collate data, interpret analysis and reviewed the manuscript; V.S., D.B., P.F., P.N., N.W., P.M., C.L.M., S.R.S., C.C.R., O.S. and R.N.M. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing Interests
CLM is an advisor to Prana Biotechnology Ltd and a consultant to Eli Lilly. PM is a full-time employee of Cogstate Ltd. DA has served on scientific advisory boards for Novartis, Eli Lilly, Janssen, and Pfizer Inc. RNM is a consultant to Alzhyme. SML has previously been a paid consultant to Alzhyme. CCR has served on scientific advisory boards for Bayer Pharma, Elan Corporation, GE Healthcare and AstraZeneca; has received speaker honoraria from Bayer Pharma and GE Healthcare; and has received research support from Bayer Pharma, GE Healthcare, Piramal Lifesciences and Avid Radiopharmaceuticals. VLV served as a consultant for Bayer Pharma; and received research support from a NEDO grant from Japan. All other authors have nothing to disclose.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Laws, S.M., Gaskin, S., Woodfield, A. et al. Insulin resistance is associated with reductions in specific cognitive domains and increases in CSF tau in cognitively normal adults. Sci Rep 7, 9766 (2017). https://doi.org/10.1038/s41598-017-09577-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09577-4
This article is cited by
-
The relationship of insulin resistance and diabetes to tau PET SUVR in middle-aged to older adults
Alzheimer's Research & Therapy (2023)
-
Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: a randomized trial
Scientific Reports (2022)
-
Network activity changes in the pathophysiology of Alzheimer’s disease: the role of aging and early entorhinal cortex dysfunction
Metabolic Brain Disease (2022)
-
Intranasal Insulin for Alzheimer’s Disease
CNS Drugs (2021)
-
A Phase II, Single-Center, Randomized, Double-Blind, Placebo-Controlled Study of the Safety and Therapeutic Efficacy of Intranasal Glulisine in Amnestic Mild Cognitive Impairment and Probable Mild Alzheimer’s Disease
Drugs & Aging (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
